Optimizing Hydroxyurea Treatment for Sickle Cell Disease Patients: The Pharmacokinetic Approach

 and
and
Abstract
1. Introduction
2. Experimental Section
2.1. Patients
2.2. Study Design
2.3. GC-MS
2.4. Modelling and Statistical Analysis
2.5. Optimal Sampling
3. Results
3.1. Characteristics of Patients
3.2. Biological Parameters and Self-Reported Compliance
3.3. Pharmacokinetic Parameters
3.4. Optimal Sampling
4. Discussion
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global burden of sickle cell anaemia in children under five, 2010–2050: Modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef]
- Ware, R.E.; Rees, R.C.; Sarnaik, S.A.; Iyer, R.V.; Alvarez, O.A.; Casella, J.F.; Shulkin, B.L.; Shalaby-Rana, E.; Strife, C.F.; Miller, J.H.; et al. Renal function in infants with sickle cell anemia: Baseline data from the BABY HUG trial. J. Pediatr. 2010, 156, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Niss, O.; Quinn, C.T.; Lane, A.; Daily, J.; Khoury, P.R.; Bakeer, N.; Kimball, T.R.; Towbin, J.A.; Malik, P.; Taylor, M.D. Cardiomyopathy with Restrictive Physiology in Sickle Cell Disease. JACC Cardiovasc. Imaging 2016, 9, 243–252. [Google Scholar] [CrossRef]
- Niss, O.; Fleck, R.; Makue, F.; Alsaied, T.; Desai, P.; Towbin, J.A.; Malik, P.; Taylor, M.D.; Quinn, C.T. Association between diffuse myocardial fibrosis and diastolic dysfunction in sickle cell anemia. Blood 2017, 130, 205–213. [Google Scholar] [CrossRef]
- Fitzhugh, C.D.; Lauder, N.; Jonassaint, J.C.; Telen, M.J.; Zhao, X.; Wright, E.C.; Gilliam, F.R.; De Castro, L.M. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. Am. J. Hematol. 2010, 85, 36–40. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Sachdev, V.; Jison, M.L.; Shizukuda, Y.; Plehn, J.F.; Minter, K.; Brown, B.; Coles, W.A.; Nichols, J.S.; Ernst, I.; et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N. Engl. J. Med. 2004, 350, 886–895. [Google Scholar] [CrossRef]
- Kwiatkowski, J.L.; Zimmerman, R.A.; Pollock, A.N.; Seto, W.; Smith-Whitley, K.; Shults, J.; Blackwood-Chirchir, A.; Ohene-Frempong, K. Silent infarcts in young children with sickle cell disease. Br. J. Haematol. 2009, 146, 300–305. [Google Scholar] [CrossRef]
- Ohene-Frempong, K.; Weiner, S.J.; Sleeper, L.A.; Miller, S.T.; Embury, S.; Moohr, J.W.; Wethers, D.L.; Pegelow, C.H.; Gill, F.M. Cerebrovascular accidents in sickle cell disease: Rates and risk factors. Blood 1998, 91, 288–294. [Google Scholar]
- Thornburg, C.D.; Files, B.A.; Luo, Z.; Miller, S.T.; Kalpatthi, R.; Iyer, R.; Seaman, P.; Lebensburger, J.; Alvarez, O.; Thompson, B.; et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood 2012, 120, 4304–4310. [Google Scholar] [CrossRef]
- Wang, W.C.; Ware, R.E.; Miller, S.T.; Iyer, R.V.; Casella, J.F.; Minniti, C.P.; Rana, S.; Thornburg, C.D.; Rogers, Z.R.; Kalpatthi, R.V.; et al. Hydroxycarbamide in very young children with sickle-cell anaemia: A multicentre, randomised, controlled trial (BABY HUG). Lancet 2011, 377, 1663–1672. [Google Scholar] [CrossRef]
- Lopes de Castro Lobo, C.; Pinto, J.F.C.; Nascimento, E.M.; Moura, P.G.; Cardoso, G.P.; Hankins, J.S. The effect of hydroxcarbamide therapy on survival of children with sickle cell disease. Br. J. Haematol. 2013, 161, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Lê, P.Q.; Gulbis, B.; Dedeken, L.; Dupont, S.; Vanderfaeillie, A.; Heijmans, C.; Huybrechts, S.; Devalck, C.; Efira, A.; Dresse, M.-F.; et al. Survival among children and adults with sickle cell disease in Belgium: Benefit from hydroxyurea treatment. Pediatr. Blood Cancer 2015, 62, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- Quarmyne, M.-O.; Dong, W.; Theodore, R.; Anand, S.; Barry, V.; Adisa, O.; Buchanan, I.D.; Bost, J.; Brown, R.C.; Joiner, C.H.; et al. Hydroxyurea effectiveness in children and adolescents with sickle cell anemia: A large retrospective, population-based cohort. Am. J. Hematol. 2017, 92, 77–81. [Google Scholar] [CrossRef]
- Steinberg, M.H.; Barton, F.; Castro, O.; Pegelow, C.H.; Ballas, S.K.; Kutlar, A.; Orringer, E.; Bellevue, R.; Olivieri, N.; Eckman, J.; et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: Risks and benefits up to 9 years of treatment. JAMA 2003, 289, 1645–1651. [Google Scholar] [CrossRef]
- Voskaridou, E.; Christoulas, D.; Bilalis, A.; Plata, E.; Varvagiannis, K.; Stamatopoulos, G.; Sinopoulou, K.; Balassopoulou, A.; Loukopoulos, D.; Terpos, E. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: Results of a 17-year, single-center trial (LaSHS). Blood 2010, 115, 2354–2363. [Google Scholar] [CrossRef]
- Ware, R.E. Optimizing hydroxyurea therapy for sickle cell anemia. Hematol. Am. Soc. Hematol. Educ. Program. 2015, 2015, 436–443. [Google Scholar] [CrossRef]
- Ware, R.E. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010, 115, 5300–5311. [Google Scholar] [CrossRef]
- Zimmerman, S.A.; Schultz, W.H.; Davis, J.S.; Pickens, C.V.; Mortier, N.A.; Howard, T.A.; Ware, R.E. Sustained long-term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease. Blood 2004, 103, 2039–2045. [Google Scholar] [CrossRef]
- Ware, R.E.; Davis, B.R.; Schultz, W.H.; Brown, R.C.; Aygun, B.; Sarnaik, S.; Odame, I.; Fuh, B.; George, A.; Owen, W.; et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): A multicentre, open-label, phase 3, non-inferiority trial. Lancet 2016, 387, 661–670. [Google Scholar] [CrossRef]
- Dong, M.; McGann, P.T.; Mizuno, T.; Ware, R.E.; Vinks, A.A. Development of a pharmacokinetic-guided dose individualization strategy for hydroxyurea treatment in children with sickle cell anaemia. Br. J. Clin. Pharmacol. 2016, 81, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; Helms, R.W. SWiTCH Investigators Stroke with Transfusions Changing to Hydroxyurea (SWiTCH). Blood 2012, 119, 3925–3932. [Google Scholar] [CrossRef] [PubMed]
- Brandow, A.M.; Jirovec, D.L.; Panepinto, J.A. Hydroxyurea in children with sickle cell disease: Practice patterns and barriers to utilization. Am. J. Hematol. 2010, 85, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, C.D.; Calatroni, A.; Telen, M.; Kemper, A.R. Adherence to hydroxyurea therapy in children with sickle cell anemia. J. Pediatr. 2010, 156, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Paule, I.; Sassi, H.; Habibi, A.; Pham, K.P.; Bachir, D.; Galactéros, F.; Girard, P.; Hulin, A.; Tod, M. Population pharmacokinetics and pharmacodynamics of hydroxyurea in sickle cell anemia patients, a basis for optimizing the dosing regimen. Orphanet J. Rare Dis. 2011. [Google Scholar] [CrossRef]
- Hydroxycarbamide—An Overview. Available online: https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/hydroxycarbamide (accessed on 29 Jul 2019).
- Hankins, J.S.; Ware, R.E.; Rogers, Z.R.; Wynn, L.W.; Lane, P.A.; Scott, J.P.; Wang, W.C. Long-term hydroxyurea therapy for infants with sickle cell anemia: The HUSOFT extension study. Blood 2005, 106, 2269–2275. [Google Scholar] [CrossRef]
- Ware, R.E.; Despotovic, J.M.; Mortier, N.A.; Flanagan, J.M.; He, J.; Smeltzer, M.P.; Kimble, A.C.; Aygun, B.; Wu, S.; Howard, T.; et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood 2011, 118, 4985–4991. [Google Scholar] [CrossRef]
- Wiczling, P.; Liem, R.I.; Panepinto, J.A.; Garg, U.; Abdel-Rahman, S.M.; Kearns, G.L.; Neville, K.A. Population pharmacokinetics of hydroxyurea for children and adolescents with sickle cell disease. J. Clin. Pharmacol. 2014, 54, 1016–1022. [Google Scholar] [CrossRef]
- Scott, D.K.; Neville, K.; Garg, U. Determination of hydroxyurea in serum or plasma using gas chromatography-mass spectrometry (GC-MS). Methods Mol. Biol. 2010, 603, 279–287. [Google Scholar]
- Garg, U.; Scott, D.; Frazee, C.; Kearns, G.; Neville, K. Isotope-dilution gas chromatography-mass spectrometry method for the analysis of hydroxyurea. Ther. Drug Monit. 2015, 37, 325–330. [Google Scholar] [CrossRef]
- Marahatta, A.; Megaraj, V.; McGann, P.T.; Ware, R.E.; Setchell, K.D.R. Stable-Isotope Dilution HPLC-Electrospray Ionization Tandem Mass Spectrometry Method for Quantifying Hydroxyurea in Dried Blood Samples. Clin. Chem. 2016, 62, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- McGann, P.T.; Niss, O.; Dong, M.; Marahatta, A.; Howard, T.A.; Mizuno, T.; Lane, A.; Kalfa, T.A.; Malik, P.; Quinn, C.T.; et al. Robust clinical and laboratory response to hydroxyurea using pharmacokinetically guided dosing for young children with sickle cell anemia. Am. J. Hematol. 2019, 94, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Bernaudin, F.; Arnaud, C.; Kamdem, A.; Hau, I.; Lelong, F.; Epaud, R.; Pondarré, C.; Pissard, S. Biological impact of α genes, β haplotypes, and G6PD activity in sickle cell anemia at baseline and with hydroxyurea. Blood Adv. 2018, 2, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Estepp, J.H.; Wiczling, P.; Moen, J.; Kang, G.; Mack, J.M.; Liem, R.; Panepinto, J.A.; Garg, U.; Kearns, G.; Neville, K.A. Hydroxycarbamide in children with sickle cell anaemia after first-dose vs. chronic therapy: Pharmacokinetics and predictive models for drug exposure: Pharmacokinetics of first vs. chronic hydroxycarbamide. Br. J. Clin. Pharmacol. 2018, 84, 1478–1485. [Google Scholar] [CrossRef]
- Leikin, S.L.; Gallagher, D.; Kinney, T.R.; Sloane, D.; Klug, P.; Rida, W. Mortality in children and adolescents with sickle cell disease. Cooperative Study of Sickle Cell Disease. Pediatrics 1989, 84, 500–508. [Google Scholar]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Steinberg, M.H.; Klug, P.P. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef]
- Miller, S.T.; Sleeper, L.A.; Pegelow, C.H.; Enos, L.E.; Wang, W.C.; Weiner, S.J.; Wethers, D.L.; Smith, J.; Kinney, T.R. Prediction of adverse outcomes in children with sickle cell disease. N. Engl. J. Med. 2000, 342, 83–89. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Demographic Characteristics | |
|---|---|
| Sex ratio M/F | 0.8 (4/5) |
| Age | |
| Mean ± standard deviation | 14.4 (±3.7) |
| Median | 16.5 |
| Weight | |
| Mean ± standard deviation | 49.9 (±20.5) |
| Median | 49.1 |
| Background (Number of Patients and Percentage) | |
| Cholecystectomy | 3 (33%) |
| Stroke | 1 (11.1%) |
| Abnormal Transcranial doppler episode | 1 (11.1%) |
| Osteonecrosis | 2 (22.2%) |
| Retinopathy | 1 (11.1%) |
| Splenic Sequestration | 0 (0%) |
| Pulmonary Hypertension | 1 (11.1%) |
| Cardiac Events | 1 (11.1%) |
| Kidney Failure | 0 (0%) |
| Events per Year: 2016–2018 Period | |
| Transfusion/Year | |
| Mean ± Standard Deviation | 0.8 |
| Median | 0.3 (0–2) |
| Hospitalization/Year | |
| Mean ± standard deviation | 1.4 |
| Median (range) | 0.6 (0–5.0) |
| VOC/Year | |
| Mean ± Standard Deviation | 1.6 |
| Median (range) | 1 (0–5.6) |
| ACS | |
| Number of Patients > 1 ACS | 4 (45%) |
| HU | |
| Dose (mg/kg/day) | |
| Mean ± Standard Deviation | 19.0 (±4.0) |
| Median (range) | 20.4 (12.9–24.6) |
| Time since Introduction of HU (Months) | |
| Mean ± Standard Deviation | 63.5 (±44.6) |
| Median (Range) | 58.8 (11.2–138.8) |
| Age at Introduction (Year) | |
| Mean ± Standard Deviation | 8.5 (±4.4) |
| Median (Range) | 6.0 (4.0–16.0) |
| Patient | Self-Reported Compliance | Hb (g/dL) | MCV (fL) | Retic. (G/L) | PNN (G/L) | Platelets (G/L) | HbF(%) | Dose HU (mg/kg/day) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Pre-HU | Post-HU | Pre-HU | Post-HU | |||||||
| 1 | poor | 8.6 | 81.2 | 87.5 | 284.0 | 6.0 | 427 | 3.3 | 7.0 | 17.1 |
| 2 | good | 9.0 | 86.3 | 111.9 | 161.2 | 4.9 | 282 | 8.8 | 20.5 | 20.9 |
| 3 | poor | 7.7 | N/A | 70.2 | 192.7 | 11.5 | 279 | N/A | 3.0 | 20.4 |
| 4 | medium | 7.1 | 83.0 | 95.8 | 242.2 | 14.2 | 81 | 4.8 | 7.9 | 21.4 |
| 5 | medium | 8.3 | 79.0 | 78.1 | 286.7 | 8.0 | 539 | N/A | 1.4 | 21.4 |
| 6 | good | 9.1 | N/A | 94.9 | 185.0 | 5.9 | 232 | N/A | 23.7 | 18.9 |
| 7 | medium | 7.8 | 88.0 | 80.4 | 325.2 | 9.2 | 589 | 5.7 | 7.4 | 24.6 |
| 8 | medium | 7.6 | 78.4 | 95.0 | 177.1 | 3.2 | 290 | 2.5 | 5.6 | 13.0 |
| 9 | good | 7.6 | N/A | 97.8 | 164.7 | 4.9 | 465 | N/A | 14.5 | 12.9 |
| Mean ± SD | 8.1 ± 0.7 | 90.2 ± 12.5 | 224.3 ± 61.5 | 7.5 ± 3.6 | 354 ± 163 | 10.1 ± 7.8 | 19.0 ± 4.0 | |||
| Median | 7.8 | 94.9 | 192.7 | 6.0 | 290.0 | 7.4 | 20.4 | |||
| Patient | Treatment Duration (Months) | Dose (mg/kg/day) | Cmax (mg/L) | Tmax (hours) | AUC (h.mg/L) |
|---|---|---|---|---|---|
| 1 | 14.2 | 17.1 | 24.0 | 1.33 | 75.1 |
| 2 | 121.6 | 20.9 | 33.9 | 1.11 | 113.5 |
| 3 | 27.3 | 20.4 | 14.9 | 2.00 | 59.5 |
| 4 | 58.8 | 21.4 | 15.2 | 2.44 | 57.3 |
| 5 | 138.8 | 21.4 | 37.5 | 1.11 | 102.0 |
| 6 | 51.6 | 18.9 | 25.8 | 0.66 | 74.0 |
| 7 | 70.1 | 24.6 | 31.0 | 2.44 | 108.2 |
| 8 | 11.2 | 13.0 | 10.8 | 1.33 | 43.3 |
| 9 | 77.8 | 12.9 | 24.0 | 1.33 | 98.2 |
| Mean ± Standard Deviation | 63.5 (±44.6) | 19.0 (±4.0) | 24.1 (±9.1) | 1.5 (±0.6) | 81.3 (±25.2) |
| Median | 58.8 | 20.4 | 24.0 | 1.33 | 75.1 |
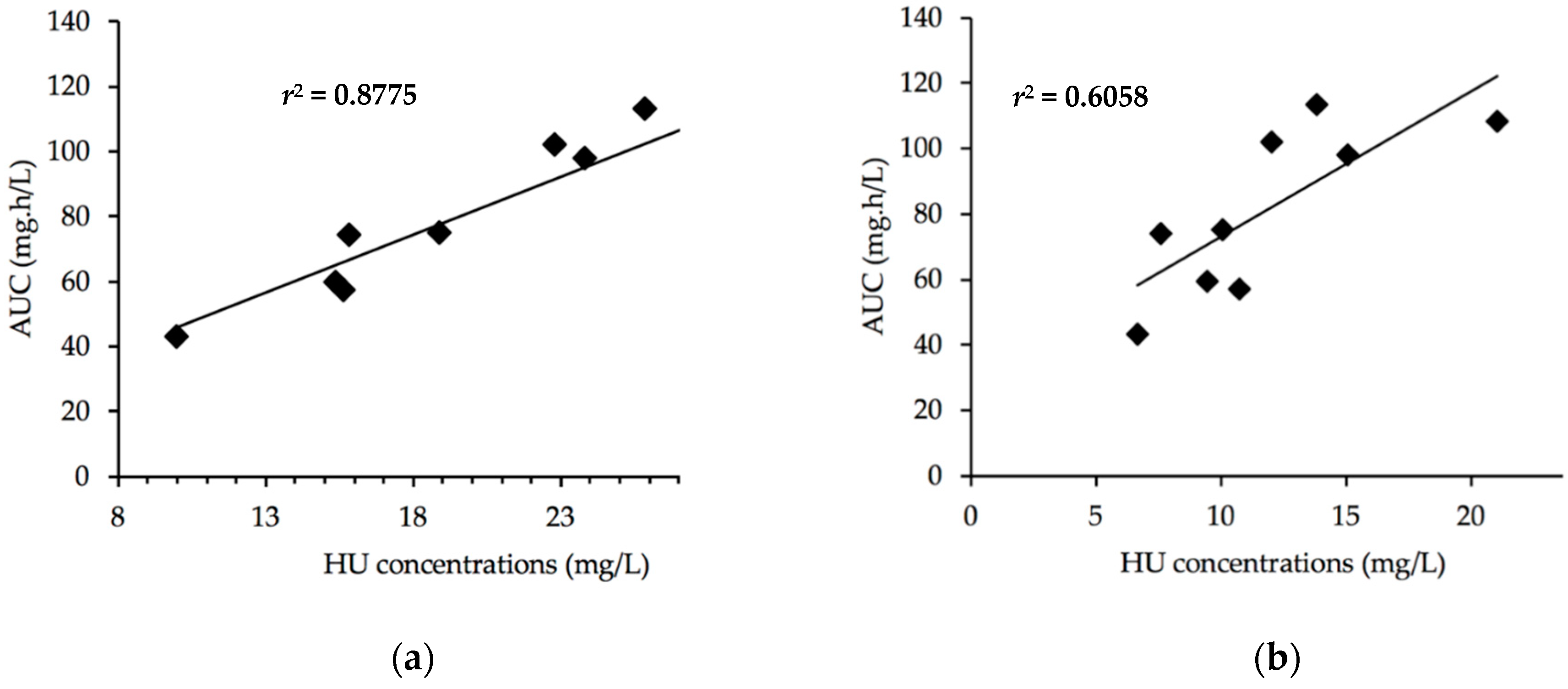
| Sampling Time | Determination Coefficient r2 |
|---|---|
| 0 | 0.0314 |
| 10 min | 0.0146 |
| 20 min | 0.0947 |
| 1 h | 0.3415 |
| 2 h | 0.8775 |
| 4 h | 0.6058 |
| 6 h | 0.4813 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazon, C.; Sabo, A.-N.; Becker, G.; Lessinger, J.-M.; Kemmel, V.; Paillard, C. Optimizing Hydroxyurea Treatment for Sickle Cell Disease Patients: The Pharmacokinetic Approach. J. Clin. Med. 2019, 8, 1701. https://doi.org/10.3390/jcm8101701
Nazon C, Sabo A-N, Becker G, Lessinger J-M, Kemmel V, Paillard C. Optimizing Hydroxyurea Treatment for Sickle Cell Disease Patients: The Pharmacokinetic Approach. Journal of Clinical Medicine. 2019; 8(10):1701. https://doi.org/10.3390/jcm8101701
Chicago/Turabian StyleNazon, Charlotte, Amelia-Naomi Sabo, Guillaume Becker, Jean-Marc Lessinger, Véronique Kemmel, and Catherine Paillard. 2019. "Optimizing Hydroxyurea Treatment for Sickle Cell Disease Patients: The Pharmacokinetic Approach" Journal of Clinical Medicine 8, no. 10: 1701. https://doi.org/10.3390/jcm8101701
APA StyleNazon, C., Sabo, A.-N., Becker, G., Lessinger, J.-M., Kemmel, V., & Paillard, C. (2019). Optimizing Hydroxyurea Treatment for Sickle Cell Disease Patients: The Pharmacokinetic Approach. Journal of Clinical Medicine, 8(10), 1701. https://doi.org/10.3390/jcm8101701