Long Term Follow-Up of 103 Untreated Adult Patients with Type 1 Gaucher Disease




Abstract
1. Introduction
2. Methods
Statistical Analysis
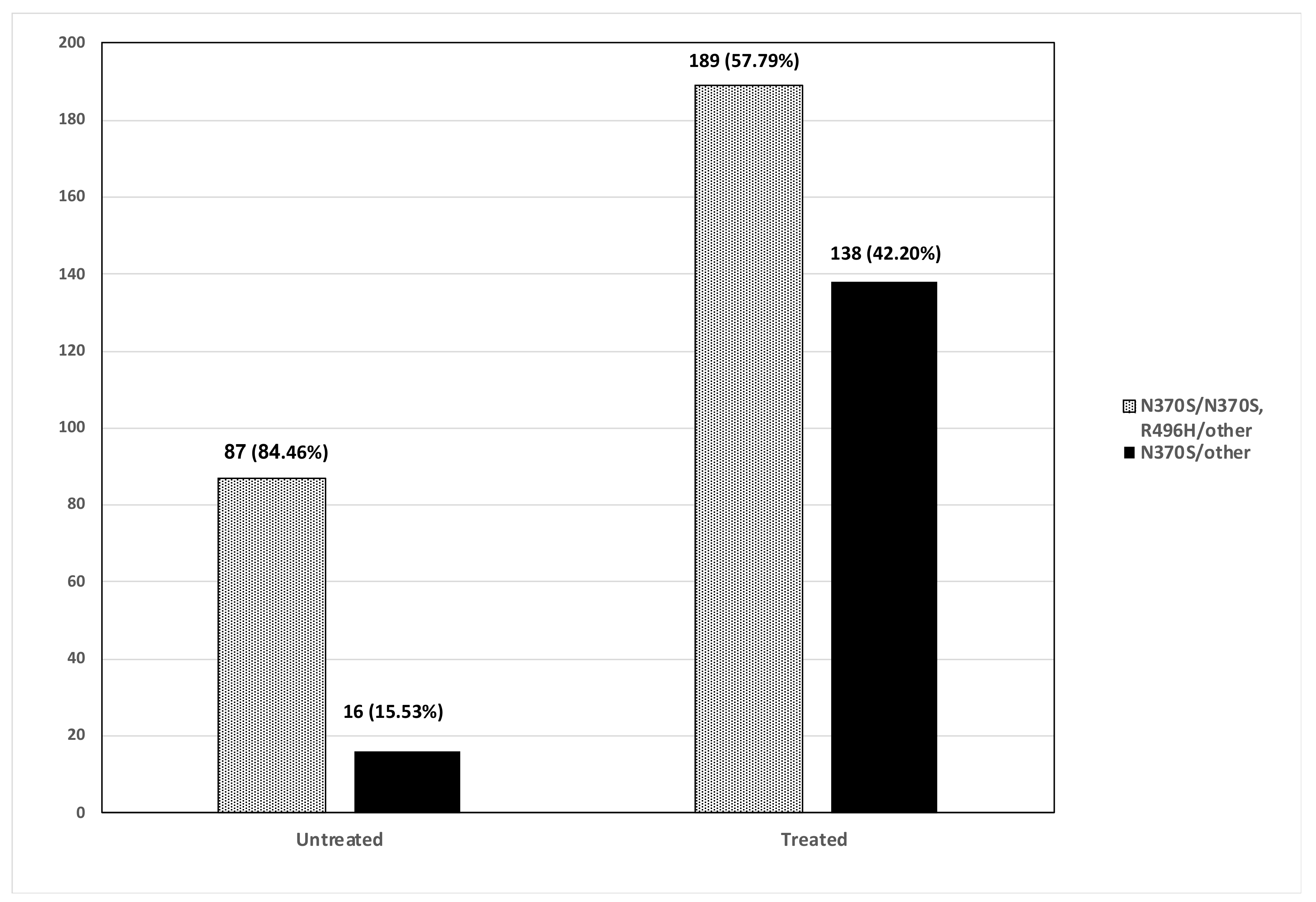
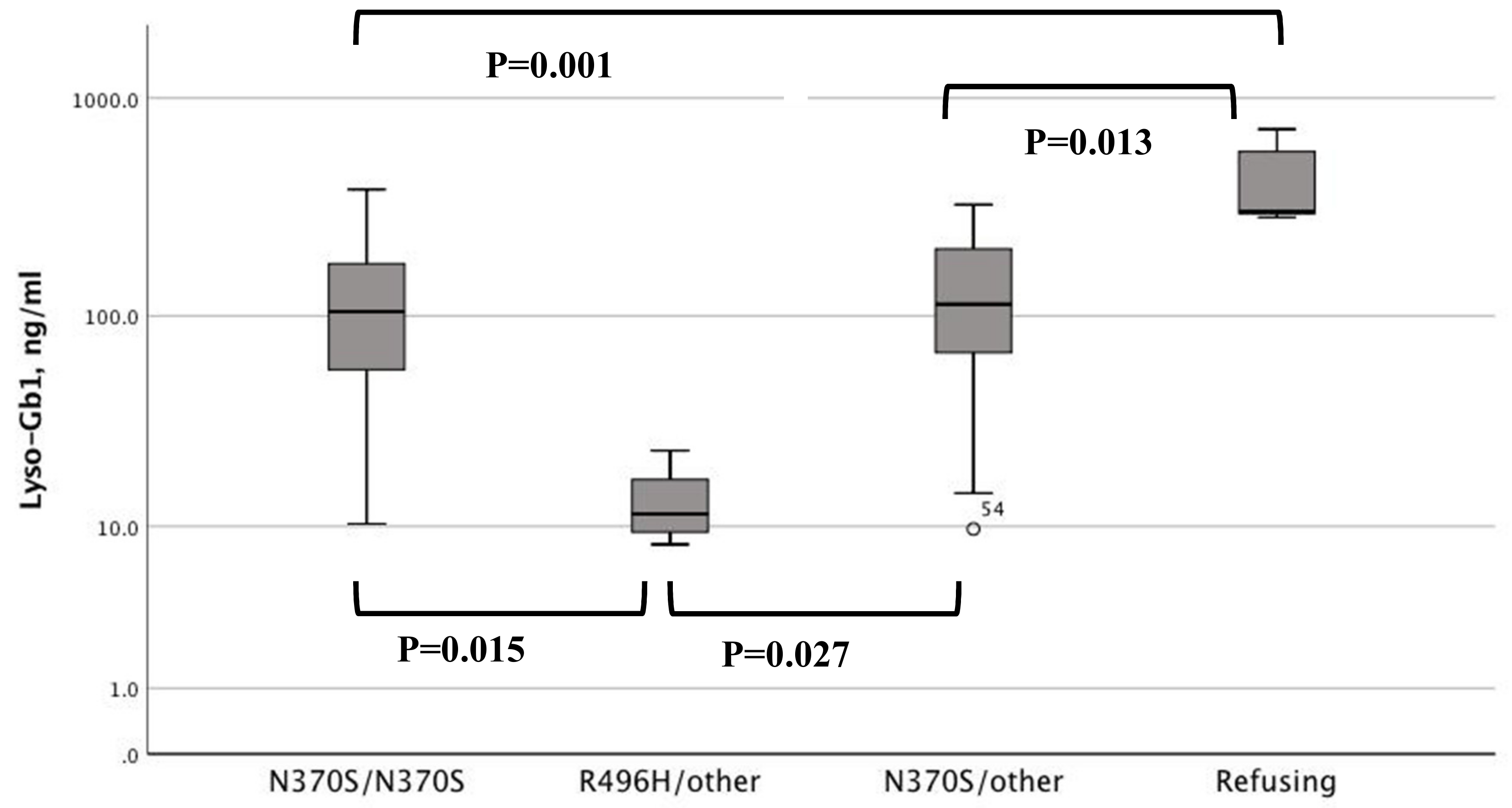
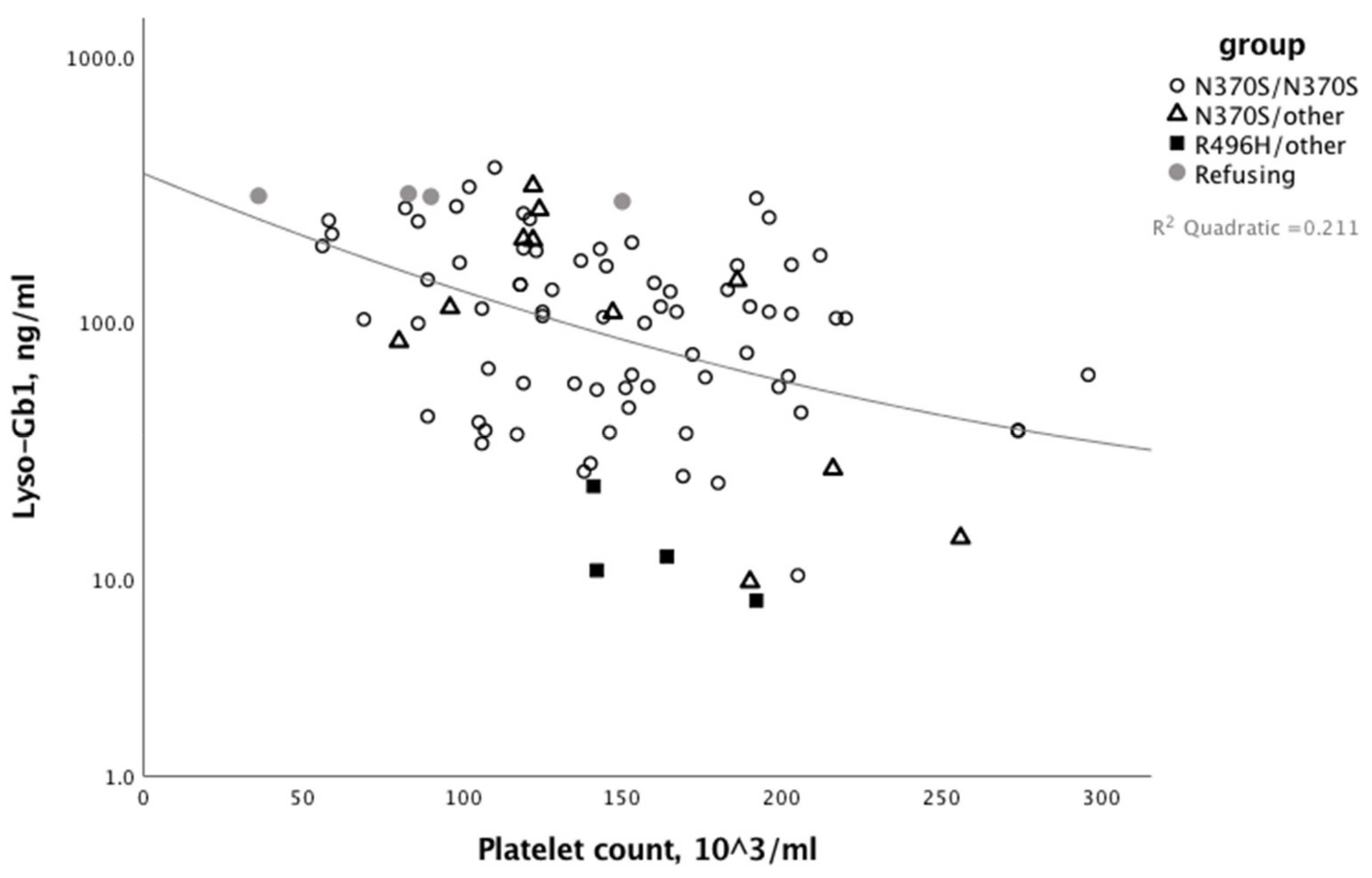
3. Results
4. Discussion
Author Contributions
Conflicts of Interest
References
- Zimran, A.; Elstein, D. Gaucher disease and related Lysosomal Storage Diseases. In Williams’ Hematology, 9th ed.; McGraw-Hill Education: New York, NY, USA, 2016; pp. 1121–1133. [Google Scholar]
- Revel-Vilk, S.; Szer, J.; Mehta, A.; Zimran, A. How we manage Gaucher Disease in the era of choices. Br. J. Haematol. 2018, 182, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement therapy for inherited enzyme deficiency--macrophage-targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Gary, S.E.; Ryan, E.; Steward, A.M.; Sidransky, E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev. Endocrinol. Metab. 2018, 13, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.; Lachmann, R.; Hollak, C.; Aerts, J.; van Weely, S.; Hrebícek, M.; Platt, F.; Butters, T.; Dwek, R.; Moyses, C.; et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000, 355, 1481–1485. [Google Scholar] [CrossRef]
- Elstein, D.; Hollak, C.; Aerts, J.M.; van Weely, S.; Maas, M.; Cox, T.M.; Lachmann, R.H.; Hrebicek, M.; Platt, F.M.; Butters, T.D.; et al. Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J. Inherit. Metab. Dis. 2004, 27, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Hirabayashi, Y. Glucosylceramide synthase and glycosphingolipid synthesis. Trends Cell Biol. 1998, 8, 198–202. [Google Scholar] [CrossRef]
- Peterschmitt, M.J.; Cox, G.F.; Ibrahim, J.; MacDougall, J.; Underhill, L.H.; Patel, P.; Gaemers, S.J.M. A pooled analysis of adverse events in 393 adults with Gaucher disease type 1 from four clinical trials of oral eliglustat: Evaluation of frequency, timing, and duration. Blood Cells Mol. Dis. 2018, 68, 185–191. [Google Scholar] [CrossRef]
- Mistry, P.K.; Lopez, G.; Schiffmann, R.; Barton, N.W.; Weinreb, N.J.; Sidransky, E. Gaucher disease: Progress and ongoing challenges. Mol. Genet. Metab. 2017, 120, 8–21. [Google Scholar] [CrossRef]
- Elstein, D.; Hadas-Halpern, I.; Azuri, Y.; Abrahamov, A.; Bar-Ziv, Y.; Zimran, A. Accuracy of ultrasonography in assessing spleen and liver size in patients with Gaucher disease: Comparison to computed tomographic measurements. J. Ultrasound Med. 1997, 16, 209–211. [Google Scholar] [CrossRef]
- Yetter, E.M.; Acosta, K.B.; Olson, M.C.; Blundell, K. Estimating Splenic Volume: Sonographic Measurements Correlated with Helical CT Determination. Am. J. Roentgenol. 2003, 181, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Badran, D.H.; Kalbouneh, H.M.; Al-Hadidi, M.T.; Shatarat, A.T.; Tarawneh, E.S.; Hadidy, A.M.; Mahafza, W.S. Ultrasonographic assessment of splenic volume and its correlation with body parameters in a Jordanian population. Saudi Med. J. 2015, 36, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Böttcher, T.; Lukas, J.; Hübner, R.; Gölnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef] [PubMed]
- EvaluatePharma-Orphan Drug Report. Available online: https://www.evaluate.com/thought-leadership/pharma/evaluatepharma-orphan-drug-report-2019 (accessed on 11 October 2019).
- Beutler, E. Economic malpractice in the treatment of Gaucher’s disease. Am. J. Med. 1994, 97, 1–2. [Google Scholar] [CrossRef]
- Zimran, A.; Ilan, Y.; Elstein, D. Enzyme replacement therapy for mild patients with Gaucher disease. Am. J. Hematol. 2009, 84, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M. Origin and uses of primum non nocere—Above all, do no harm! J. Clin. Pharmacol. 2005, 45, 371–377. [Google Scholar] [CrossRef]
- Mistry, P.K.; Weinreb, N.J.; Brady, R.O.; Grabowski, G.A. Gaucher disease: Resetting the clinical and scientific agenda. Am. J. Hematol. 2009, 84, 205–207. [Google Scholar] [CrossRef]
- Beutler, E.; Demina, A.; Laubscher, K.; Garver, P.; Gelbart, T.; Balicki, D.; Vaughan, L. The clinical course of treated and untreated Gaucher disease. A study of 45 patients. Blood Cells Mol. Dis. 1995, 21, 86–108. [Google Scholar] [CrossRef]
- Piran, S.; Roberts, A.; Patterson, M.A.; Amato, D. The clinical course of untreated Gaucher disease in 22 patients over 10 years: Hematological and skeletal manifestations. Blood Cells Mol. Dis. 2009, 43, 289–293. [Google Scholar] [CrossRef]
- Azuri, J.; Elstein, D.; Lahad, A.; Abrahamov, A.; Hadas-Halpern, I.; Zimran, A. Asymptomatic Gaucher disease implications for large-scale screening. Genet. Test. 1998, 2, 297–299. [Google Scholar] [CrossRef]
- Elstein, D.; Altarescu, G.; Abrahamov, A.; Zimran, A. Children with type 1 Gaucher disease: Changing profiles in the 21st century. Blood Cells Mol. Dis. 2018, 68, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Bier, L.; Overbey, J.R.; Cohen-Pfeffer, J.; Desai, K.; Desnick, R.J.; Balwani, M. Early manifestations of type 1 Gaucher disease in presymptomatic children diagnosed after parental carrier screening. Genet. Med. 2017, 19, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Schielen, P.; Kemper, E.A.; Gelb, M.H. Newborn Screening for Lysosomal Storage Diseases: A Concise Review of the Literature on Screening Methods, Therapeutic Possibilities and Regional Programs. Int. J. Neonatal Screen 2017, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, S.; Lahad, A.; Shmueli, A.; Zimran, A.; Peleg, L.; Orr-Urtreger, A.; Levy-Lahad, E.; Sagi, M. Carrier screening for Gaucher disease: Lessons for low-penetrance, treatable diseases. JAMA 2007, 298, 1281–1290. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hurvitz, N.; Dinur, T.; Becker-Cohen, M.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Abramov, A.; Zimran, A.; et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. Int. J. Mol. Sci. 2019, 20, 3033. [Google Scholar] [CrossRef] [PubMed]
- Arkadir, D.; Dinur, T.; Revel-Vilk, S.; Becker, C.M.; Cozma, C.; Hovakimyan, M.; Eichler, S.; Rolfs, A.; Zimran, A. Glucosylsphingosine is a reliable response biomarker in Gaucher disease. Am. J. Hematol. 2018, 93, E140. [Google Scholar] [CrossRef]
- Weinreb, N.J.; Barbouth, D.S.; Lee, R.E. Causes of death in 184 patients with type 1 Gaucher disease from the United States who were never treated with enzyme replacement therapy. Blood Cells Mol. Dis. 2018, 68, 211–217. [Google Scholar] [CrossRef]
- Regenboog, M.; van Dussen, L.; Verheij, J.; Weinreb, N.J.; Santosa, D.; vom Dahl, S.; Häussinger, D.; Müller, M.N.; Canbay, A.; Rigoldi, M.; et al. Hepatocellular carcinoma in Gaucher disease: An international case series. J. Inherit. Metab. Dis. 2018, 41, 819–827. [Google Scholar] [CrossRef]
- Sonder, S.U.; Limgala, R.P.; Ivanova, M.M.; Ioanou, C.; Plassmeyer, M.; Marti, G.E.; Alpan, O.; Goker-Alpan, O. Persistent immune alterations and comorbidities in splenectomized patients with Gaucher disease. Blood Cells Mol. Dis. 2016, 59, 8–15. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| N370S/N370S | R496H/Other | N370S/Other | Refusing Tx ** | |
|---|---|---|---|---|
| Number | 80 | 4 | 13 | 6 |
| Female | 49 (60.5%) | 1 (25%) | 7 (50%) | 6 (100%) |
| Diagnosis age, Y * | 22 (0–60) | 6 (2–19) | 25 (3–40) | 13 (5–23) |
| Last F/U age, Y * | 45.5 (22–83) | 27 (24–43) | 56 (23–72) | 46 (22–55) |
| Time of F/U, Y * | 20 (5–58) | 22 (5–41) | 25 (6–40) | 27 (10–46) |
| PLT, ×103/mL * | 163 (56–408) | 160 (141–192) | 176 (80–364) | 103 (36–171) |
| Hb, ×103/mL * | 13 (7.9–16.8) | 14.4 (12.9–16) | 13.6 (10–16.2) | 12.1 (10–14.1) |
| Spleen, MN * | 2.13 (0.51–9.55) | 2.39 (2.02–3.41) | 2.25 (1.03–9.96) | 5.86 (3.57–9.92) |
| Liver size, MN * | 1.07 (0–4.68) | 1.22 (1.1–1.63) | 1.24 (0.94–5) | 3.78 (1.06–8) |
| T-score lumbar * | −0.75 (−2.9 to 2.8) | −0.5 (−0.8 to −0.2) | −1.4 (−2.5 to 1.3) | −2 (−3.0 to −0.4) |
| Lyso-Gb1, ng/mL * | 104.5 (10.3–381) | 11.5 (8.1–23.4) | 113 (9.7–325) | 301 (284–719) |
| Splenectomy | 5 | 0 | 2 | 2 |
| Parkinson | 2 | 0 | 2 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinur, T.; Zimran, A.; Becker-Cohen, M.; Arkadir, D.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Revel-Vilk, S. Long Term Follow-Up of 103 Untreated Adult Patients with Type 1 Gaucher Disease. J. Clin. Med. 2019, 8, 1662. https://doi.org/10.3390/jcm8101662
Dinur T, Zimran A, Becker-Cohen M, Arkadir D, Cozma C, Hovakimyan M, Oppermann S, Demuth L, Rolfs A, Revel-Vilk S. Long Term Follow-Up of 103 Untreated Adult Patients with Type 1 Gaucher Disease. Journal of Clinical Medicine. 2019; 8(10):1662. https://doi.org/10.3390/jcm8101662
Chicago/Turabian StyleDinur, Tama, Ari Zimran, Michal Becker-Cohen, David Arkadir, Claudia Cozma, Marina Hovakimyan, Sebastian Oppermann, Laura Demuth, Arndt Rolfs, and Shoshana Revel-Vilk. 2019. "Long Term Follow-Up of 103 Untreated Adult Patients with Type 1 Gaucher Disease" Journal of Clinical Medicine 8, no. 10: 1662. https://doi.org/10.3390/jcm8101662
APA StyleDinur, T., Zimran, A., Becker-Cohen, M., Arkadir, D., Cozma, C., Hovakimyan, M., Oppermann, S., Demuth, L., Rolfs, A., & Revel-Vilk, S. (2019). Long Term Follow-Up of 103 Untreated Adult Patients with Type 1 Gaucher Disease. Journal of Clinical Medicine, 8(10), 1662. https://doi.org/10.3390/jcm8101662