Allogeneic Hematopoietic Stem Cell Transplantation for Adults with Sickle Cell Disease


Abstract
1. Morbidity and Mortality of Sickle Cell Disease
1.1. Contemporary Survival Patterns in Sickle Cell Disease
1.2. Acute Complications in Adults with Sickle Cell Disease
1.3. Cardiovascular Complications in Sickle Cell Disease
1.4. Health-Related Quality of Life
1.5. Healthcare Utilization and Healthcare Costs
2. Currently Accepted Indications for Allogeneic hematopoietic stem cell transplantation (HSCT) in Sickle Cell Disease
3. Allogeneic HSCT in Adults with Sickle Cell Disease
3.1. Global Experience
3.2. Myeloablative Conditioning Regimens
3.3. Reduced-Intensity Conditioning Regimens
3.4. Nonmyeloablative Conditioning Regimens
3.5. Alternative Donor Approaches
4. Evaluating Risks versus Benefits of Allogeneic HSCT in Adults with Sickle Cell Disease
4.1. Acute Complications after HSCT
4.2. Cardiovascular Function after HSCT
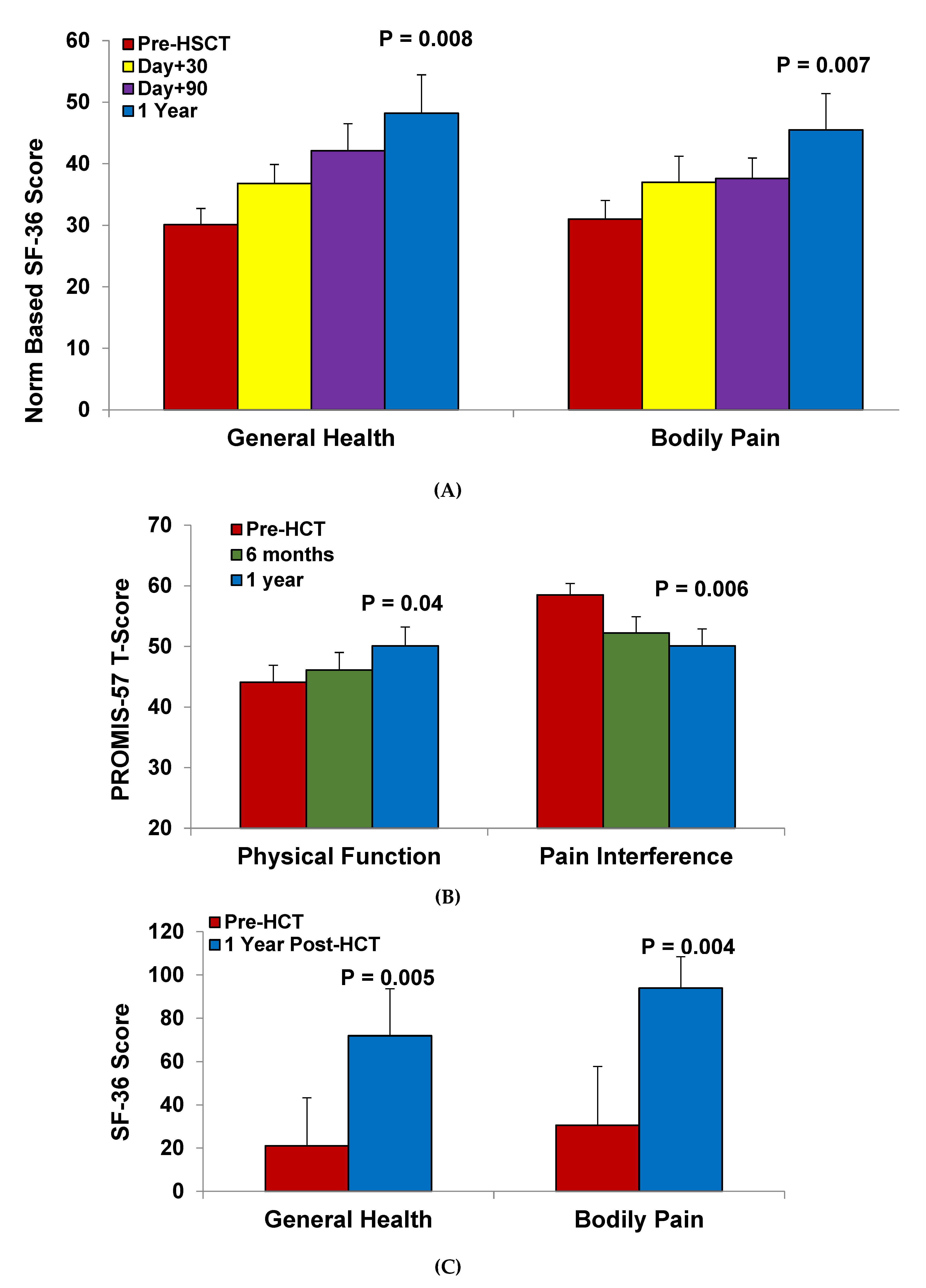
4.3. Health-Related Quality of Life after HSCT
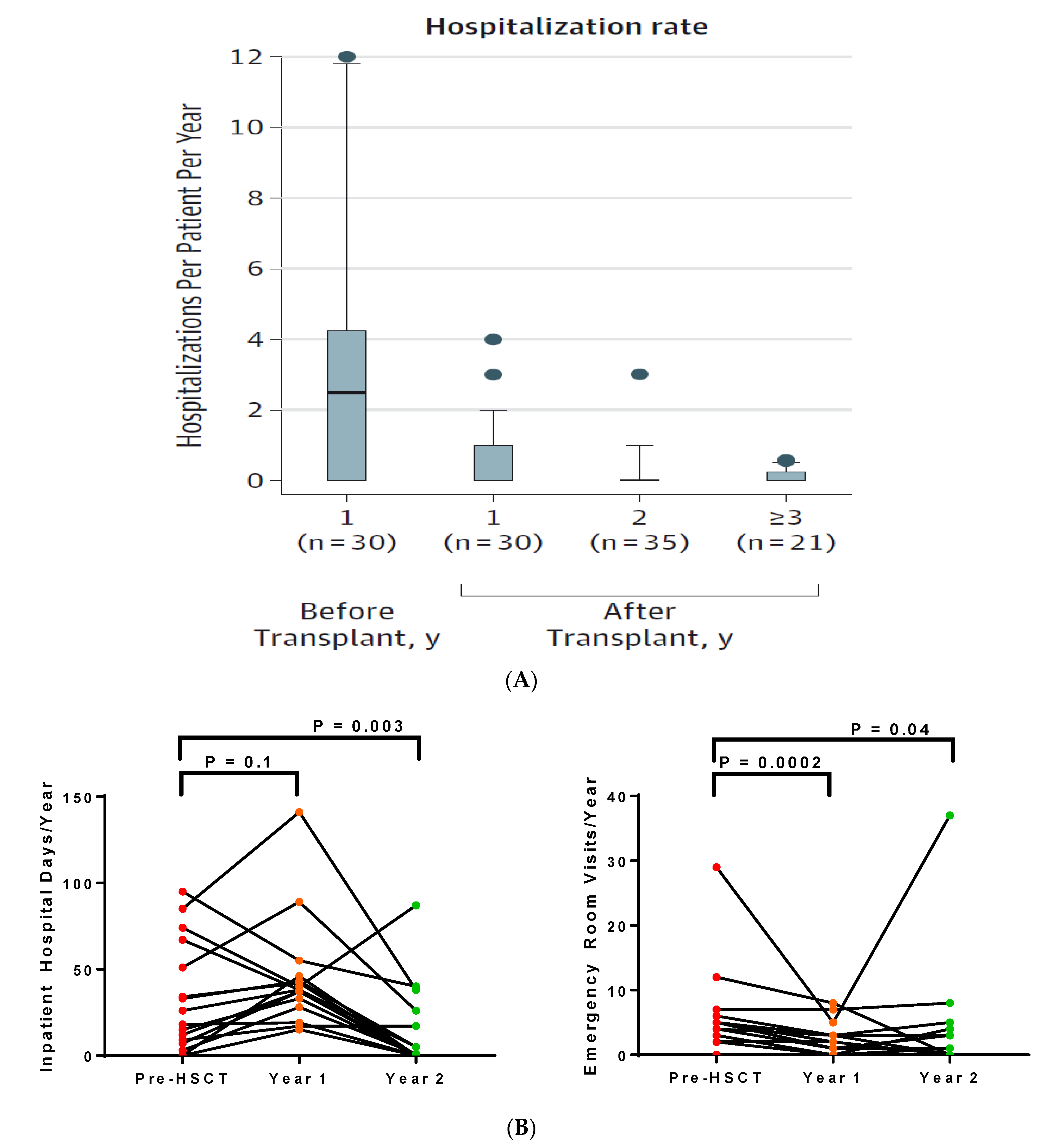
4.4. Healthcare Utilization after HSCT
5. Future Directions
Author Contributions
Conflicts of Interest
References
- Modell, B.; Darlison, M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, E.; Cappelli, B.; Bernaudin, F.; Labopin, M.; Volt, F.; Carreras, J.; Simões, B.P.; Ferster, A.; Dupont, S.; de la Fuente, J.; et al. Sickle cell disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017, 129, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Klug, P.P.; Steinberg, M.H. Mortality In Sickle Cell Disease—Life Expectancy and Risk Factors for Early Death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.T.; Rogers, Z.R.; McCavit, T.L.; Buchanan, G.R. Improved survival of children and adolescents with sickle cell disease. Blood 2010, 115, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Hamideh, D.; Alvarez, O. Sickle cell disease related mortality in the United States (1999–2009). Pediatr. Blood Cancer 2013, 60, 1482–1486. [Google Scholar] [CrossRef]
- Lanzkron, S.; Carroll, C.P.; Haywood, C., Jr. Mortality Rates and Age at Death from Sickle Cell Disease: U.S.; 1979–2005. Public Health Rep. 2013, 128, 110–116. [Google Scholar] [CrossRef] [PubMed]
- DeBaun, M.R.; Ghafuri, D.L.; Rodeghier, M.; Maitra, P.; Chaturvedi, S.; Kassim, A.; Ataga, K.I. Decreased median survival of adults with sickle cell disease after adjusting for left truncation bias: A pooled analysis. Blood 2019, 133, 615–617. [Google Scholar] [CrossRef]
- Elmariah, H.; Garrett, M.E.; De Castro, L.M.; Jonassaint, J.C.; Ataga, K.I.; Eckman, J.R.; Ashley-Koch, A.E.; Telen, M.J. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am. J. Hematol. 2014, 89, 530–535. [Google Scholar] [CrossRef]
- Gardner, K.; Douiri, A.; Drasar, E.; Allman, M.; Mwirigi, A.; Awogbade, M.; Thein, S.L. Survival in adults with sickle cell disease in a high-income setting. Blood 2016, 128, 1436–1438. [Google Scholar] [CrossRef]
- Lobo, C.L.C.; Nascimento, E.M.D.; Jesus, L.J.C.; Freitas, T.G.; Lugon, J.R.; Ballas, S.K. Mortality in children, adolescents and adults with sickle cell anemia in Rio de Janeiro, Brazil. Hematol. Transfus. Cell Ther. 2018, 40, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Powars, D.R.; Chan, L.S.; Hiti, A.; Ramicone, E.; Johnson, C. Outcome of sickle cell anemia: A 4-decade observational study of 1056 patients. Medicine 2005, 84, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Barton, F.; Castro, O.; Pegelow, C.H.; Ballas, S.K.; Kutlar, A.; Orringer, E.; Bellevue, R.; Olivieri, N.; Eckman, J.; et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: Risks and benefits up to 9 years of treatment. JAMA 2003, 289, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Darbari, D.S.; Wang, Z.; Kwak, M.; Hildesheim, M.; Nichols, J.; Allen, D.; Seamon, C.; Peters-Lawrence, M.; Conrey, A.; Hall, M.K.; et al. Severe Painful Vaso-Occlusive Crises and Mortality in a Contemporary Adult Sickle Cell Anemia Cohort Study. PLoS ONE 2013, 8, e79923. [Google Scholar] [CrossRef] [PubMed]
- Maitra, P.; Caughey, M.; Robinson, L.; Desai, P.C.; Jones, S.; Nouraie, M.; Gladwin, M.T.; Hinderliter, A.; Cai, J.; Ataga, K.I. Risk factors for mortality in adult patients with sickle cell disease: A meta-analysis of studies in North America and Europe. Haematologica 2017, 102, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, C.D.; Lauder, N.; Jonassaint, J.C.; Telen, M.J.; Zhao, X.; Wright, E.C.; Gilliam, F.R.; De Castro, L.M. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. Am. J. Hematol. 2010, 85, 36–40. [Google Scholar] [CrossRef]
- Lanzkron, S.; Little, J.; Field, J.; Shows, J.R.; Wang, H.; Seufert, R.; Brooks, J.; Varadhan, R.; Haywood, C.; Saheed, M.; et al. Increased acute care utilization in a prospective cohort of adults with sickle cell disease. Blood Adv. 2018, 2, 2412–2417. [Google Scholar] [CrossRef]
- Jordan, L.C.; Kassim, A.A.; Donahue, M.J.; Juttukonda, M.R.; Pruthi, S.; Davis, L.T.; Rodeghier, M.; Lee, C.A.; Patel, N.J.; DeBaun, M.R. Silent infarct is a risk factor for infarct recurrence in adults with sickle cell anemia. Neurology 2018, 91, e781–e784. [Google Scholar] [CrossRef]
- Wong, W.-Y.; Powars, D.R. Overt and Incomplete (Silent) Cerebral Infarction in Sickle Cell Anemia: Diagnosis and Management. Hematol. Clin. N. Am. 2005, 19, 839–855. [Google Scholar] [CrossRef]
- Adams, R.J.; McKie, V.C.; Hsu, L.; Files, B.; Vichinsky, E.; Pegelow, C.; Abboud, M.; Gallagher, D.; Kutlar, A.; Nichols, F.T.; et al. Prevention of a First Stroke by Transfusions in Children with Sickle Cell Anemia and Abnormal Results on Transcranial Doppler Ultrasonography. N. Engl. J. Med. 1998, 339, 5–11. [Google Scholar] [CrossRef]
- Valadi, N.; Silva, G.S.; Bowman, L.S.; Ramsingh, D.; Vicari, P.; Filho, A.C.; Massaro, A.R.; Kutlar, A.; Nichols, F.T.; Adams, R.J. Transcranial Doppler ultrasonography in adults with sickle cell disease. Neurology 2006, 67, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Powars, D.; Wilson, B.; Imbus, C.; Pegelow, C.; Allen, J. The natural history of stroke in sickle cell disease. Am. J. Med. 1978, 65, 461–471. [Google Scholar] [CrossRef]
- Kassim, A.A.; Galadanci, N.A.; Pruthi, S.; DeBaun, M.R. How I treat and manage strokes in sickle cell disease. Blood 2015, 125, 3401–3410. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, M.L.; McKinstry, R.C.; Lacey, J.L.; Moran, C.J.; Panepinto, J.A.; Thompson, A.A.; Sarnaik, S.A.; Woods, G.M.; Casella, J.F.; Inusa, B.; et al. Silent cerebral infarcts occur despite regular blood transfusion therapy after first strokes in children with sickle cell disease. Blood 2011, 117, 772–779. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Sachdev, V.; Jison, M.L.; Minter, K.; Brown, B.; Coles, W.A.; Nichols, J.S.; Ernst, I.; Hunter, L.A.; Ognibene, F.P.; et al. Pulmonary Hypertension as a Risk Factor for Death in Patients with Sickle Cell Disease. N. Engl. J. Med. 2004, 350, 886–895. [Google Scholar] [CrossRef] [PubMed]
- De Castro, L.M.; Jonassaint, J.C.; Graham, F.L.; Ashley-Koch, A.; Telen, M.J. Pulmonary hypertension associated with sickle cell disease: Clinical and laboratory endpoints and disease outcomes. Am. J. Hematol. 2008, 83, 19–25. [Google Scholar] [CrossRef]
- Ataga, K.I.; Moore, C.G.; Jones, S.; Olajide, O.; Strayhorn, D.; Hinderliter, A.; Orringer, E.P. Pulmonary hypertension in patients with sickle cell disease: A longitudinal study. Br. J. Haematol. 2006, 134, 109–115. [Google Scholar] [CrossRef]
- Damy, T.; Bodez, D.; Habibi, A.; Guellich, A.; Rappeneau, S.; Inamo, J.; Guendouz, S.; Gellen-Dautremer, J.; Pissard, S.; Loric, S. Haematological determinants of cardiac involvement in adults with sickle cell disease. Eur. Heart J. 2016, 37, 1158–1167. [Google Scholar] [CrossRef]
- Machado, R.F.; Hildesheim, M.; Mendelsohn, L.; Remaley, A.T.; Kato, G.J.; Gladwin, M.T. NT-pro brain natriuretic peptide levels and the risk of death in the cooperative study of sickle cell disease. Br. J. Haematol. 2011, 154, 512–520. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Barst, R.J.; Gibbs, J.S.R.; Hildesheim, M.; Sachdev, V.; Nouraie, M.; Hassell, K.L.; Little, J.A.; Schraufnagel, D.E.; Krishnamurti, L.; et al. Risk Factors for Death in 632 Patients with Sickle Cell Disease in the United States and United Kingdom. PLoS ONE 2014, 9, e99489. [Google Scholar] [CrossRef]
- McClish, D.K.; Penberthy, L.T.; Bovbjerg, V.E.; Roberts, J.D.; Aisiku, I.P.; Levenson, J.L.; Roseff, S.D.; Smith, W.R. Health related quality of life in sickle cell patients: The PiSCES project. Health Qual. Life Outcomes 2005, 3, 50. [Google Scholar] [CrossRef] [PubMed]
- Kauf, T.L.; Coates, T.D.; Huazhi, L.; Hartzema, A.G.; Mody-Patel, N.; Mody-Patel, N. The cost of health care for children and adults with sickle cell disease. Am. J. Hematol. 2009, 84, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Ballas, S.K. The cost of health care for patients with sickle cell disease. Am. J. Hematol. 2009, 84, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, E. Allogeneic transplantation strategies including haploidentical transplantation in sickle cell disease. Hematology 2013, 2013, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurti, L.; Neuberg, D.S.; Sullivan, K.M.; Kamani, N.R.; Abraham, A.; Campigotto, F.; Zhang, W.; Dahdoul, T.; De Castro, L.; Parikh, S.; et al. Bone Marrow Transplantation for Adolescents and Young Adults with Sickle Cell Disease: Results of a Prospective Multicenter Pilot Study. Am. J. Hematol. 2019, 94, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Guilcher, G.M.; Truong, T.H.; Saraf, S.L.; Joseph, J.J.; Rondelli, D.; Hsieh, M.M. Curative therapies - allogeneic hematopoietic cell transplantation from matched related donors using myeloablative, reduced intensity, and non-myeloablative conditioning in sickle cell disease. Semin. Hematol. 2018, 55, 87–93. [Google Scholar] [CrossRef]
- Walters, M.C.; Patience, M.; Leisenring, W.; Eckman, J.R.; Scott, J.P.; Mentzer, W.C.; Davies, S.C.; Ohene-Frempong, K.; Bernaudin, F.; Matthews, D.C.; et al. Bone Marrow Transplantation for Sickle Cell Disease. N. Engl. J. Med. 1996, 335, 369–376. [Google Scholar] [CrossRef]
- McGann, P.T.; Ware, R.E. Hydroxyurea therapy for sickle cell anemia. Expert Opin. Drug Saf. 2015, 14, 1749–1758. [Google Scholar] [CrossRef]
- Charache, S.; Terrin, M.L.; Moore, R.D.; Barton, F.B.; Eckert, S.V.; Bonds, D.R.; Dover, G.J.; McMahon, R.P. Effect of Hydroxyurea on the Frequency of Painful Crises in Sickle Cell Anemia. N. Engl. J. Med. 1995, 332, 1317–1322. [Google Scholar] [CrossRef]
- Fitzhugh, C.D.; Hsieh, M.M.; Allen, D.; Coles, W.A.; Seamon, C.; Ring, M.; Zhao, X.; Minniti, C.P.; Rodgers, G.P.; Schechter, A.N.; et al. Hydroxyurea-Increased Fetal Hemoglobin Is Associated with Less Organ Damage and Longer Survival in Adults with Sickle Cell Anemia. PLoS ONE 2015, 10, e0141706. [Google Scholar] [CrossRef]
- Voskaridou, E.; Christoulas, D.; Bilalis, A.; Plata, E.; Varvagiannis, K.; Stamatopoulos, G.; Sinopoulou, K.; Balassopoulou, A.; Loukopoulos, D.; Terpos, E. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: Results of a 17-year, single-center trial (LaSHS). Blood 2010, 115, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Bernaudin, F.; Socie, G.; Kuentz, M.; Chevret, S.; Duval, M.; Bertrand, Y.; Vannier, J.-P.; Yakouben, K.; Thuret, I.; Bordigoni, P.; et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 2007, 110, 2749–2756. [Google Scholar] [CrossRef] [PubMed]
- Panepinto, J.A.; Walters, M.C.; Carreras, J.; Marsh, J.; Bredeson, C.N.; Gale, R.P.; Hale, G.A.; Horan, J.; Hows, J.M.; Klein, J.P.; et al. Matched-related donor transplantation for sickle cell disease: Report from the Center for International Blood and Transplant Research. Br. J. Haematol. 2007, 137, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Vermylen, C.; Cornu, G.; Ferster, A.; Brichard, B.; Ninane, J.; Ferrant, A.; Zenebergh, A.; Maes, P.; Dhooge, C.; Benoit, Y.; et al. Haematopoietic stem cell transplantation for sickle cell anaemia: The first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998, 22, 1–6. [Google Scholar] [CrossRef][Green Version]
- Kuentz, M.; Robin, M.; Dhedin, N.; Hicheri, Y.; de Latour, R.P.; Rohrlich, P.; Bordigoni, P.; Bruno, B.; Socié, G.; Bernaudin, F. Is there still a place for myeloablative regimen to transplant young adults with sickle cell disease? Blood 2011, 118, 4491–4492. [Google Scholar] [CrossRef] [PubMed]
- Van Besien, K.; Bartholomew, A.; Stock, W.; Peace, D.; Devine, S.; Sher, D.; Sosman, J.; Chen, Y.; Koshy, M.; Hoffman, R. Fludarabine-based conditioning for allogeneic transplantation in adults with sickle cell disease. Bone Marrow Transplant. 2000, 26, 445–449. [Google Scholar] [CrossRef]
- Özdoğu, H.; Boga, C.; Yeral, M.; Kozanoglu, I.; Gereklioglu, Ç.; Aytan, P.; Kasar, M.; Asma, S.; Buyukkurt, N.; Solmaz, S.; et al. Allogenic peripheral stem cell transplantation from HLA-matched related donors for adult sickle cell disease: Remarkable outcomes from a single-center trial. Bone Marrow Transplant. 2018, 53, 880–890. [Google Scholar] [CrossRef]
- Walters, M.; Patience, M.; Leisenring, W.; Rogers, Z.; Aquino, V.; Buchanan, G.; Roberts, I.; Yeager, A.; Hsu, L.; Adamkiewicz, T.; et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol. Blood Marrow Transplant. 2001, 7, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Sachs, D.H.; Kawai, T.; Sykes, M. Induction of Tolerance through Mixed Chimerism. Cold Spring Harb. Perspect. Med. 2014, 4, a015529. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Hsieh, M.; Eapen, M.; Fitzhugh, C.; Carreras, J.; Keesler, D.; Guilcher, G.; Kamani, N.; Walters, M.C.; Boelens, J.J.; et al. Relationship between Mixed Donor–Recipient Chimerism and Disease Recurrence after Hematopoietic Cell Transplantation for Sickle Cell Disease. Biol. Blood Marrow Transplant. 2017, 23, 2178–2183. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, C.D.; Cordes, S.; Taylor, T.; Coles, W.; Roskom, K.; Link, M.; Hsieh, M.M.; Tisdale, J.F. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood 2017, 130, 1946–1948. [Google Scholar] [CrossRef]
- Iannone, R.; Luznik, L.; Engstrom, L.W.; Tennessee, S.L.; Askin, F.B.; Casella, J.F.; Kickler, T.S.; Goodman, S.N.; Hawkins, A.L.; Griffin, C.A.; et al. Effects of mixed hematopoietic chimerism in a mouse model of bone marrow transplantation for sickle cell anemia. Blood 2001, 97, 3960–3965. [Google Scholar] [CrossRef]
- Hsieh, M.M.; Kang, E.M.; Fitzhugh, C.D.; Link, M.B.; Bolan, C.D.; Kurlander, R.; Childs, R.W.; Rodgers, G.P.; Powell, J.D.; Tisdale, J.F. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N. Engl. J. Med. 2009, 361, 2309–2317. [Google Scholar] [CrossRef]
- Hsieh, M.M.; Fitzhugh, C.D.; Weitzel, R.P.; Link, M.E.; Coles, W.A.; Zhao, X.; Rodgers, G.P.; Powell, J.D.; Tisdale, J.F. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 2014, 312, 48–56. [Google Scholar] [CrossRef]
- Saraf, S.L.; Oh, A.L.; Patel, P.R.; Jalundhwala, Y.; Sweiss, K.; Koshy, M.; Campbell-Lee, S.; Gowhari, M.; Hassan, J.; Peace, D.; et al. Nonmyeloablative Stem Cell Transplantation with Alemtuzumab/Low-Dose Irradiation to Cure and Improve the Quality of Life of Adults with Sickle Cell Disease. Biol. Blood Marrow Transplant. 2016, 22, 441–448. [Google Scholar] [CrossRef]
- Al-Zahrani, M.; Alaskar, A.; Damlaj, M.; Abuelgasim, K.; Gmati, G.; Salama, H.A.; Ghazi, S.; Ali, O.; Alahmari, B.; Hejazi, A. Non-myeloablative transplant in severe sickel cell disease is safe & effective. Single center experience from Saudi Arabia. Blood 2017, 130, 5549. [Google Scholar]
- Walters, M.C.; Patience, M.; Leisenring, W.; Eckman, J.R.; Buchanan, G.R.; Rogers, Z.R.; Olivieri, N.E.; Vichinsky, E.; Davies, S.C.; Mentzer, W.C.; et al. Barriers to bone marrow transplantation for sickle cell anemia. Biol. Blood Marrow Transplant. 1996, 2, 100–104. [Google Scholar]
- Mentzer, W.C.; Heller, S.; Pearle, P.R.; Hackney, E.; Vichinsky, E. Availability of related donors for bone marrow transplantation in sickle cell anemia. J. Pediatr. Hematol. 1994, 16, 27–29. [Google Scholar]
- Gragert, L.; Eapen, M.; Williams, E.; Freeman, J.; Spellman, S.; Baitty, R.; Hartzman, R.; Rizzo, J.D.; Horowitz, M.; Confer, D.; et al. HLA Match Likelihoods for Hematopoietic Stem-Cell Grafts in the U.S. Registry. N. Engl. J. Med. 2014, 371, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Luznik, L.; Engstrom, L.W.; Iannone, R.; Fuchs, E.J. Posttransplantation cyclophosphamide facilitates engraftment of major histocompatibility complex-identical allogeneic marrow in mice conditioned with low-dose total body irradiation. Biol. Blood Marrow Transplant. 2002, 8, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Bolaños-Meade, J.; Fuchs, E.J.; Luznik, L.; Lanzkron, S.M.; Gamper, C.J.; Jones, R.J.; Brodsky, R.A. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood 2012, 120, 4285–4291. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.L.; Oh, A.L.; Patel, P.R.; Sweiss, K.; Koshy, M.; Campbell-Lee, S.; Gowhari, M.; Jain, S.; Peace, D.; Quigley, J.G.; et al. Haploidentical Peripheral Blood Stem Cell Transplantation Demonstrates Stable Engraftment in Adults with Sickle Cell Disease. Biol. Blood Marrow Transplant. 2018, 24, 1759–1765. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, J.; Dhedin, N.; Koyama, T.; Bernaudin, F.; Kuentz, M.; Karnik, L.; Socié, G.; Culos, K.A.; Brodsky, R.A.; DeBaun, M.R.; et al. Haploidentical Bone Marrow Transplantation with Post-Transplantation Cyclophosphamide Plus Thiotepa Improves Donor Engraftment in Patients with Sickle Cell Anemia: Results of an International Learning Collaborative. Biol. Blood Marrow Transplant. 2019, 25, 1197–1209. [Google Scholar] [CrossRef]
- Pawlowska, A.B.; Cheng, J.C.; Karras, N.A.; Sun, W.; Wang, L.D.; Bell, A.D.; Gutierrez, L.; Rosenthal, J. HLA Haploidentical Stem Cell Transplant with Pretransplant Immunosuppression for Patients with Sickle Cell Disease. Biol. Blood Marrow Transplant. 2018, 24, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, C.D.; Hsieh, M.M.; Taylor, T.; Coles, W.; Roskom, K.; Wilson, D.; Wright, E.; Jeffries, N.; Gamper, C.J.; Powell, J.; et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv. 2017, 1, 652–661. [Google Scholar] [CrossRef]
- Sorror, M.; Maris, M.; Storb, R.; Baron, F.; Sandmaier, B.; Maloney, D.; Barry, S. Hematopoietic cell transplantation (HCT)-specific comorbidity index: A new tool for risk assessment before allogeneic HCT. Blood 2005, 106, 2912–2919. [Google Scholar] [CrossRef]
- Thakar, M.S.; Broglie, L.; Logan, B.; Artz, A.; Bunin, N.; Burroughs, L.M.; Fretham, C.; Jacobsohn, D.A.; Loren, A.W.; Kurtzberg, J.; et al. The Hematopoietic Cell Transplant Comorbidity Index predicts survival after allogeneic transplant for nonmalignant diseases. Blood 2019, 133, 754–762. [Google Scholar] [CrossRef]
- Van Besien, K.; Koshy, M.; Anderson-Shaw, L.; Talishy, N.; Dorn, L.; Devine, S.; Yassine, M.; Kodish, E. Allogeneic stem cell transplantation for sickle cell disease. A study of patients’ decisions. Bone Marrow Transplant. 2001, 28, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Bareford, D. A survey on patient perception of reduced-intensity transplantation in adults with sickle cell disease. Bone Marrow Transplant. 2007, 39, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Darbari, D.S.; Liljencrantz, A.I.J.; Martin, S.; Roderick, M.C.; Fitzhugh, C.D.; Tisdale, J.F.; Thein, S.L.; Hsieh, M. Pain and opioid use after reversal of sickle cell disease following HLA-matched sibling haematopoietic stem cell transplant. Br. J. Haematol. 2019, 184, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.L.; Ghimire, K.; Patel, P.; Sweiss, K.; Quigley, J.G.; Khan, I.; Peace, D.; Gowhari, M.; Jain, S.; Galvin, J.P.; et al. Health care utilization in transplanted versus non-transplanted sickle cell disease patients. Blood 2018, 132, 313. [Google Scholar]
- Mehari, A.; Gladwin, M.T.; Tian, X.; Machado, R.F.; Kato, G.J. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA 2012, 307, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Pittman, C.; Hsieh, M.M.; Coles, W.; Tisdale, J.F.; Weir, N.A.; Fitzhugh, C.D. Reversal of pre-capillary pulmonary hypertension in a patient with sickle cell anemia who underwent haploidentical peripheral blood stem cell transplantation. Bone Marrow Transplant. 2017, 52, 641–642. [Google Scholar] [CrossRef]
- Kelly, M.J.; Pennarola, B.W.; Rodday, A.M.; Parsons, S.K. Journeys to Recovery Study H-CS. Health-related quality of life (HRQL) in children with sickle cell disease and thalassemia following hematopoietic stem cell transplant (HSCT). Pediatr. Blood Cancer 2012, 59, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Kolva, E.; Cimini, L.; Jin, Z.; Satwani, P.; Savone, M.; George, D.; Garvin, J.; Paz, M.L.; Briamonte, C.; et al. Health-Related Quality of Life after Allogeneic Hematopoietic Stem Cell Transplantation for Sickle Cell Disease. Biol. Blood Marrow Transplant. 2015, 21, 666–672. [Google Scholar] [CrossRef]
- Angelucci, E.; Matthes-Martin, S.; Baronciani, D.; Bernaudin, F.; Bonanomi, S.; Cappellini, M.D.; Dalle, J.-H.; Di Bartolomeo, P.; De Heredia, C.D.; Dickerhoff, R.; et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: Indications and management recommendations from an international expert panel. Haematologica 2014, 99, 811–820. [Google Scholar] [CrossRef]
- Rotz, S.J.; O’Riordan, M.A.; Kim, C.; De Lima, M.; Gladwin, M.T.; Little, J.A.; Rotz, S. Traffic Light: Prognosis-based eligibility for clinical trials of hematopoietic SCT in adults with sickle cell anemia. Bone Marrow Transplant. 2015, 50, 918–923. [Google Scholar] [CrossRef]
- Saraf, S.L.; Akingbola, T.S.; Shah, B.N.; Ezekekwu, C.A.; Sonubi, O.; Zhang, X.; Hsu, L.L.; Gladwin, M.T.; Machado, R.F.; Cooper, R.S.; et al. Associations of alpha-thalassemia and BCL11A with stroke in Nigerian, United States, and United Kingdom sickle cell anemia cohorts. Blood Adv. 2017, 1, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.A.; Lei, Z.; Bahroos, N.; Maienschein-Cline, M.; Saraf, S.L.; Zhang, X.; Shah, B.N.; Nouraie, S.M.; Abbasi, T.; Patel, A.R.; et al. Association of circulating transcriptomic profiles with mortality in sickle cell disease. Blood 2017, 129, 3009–3016. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Indication | HLA-Matched Related Donor | Alternative Donor* | |
|---|---|---|---|
| Predictors for Increased Mortality | ≥2 VOC/year | X | X |
| Recurrent acute chest syndrome | X | X | |
| Stroke | X | X | |
| Cognitive impairment + Abnormal cerebral MRI | X | X | |
| TRJV ≥ 2.7 m/s | X | X | |
| Sickle nephropathy | X | ||
| Predictors for Increased Morbidity | Red blood cell alloimmunization | X | |
| Recurrent priapism | X | ||
| Osteonecrosis of multiple joints | X | ||
| Conditioning Regimen | GVHD Prophylaxis | N | Age (years) | Graft Source | Event-free Survival | Overall Survival | aGVHD (≥grade 2) | cGVHD (≥moderate) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Fludarabine (150 mg/kg) Busulfan (3.2 mg/kg) Cy (29 mg/kg) ATG (15 mg/kg) Total body irradiation (200 cGy) |
| 20 | 20–45 | 20 MRD | 100% | 100% | 5% | 0% | [50] |
| Fludarabine (175 mg/m2) Busulfan (13.2 mg/kg) ATG (6 mg/kg) |
| 22 | 17–36 | 17 MRD 5 MUD | 82% | 91% | 18% | 14% | [35] |
| Conditioning Regimen | GVHD Prophylaxis | N | Age (years) | Graft Source | Event-free Survival | Overall Survival | aGVHD (≥grade 2) | cGVHD (≥moderate) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Alemtuzumab 1 mg/kg Total body irradiation (300 cGy) | Sirolimus | 30 | 17–65 | MRD | 87% | 97% | 0% | 0% | [57] |
| 13 | 17–40 | 92% | 100% | 0% | 0% | [58] | |||
| 17 | 14–39 | 89% | 100% | 0% | 0% | [59] |
| Conditioning Regimen | GVHD Prophylaxis | N | Age (years) | Graft Source | Event-free Survival | Overall Survival | aGVHD (≥grade2) | cGVHD (≥moderate) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Fludarabine (150 mg/m2) Cy (29 mg/kg) ATG (4.5 mg/kg) Total body irradiation (200 cGy) |
| 14 | 15–42 | Bone Marrow | 57% | 100% | 0% | 0% | [64] |
| Alemtuzumab (1 mg/kg) Total body irradiation (400 cGy) |
| 12 | 20–56 | PBSC | 50% | 92% | 0% | 0% | [68] |
| Fludarabine (150 mg/m2) Cy (29 mg/kg) ATG (4.5 mg/kg) Total body irradiation (300 cGy) |
| 8 | 20–38 | PBSC | 75% | 88% | 25% | 13% | [65] |
| Pre-HSCT Immunosuppression: Fludarabine (200 mg/m2) Dexamethasone (125 mg/m2) Conditioning: Fludarabine (210 mg/m2) Busulfan (520 mg/m2) ATG (4.5 mg/kg) |
| 4 | 12–24 | Bone Marrow (n = 3) PBSC (n = 1) | 100% | 100% | 0% | 0% | [67] |
| Fludarabine (150 mg/m2) Cy (29 mg/kg) Thiotepa (10 mg/kg) ATG (4.5 mg/kg) Total body irradiation (300 cGy) |
| 15 | 7–40 | Bone Marrow | 93% | 100% | 13% | 0% | [66] |
| HSCT Type | Stroke Recurrence | Brain MRI Findings | Reference |
|---|---|---|---|
| • Nonmyeloablative, • Matched-related donor | 0 out of 9 | Stable brain MRI and angiography findings | [57] |
| • Nonmyeloablative • Haploidentical related donor | 0 out of 3 | – | [64] |
| • Nonmyeloablative • Matched-related donor | 1 out of 4 | – | [58] |
| • Nonmyeloablative • Haploidentical | 0 out of 2 | – | [65] |
| • Nonmyeloablative • Haploidentical | 0 out of 9 | – | [66] |
| • Reduced-intensity • Matched-related and unrelated donor | 0 out of 2 | Stable brain MRI in 17 patients | [35] |
| TOTAL | 1 out of 29 (3%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saraf, S.L.; Rondelli, D. Allogeneic Hematopoietic Stem Cell Transplantation for Adults with Sickle Cell Disease. J. Clin. Med. 2019, 8, 1565. https://doi.org/10.3390/jcm8101565
Saraf SL, Rondelli D. Allogeneic Hematopoietic Stem Cell Transplantation for Adults with Sickle Cell Disease. Journal of Clinical Medicine. 2019; 8(10):1565. https://doi.org/10.3390/jcm8101565
Chicago/Turabian StyleSaraf, Santosh L., and Damiano Rondelli. 2019. "Allogeneic Hematopoietic Stem Cell Transplantation for Adults with Sickle Cell Disease" Journal of Clinical Medicine 8, no. 10: 1565. https://doi.org/10.3390/jcm8101565
APA StyleSaraf, S. L., & Rondelli, D. (2019). Allogeneic Hematopoietic Stem Cell Transplantation for Adults with Sickle Cell Disease. Journal of Clinical Medicine, 8(10), 1565. https://doi.org/10.3390/jcm8101565