Targeting Cellular Trafficking of Fibroblast Growth Factor Receptors as a Strategy for Selective Cancer Treatment

 , and
, and 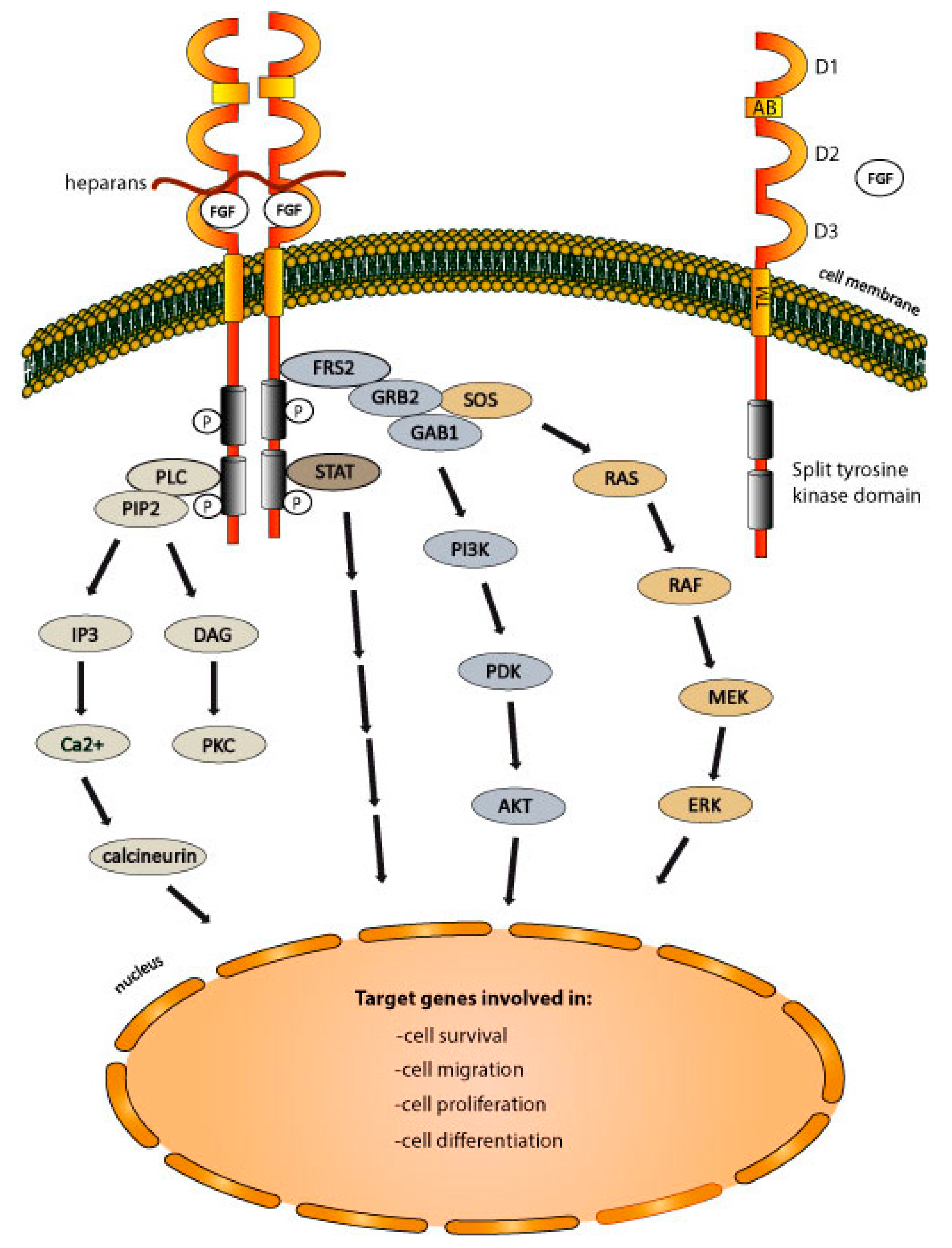{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Fibroblast Growth Factor Receptors
2.1. Structure of FGFRs
2.2. The Mechanism of FGFRs Activation
2.3. FGFRs-Dependent Signaling Pathways
3. Dysregulation of FGFRs in Cancers
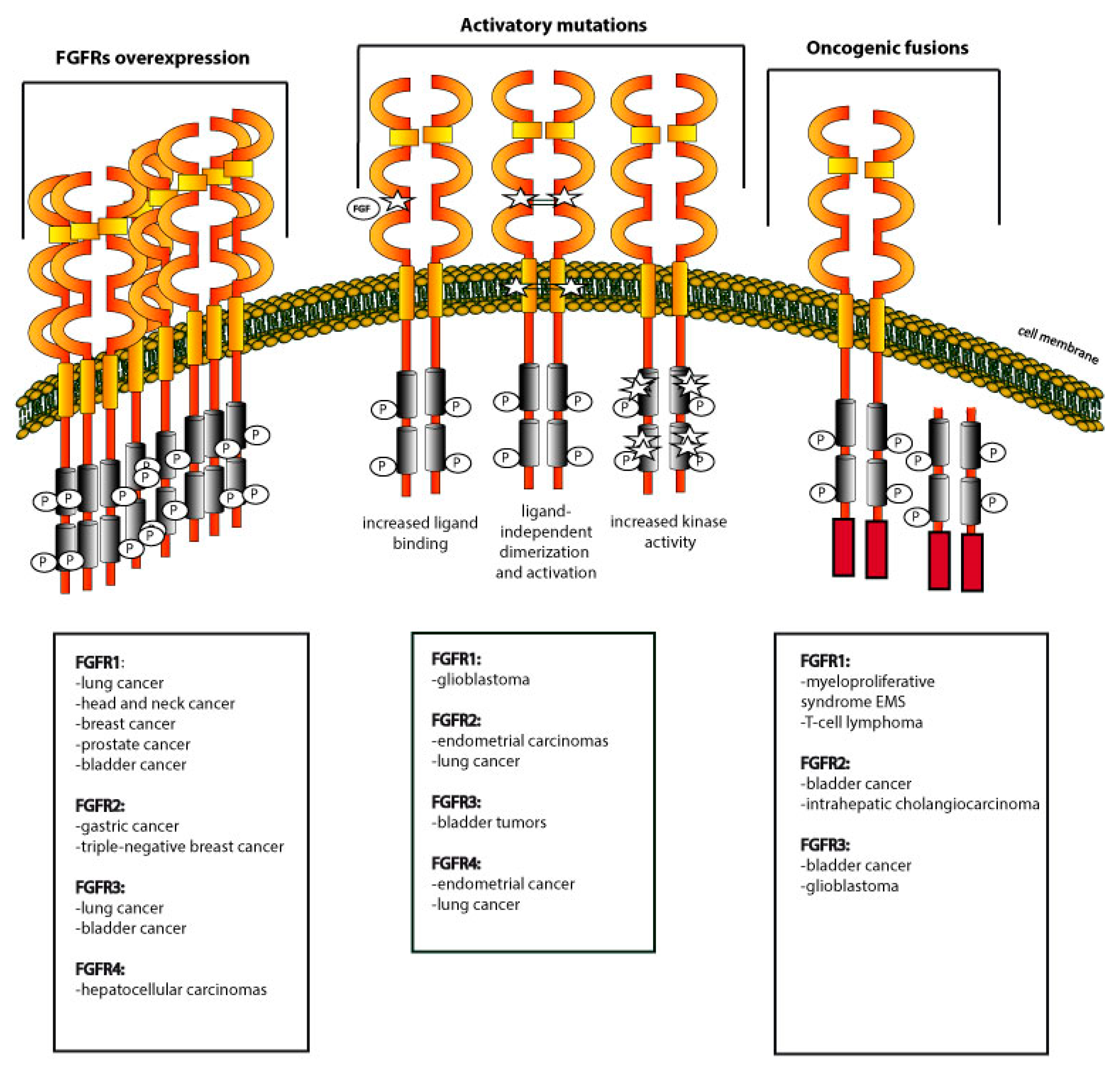
3.1. Overexpression of FGFRs
3.2. Activating Mutations within FGFRs
3.3. Oncogenic Fusions of FGFRs
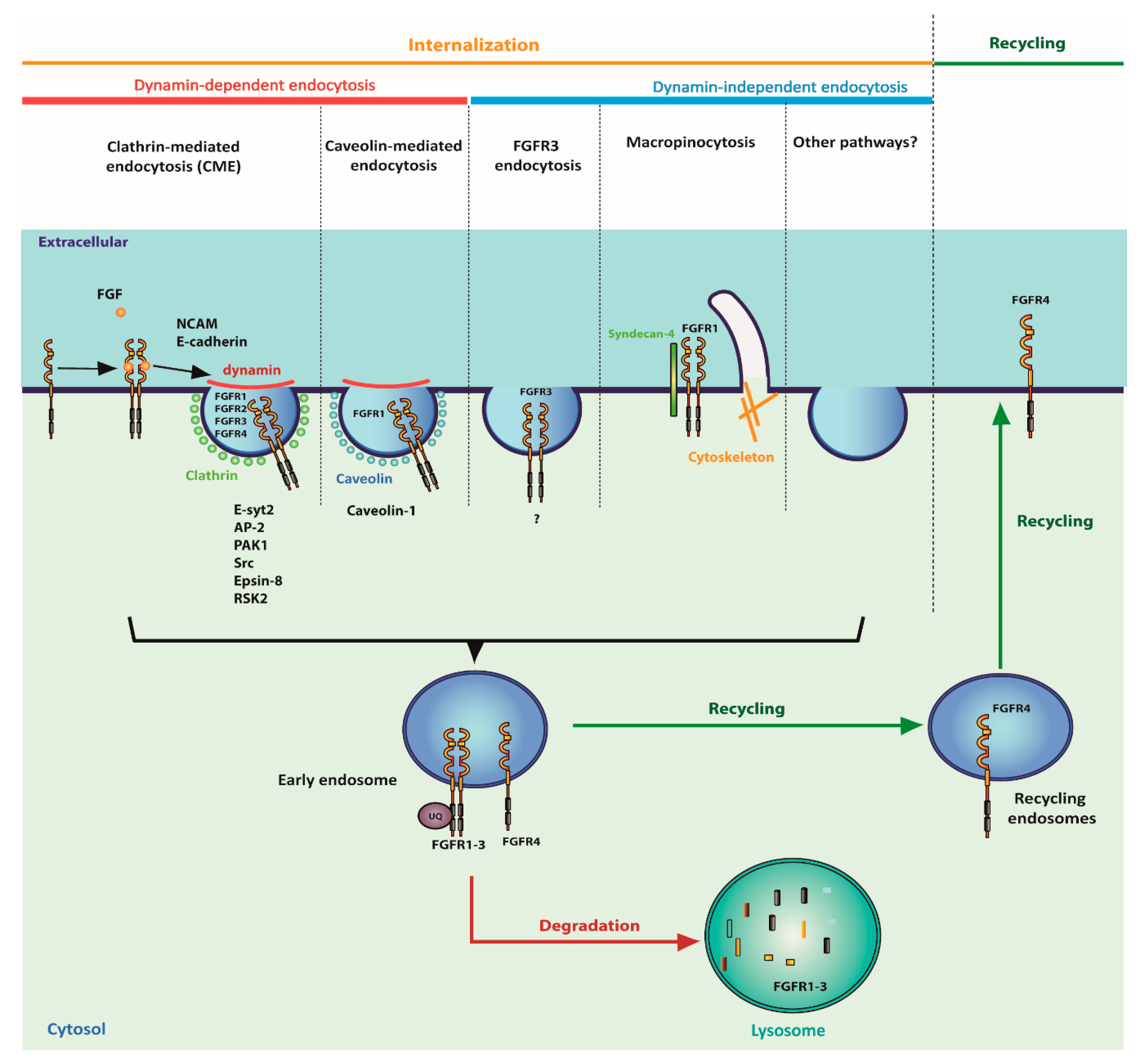
4. Cellular Trafficking of FGFRs
4.1. Internalization of FGFRs
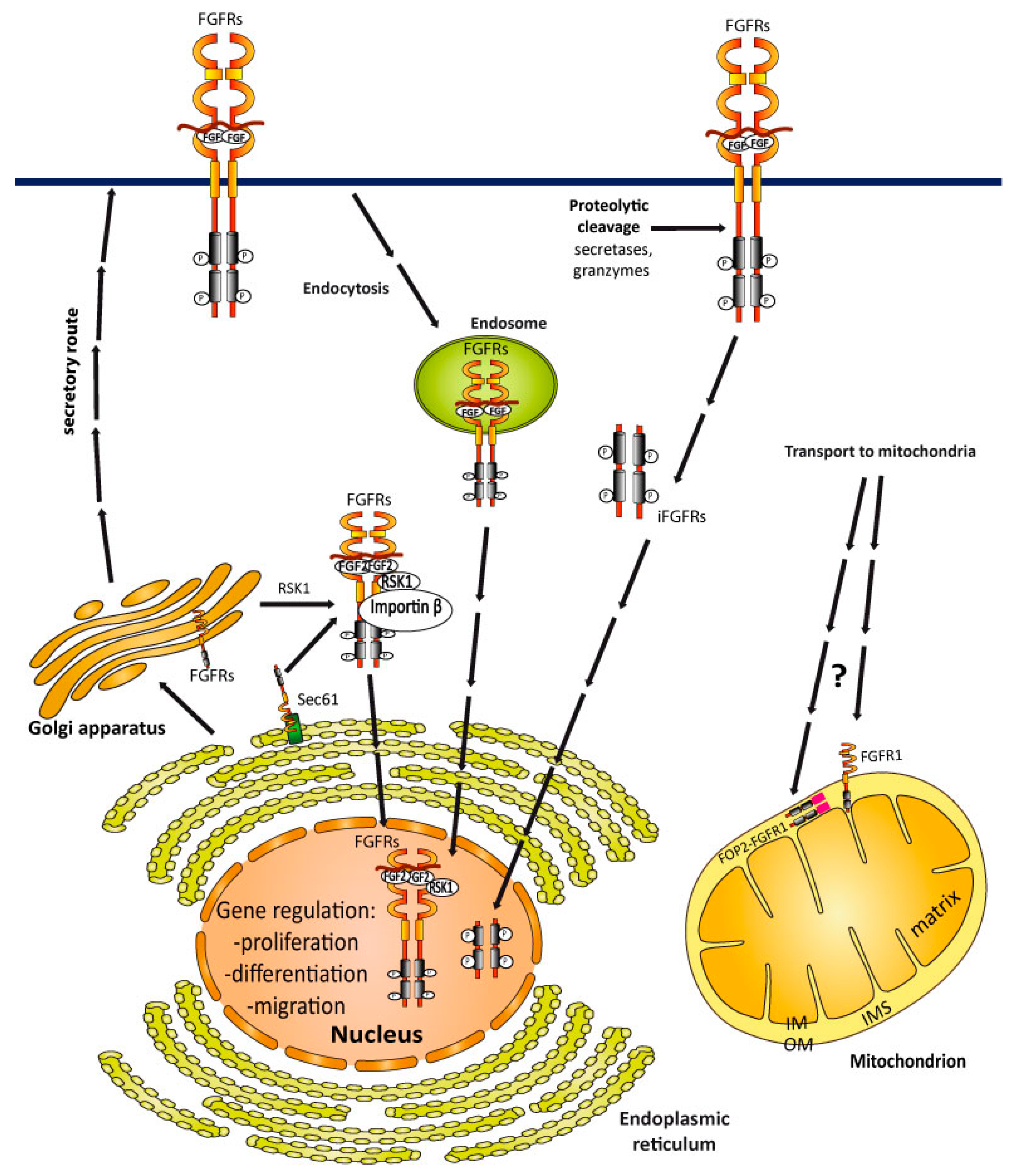
4.2. Transport of FGFRs to the Nucleus
4.3. FGFRs Sorting into Mitochondria
5. Therapeutic Strategies against Cancers with Abnormal FGFRs
5.1. Employing Internalization of FGFRs for Selective Treatment of FGFR-Dependent Cancers
5.2. Targeting the Intracellular Sorting of FGFRs into Nucleus and Mitochondria in Cancer
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AB | acidic box |
| ADCs | antibody-drug conjugates |
| AP-2 | adaptin-2 complex |
| CCPs | clathrin-coated pits |
| CIE | clathrin-independent endocytosis |
| CLIC | clathrin-independent carriers |
| CME | clathrin-mediated endocytosis |
| DAG | diacylglycerol |
| EMS | 8p11 myeloproliferative syndrome |
| ERAD | ER-associated protein degradation |
| ESCRT-0 | endosomal sorting complex required for transport-0 |
| Esyt2 | extended synaptotagmin-2 |
| FGFs | fibroblast growth factors |
| FGFRs | fibroblast growth factor receptors |
| GPI | glycosylphosphatidylinositol |
| GRB2 | growth factor receptor bound-2 |
| HPV | human papilloma virus |
| Hrs | hepatocyte growth factor-regulated tyrosine kinase substrate |
| IL2R | interleukin-2 receptor |
| IM | mitochondrial inner membrane |
| IMS | mitochondrial intermembrane space |
| IP3 | inositol triphosphate |
| LDCs | ligand-drug conjugates |
| MMAE | monomethyl auristatin E |
| mTOR | mammalian target of rapamycin |
| NCAM | neural cell adhesion molecule |
| OM | mitochondrial outer membrane |
| PAK1 | p21-GTPase Activated Kinase |
| PDC | pyruvate dehydrogenase complex |
| PDHK1 | pyruvate dehydrogenase kinase 1 |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| PLCγ | phospholipase C-gamma |
| PSCs | pancreatic stellate cells |
| RSK1 | p90 ribosomal S6 kinase 1 |
| RSK2 | p90 ribosomal S6 kinase 2 |
| RTKs | receptor tyrosine kinases |
| S4 | syndecan-4 |
| SOS | son of sevenless |
| STAT | signal transducer and activator of transcription |
| TACC3 | transforming acidic coiled-coil containing 3 |
| TKIs | tyrosine kinase inhibitors |
References
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [PubMed]
- Sclessinger, J. Receptor tyrosine kinases: Legacy of the first two decades. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Zinkle, A.; Mohammadi, M. A threshold model for receptor tyrosine kinase signaling specificity and cell fate determination. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T. Regulation and targets of receptor tyrosine kinases. Eur. J Cancer 2002, 38 (Suppl. S5), S3–S10. [Google Scholar] [CrossRef]
- Du, J.; Yu, Y.; Zhan, J.; Zhang, H. Targeted Therapies Against Growth Factor Signaling in Breast Cancer. Adv. Exp. Med. Biol. 2017, 1026, 125–146. [Google Scholar] [CrossRef]
- Takeuchi, K.; Ito, F. Receptor tyrosine kinases and targeted cancer therapeutics. Biol. Pharm. Bull. 2011, 34, 1774–1780. [Google Scholar] [CrossRef]
- Hojjat-Farsangi, M. Small-molecule inhibitors of the receptor tyrosine kinases: Promising tools for targeted cancer therapies. Int. J. Mol. Sci. 2014, 15, 13768–13801. [Google Scholar] [CrossRef]
- Bennasroune, A.; Gardin, A.; Aunis, D.; Crémel, G.; Hubert, P. Tyrosine kinase receptors as attractive targets of cancer therapy. Crit. Rev. Oncol. Hematol. 2004, 50, 23–38. [Google Scholar] [CrossRef]
- Kalinina, J.; Dutta, K.; Ilghari, D.; Beenken, A.; Goetz, R.; Eliseenkova, A.V.; Cowburn, D.; Mohammadi, M. The Alternatively Spliced Acidic Box Region Plays a Key Role in FGF Receptor Autoinhibition. Structure 2012, 20, 77–88. [Google Scholar] [CrossRef]
- Olsen, S.K.; Li, J.Y.; Bromleigh, C.; Eliseenkova, A.V.; Ibrahimi, O.A.; Lao, Z.; Zhang, F.; Linhardt, R.J.; Joyner, A.L.; Mohammadi, M. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 2006, 20, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Yie, J.; Wang, W.; Deng, L.; Tam, L.T.; Stevens, J.; Chen, M.M.; Li, Y.; Xu, J.; Lindberg, R.; Hecht, R.; et al. Understanding the physical interactions in the FGF21/FGFR/β-Klotho complex: Structural requirements and implications in FGF21 signaling. Chem. Biol. Drug Des. 2012, 79, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Opaliński, Ł.; Sokołowska-Wędzina, A.; Szczepara, M.; Zakrzewska, M.; Otlewski, J. Antibody-induced dimerization of FGFR1 promotes receptor endocytosis independently of its kinase activity. Sci. Rep. 2017, 7, 7121. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Heras, E.; Howell, F.V.; Williams, G.; Doherty, P. The fibroblast growth factor receptor acid box is essential for interactions with N-cadherin and all of the major isoforms of neural cell adhesion molecule. J. Biol. Chem. 2006, 281, 35208–35216. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005, 16, 107–137. [Google Scholar] [CrossRef]
- Peng, W.C.; Lin, X.; Torres, J. The strong dimerization of the transmembrane domain of the fibroblast growth factor receptor (FGFR) is modulated by C-terminal juxtamembrane residues. Protein Sci. 2009, 18, 450–459. [Google Scholar] [CrossRef]
- Bocharov, E.V.; Lesovoy, D.M.; Goncharuk, S.A.; Goncharuk, M.V.; Hristova, K.; Arseniev, A.S. Structure of FGFR3 transmembrane domain dimer: Implications for signaling and human pathologies. Structure 2013, 21, 2087–2093. [Google Scholar] [CrossRef]
- Sarabipour, S.; Hristova, K. FGFR3 unliganded dimer stabilization by the juxtamembrane domain. J. Mol. Biol. 2015, 427, 1705–1714. [Google Scholar] [CrossRef]
- Lin, H.Y.; Xu, J.; Ischenko, I.; Ornitz, D.M.; Halegoua, S.; Hayman, M.J. Identification of the cytoplasmic regions of fibroblast growth factor (FGF) receptor 1 which play important roles in induction of neurite outgrowth in PC12 cells by FGF-1. Mol. Cell. Biol. 1998, 18, 3762–3770. [Google Scholar] [CrossRef]
- Burgar, H.R.; Burns, H.D.; Elsden, J.L.; Lalioti, M.D.; Heath, J.K. Association of the signaling adaptor FRS2 with fibroblast growth factor receptor 1 (Fgfr1) is mediated by alternative splicing of the juxtamembrane domain. J. Biol. Chem. 2002, 277, 4018–4023. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Marie, P.J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 2015, 29, 1463–1486. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, M.; Trueb, B. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics 2000, 69, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, M.; Fraser, J.; McDonald, M.; Yuan, S.; White, D.; Grandison, P.; Kumble, K.; Watson, J.D.; Murison, J.G. Identification of a new fibroblast growth factor receptor, FGFR5. Gene 2001, 271, 171–182. [Google Scholar] [CrossRef]
- Trueb, B.; Zhuang, L.; Taeschler, S.; Wiedemann, M. Characterization of FGFRL1, a novel fibroblast growth factor (FGF) receptor preferentially expressed in skeletal tissues. J. Biol. Chem. 2003, 278, 33857–33865. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.N.; Altamentova, S.M.; Kilkenny, D.M.; Rocheleau, J.V. Fibroblast growth factor receptor like-1 (FGFRL1) interacts with SHP-1 phosphatase at insulin secretory granules and induces beta-cell ERK1/2 protein activation. J. Biol. Chem. 2013, 288, 17859–17870. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Pandey, A.V.; Villiger, P.M.; Trueb, B. Cell-cell fusion induced by the Ig3 domain of receptor FGFRL1 in CHO cells. Biochim. Biophys. Acta 2015, 1853, 2273–2285. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Steinberg, F.; Zhuang, L.; Bessey, R.; Trueb, B. Receptor FGFRL1 does not promote cell proliferation but induces cell adhesion. Int. J. Mol. Med. 2016, 38, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Trueb, B. Evolution of the fusogenic activity of the receptor FGFRL1. Arch. Biochem. Biophys. 2017, 625–626, 54–64. [Google Scholar] [CrossRef]
- Kähkönen, T.E.; Ivaska, K.K.; Jiang, M.; Büki, K.G.; Väänänen, H.K.; Härkönen, P.L. Role of fibroblast growth factor receptors (FGFR) and FGFR like-1 (FGFRL1) in mesenchymal stromal cell differentiation to osteoblasts and adipocytes. Mol. Cell. Endocrinol. 2018, 461, 194–204. [Google Scholar] [CrossRef]
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.K.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 2006, 281, 15694–15700. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Bottaro, D.P.; Fleming, T.P.; Smith, C.L.; Burgess, W.H.; Chan, A.M.; Aaronson, S.A. Determination of ligand-binding specificity by alternative splicing: Two distinct growth factor receptors encoded by a single gene. Proc. Natl. Acad. Sci. USA 1992, 89, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Chellaiah, A.T.; McEwen, D.G.; Werner, S.; Xu, J.; Ornitz, D.M. Fibroblast growth factor receptor (FGFR) 3. Alternative splicing in immunoglobulin-like domain III creates a receptor highly specific for acidic FGF/FGF-1. J. Biol. Chem. 1994, 269, 11620–11627. [Google Scholar] [PubMed]
- Ishiwata, T. Role of fibroblast growth factor receptor-2 splicing in normal and cancer cells. Front. Biosci. 2018, 23, 626–639. [Google Scholar] [CrossRef]
- Yeh, B.K.; Igarashi, M.; Eliseenkova, A.V.; Plotnikov, A.N.; Sher, I.; Ron, D.; Aaronson, S.A.; Mohammadi, M. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 2266–2271. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 2000, 101, 413–424. [Google Scholar] [CrossRef]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180. [Google Scholar] [CrossRef]
- Partanen, J.; Mäkelä, T.P.; Eerola, E.; Korhonen, J.; Hirvonen, H.; Claesson-Welsh, L.; Alitalo, K. FGFR-4, a novel acidic fibroblast growth factor receptor with a distinct expression pattern. EMBO J. 1991, 10, 1347–1354. [Google Scholar] [CrossRef]
- Duan, D.S.; Werner, S.; Williams, L.T. A naturally occurring secreted form of fibroblast growth factor (FGF) receptor 1 binds basic FGF in preference over acidic FGF. J. Biol. Chem. 1992, 267, 16076–16080. [Google Scholar]
- Gong, S.G. Isoforms of receptors of fibroblast growth factors. J. Cell. Physiol. 2014, 229, 1887–1895. [Google Scholar] [CrossRef]
- Tomlinson, D.C.; L’Hôte, C.G.; Kennedy, W.; Pitt, E.; Knowles, M.A. Alternative splicing of fibroblast growth factor receptor 3 produces a secreted isoform that inhibits fibroblast growth factor-induced proliferation and is repressed in urothelial carcinoma cell lines. Cancer Res. 2005, 65, 10441–10449. [Google Scholar] [CrossRef] [PubMed]
- Sarabipour, S.; Hristova, K. Mechanism of FGF receptor dimerization and activation. Nat. Commun. 2016, 7, 10262. [Google Scholar] [CrossRef]
- Comps-Agrar, L.; Dunshee, D.R.; Eaton, D.L.; Sonoda, J. Unliganded fibroblast growth factor receptor 1 forms density-independent dimers. J. Biol. Chem. 2015, 290, 24166–24177. [Google Scholar] [CrossRef] [PubMed]
- Del Piccolo, N.; Sarabipour, S.; Hristova, K. A New Method to Study Heterodimerization of Membrane Proteins and Its Application to Fibroblast Growth Factor Receptors. J. Biol. Chem. 2017, 292, 1288–1301. [Google Scholar] [CrossRef] [PubMed]
- Gómez, A.; Wellbrock, C.; Gutbrod, H.; Dimitrijevic, N.; Schartl, M. Ligand-independent dimerization and activation of the oncogenic Xmrk receptor by two mutations in the extracellular domain. J. Biol. Chem. 2001, 276, 3333–3340. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Gong, K.; Wohlfeld, B.; Hatanpaa, K.J.; Zhao, D.; Habib, A.A. Ligand-Independent EGFR Signaling. Cancer Res. 2015, 75, 3436–3441. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Sharma, K.D.; Takahashi, T.; Iwamoto, R.; Mekada, E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol. Biol. Cell 2002, 13, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Furdui, C.M.; Lew, E.D.; Schlessinger, J.; Anderson, K.S. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol. Cell 2006, 21, 711–717. [Google Scholar] [CrossRef]
- Mohammadi, M.; Honegger, A.M.; Rotin, D.; Fischer, R.; Bellot, F.; Li, W.; Dionne, C.A.; Jaye, M.; Rubinstein, M.; Schlessinger, J. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-gamma 1. Mol. Cell. Biol. 1991, 11, 5068–5078. [Google Scholar] [CrossRef]
- Mohammadi, M.; Dionne, C.A.; Li, W.; Li, N.; Spivak, T.; Honegger, A.M.; Jaye, M.; Schlessinger, J. Point mutation in FGF receptor eliminates phosphatidylinositol hydrolysis without affecting mitogenesis. Nature 1992, 358, 681–684. [Google Scholar] [CrossRef]
- Dudka, A.A.; Sweet, M.M.; Heath, J.K. STAT3 binding to the FGF receptor is activated by receptor amplification. Cancer Res. 2010, 70, 3391–3401. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.H.; Guy, G.R.; Hadari, Y.R.; Laks, S.; Gotoh, N.; Schlessinger, J.; Lax, I. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol. Cell. Biol. 2000, 20, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Fukami, K.; Inanobe, S.; Kanemaru, K.; Nakamura, Y. Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Prog. Lipid Res. 2010, 49, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Kadamur, G.; Ross, E.M. Mammalian phospholipase C. Annu. Rev. Physiol. 2013, 75, 127–154. [Google Scholar] [CrossRef] [PubMed]
- Black, A.R.; Black, J.D. Protein kinase C signaling and cell cycle regulation. Front. Immunol. 2012, 3, 423. [Google Scholar] [CrossRef]
- Hart, K.C.; Robertson, S.C.; Kanemitsu, M.Y.; Meyer, A.N.; Tynan, J.A.; Donoghue, D.J. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene 2000, 19, 3309–3320. [Google Scholar] [CrossRef]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef]
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Buscà, R.; Pouysségur, J.; Lenormand, P. ERK1 and ERK2 Map Kinases: Specific Roles or Functional Redundancy? Front. Cell Dev. Biol. 2016, 4, 53. [Google Scholar] [CrossRef]
- Tateossian, H.; Powles, N.; Dickinson, R.; Ficker, M.; Maconochie, M. Determination of downstream targets of FGF signalling using gene trap and cDNA subtractive approaches. Exp. Cell Res. 2004, 292, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.A.; Hyodo-Miura, J.; Kitayama, A.; Terasaka, C.; Nagamune, T.; Ueno, N. Screening of FGF target genes in Xenopus by microarray: Temporal dissection of the signalling pathway using a chemical inhibitor. Genes Cells 2004, 9, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.; Tambalo, M.; Ranganathan, R.; Grocott, T.; Streit, A. A gene network regulated by FGF signaling during ear development. Sci. Rep. 2017, 7, 612. [Google Scholar] [CrossRef]
- Raju, R.; Palapetta, S.M.; Sandhya, V.K.; Sahu, A.; Alipoor, A.; Balakrishnan, L.; Advani, J.; George, B.; Kini, K.R.; Geetha, N.P.; et al. A Network Map of FGF-1/FGFR Signaling System. J. Signal Transduct. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hallinan, N.; Finn, S.; Cuffe, S.; Rafee, S.; O’Byrne, K.; Gately, K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat. Rev. 2016, 46, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar] [CrossRef]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Koole, K.; Brunen, D.; van Kempen, P.M.; Noorlag, R.; de Bree, R.; Lieftink, C.; van Es, R.J.; Bernards, R.; Willems, S.M. FGFR1 Is a Potential Prognostic Biomarker and Therapeutic Target in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2016, 22, 3884–3893. [Google Scholar] [CrossRef]
- Koole, K.; van Kempen, P.M.; Swartz, J.E.; Peeters, T.; van Diest, P.J.; Koole, R.; van Es, R.J.; Willems, S.M. Fibroblast growth factor receptor 3 protein is overexpressed in oral and oropharyngeal squamous cell carcinoma. Cancer Med. 2016, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Reis-Filho, J.S.; Simpson, P.T.; Turner, N.C.; Lambros, M.B.; Jones, C.; Mackay, A.; Grigoriadis, A.; Sarrio, D.; Savage, K.; et al. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin. Cancer Res. 2006, 12, 6652–6662. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Seo, A.N.; Park, S.Y.; Kim, J.Y.; Park, J.Y.; Yu, J.H.; Ahn, J.H.; Gong, G. Low prognostic implication of fibroblast growth factor family activation in triple-negative breast cancer subsets. Ann. Surg. Oncol. 2014, 21, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Gelsi-Boyer, V.; Orsetti, B.; Cervera, N.; Finetti, P.; Sircoulomb, F.; Rougé, C.; Lasorsa, L.; Letessier, A.; Ginestier, C.; Monville, F.; et al. Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol. Cancer Res. 2005, 3, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.H.; Kim, E.J.; Choi, Y.; Lee, H.E.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Kim, I.A.; Parl, S.Y. FGFR1 is amplified during the progression of in situ to invasive breast carcinoma. Breast Cancer Res. 2012, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Elbauomy Elsheikh, S.; Green, A.R.; Lambros, M.B.; Turner, N.C.; Grainge, M.J.; Powe, D.; Ellis, I.O.; Reis-Filho, J.S. FGFR1 amplification in breast carcinomas: A chromogenic in situ hybridisation analysis. Breast Cancer Res. 2007, 9, R23. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.; Darby, S.; Mathers, M.E.; Gnanapragasam, V.J. Evidence for distinct alterations in the FGF axis in prostate cancer progression to an aggressive clinical phenotype. J. Pathol. 2010, 220, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, A.; Tooyama, I.; Yoshiki, T.; Kimura, H. Demonstration of fibroblast growth factor receptor-I in human prostate by polymerase chain reaction and immunohistochemistry. Prostate 1995, 27, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Kauffmann, A.; Wöhrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.C.; Lamont, F.R.; Shnyder, S.D.; Knowles, M.A. Fibroblast growth factor receptor 1 promotes proliferation and survival via activation of the mitogen-activated protein kinase pathway in bladder cancer. Cancer Res. 2009, 69, 4613–4620. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Arao, T.; Hamaguchi, T.; Shimada, Y.; Kato, K.; Oda, I.; Taniguchi, H.; Koizumi, F.; Yanagihara, K.; Sasaki, H.; et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br. J. Cancer 2012, 106, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Lambros, M.B.; Horlings, H.M.; Pearson, A.; Sharpe, R.; Natrajan, R.; Geyer, F.C.; van Kouwenhove, M.; Kreike, B.; Mackay, A.; et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 2010, 29, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Zhan, P.; Gavine, P.R.; Morgan, S.; Womack, C.; Ni, X.; Shen, D.; Bang, Y.J.; Im, S.A.; Ho Kim, W.; Jung, E.J.; Grabsch, H.I.; Kilgour, E. FGFR2 amplification has prognostic significance in gastric cancer: Results from a large international multicentre study. Br. J. Cancer 2014, 110, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Metzner, T.; Bedeir, A.; Held, G.; Peter-Vörösmarty, B.; Ghassemi, S.; Heinzle, C.; Spiegl-Kreinecker, S.; Marian, B.; Holzmann, K.; Grasl-Kraupp, B.; et al. Fibroblast Growth Factor Receptors as Therapeutic Targets in Human Melanoma: Synergism with BRAF Inhibition. J. Investig. Dermatol. 2011, 131, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Theelen, W.S.; Mittempergher, L.; Willems, S.M.; Bosma, A.J.; Peters, D.D.; van der Noort, V.; Japenga, E.J.; Peeters, T.; Koole, K.; Šuštić, T.; et al. FGFR1, 2 and 3 protein overexpression and molecular aberrations of FGFR3 in early stage non-small cell lung cancer. J. Pathol. Clin. Res. 2016, 2, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Baldia, P.H.; Maurer, A.; Heide, T.; Rose, M.; Stoehr, R.; Hartmann, A.; Williams, S.V.; Knowles, M.A.; Knuechel, R.; Gaisa, N.T. Fibroblast growth factor receptor (FGFR) alterations in squamous differentiated bladder cancer: A putative therapeutic target for a small subgroup. Oncotarget 2016, 7, 71429–71439. [Google Scholar] [CrossRef] [PubMed]
- Gauglhofer, C.; Sagmeister, S.; Schrottmaier, W.; Fischer, C.; Rodgarkia-Dara, C.; Mohr, T.; Stättner, S.; Bichler, C.; Kandioler, D.; Wrba, F.; et al. Up-regulation of the fibroblast growth factor 8 subfamily in human hepatocellular carcinoma for cell survival and neoangiogenesis. Hepatology 2011, 53, 854–864. [Google Scholar] [CrossRef]
- Gauglhofer, C.; Paur, J.; Schrottmaier, W.C.; Wingelhofer, B.; Huber, D.; Naegelen, I.; Pirker, C.; Mohr, T.; Heinzle, C.; Holzmann, K.; et al. Fibroblast growth factor receptor 4: A putative key driver for the aggressive phenotype of hepatocellular carcinoma. Carcinogenesis 2014, 35, 2331–2338. [Google Scholar] [CrossRef]
- Ho, H.K.; Pok, S.; Streit, S.; Ruhe, J.E.; Hart, S.; Lim, K.S.; Loo, H.L.; Aung, M.O.; Lim, S.G.; Ullrich, A. Fibroblast growth factor receptor 4 regulates proliferation, anti-apoptosis and alpha-fetoprotein secretion during hepatocellular carcinoma progression and represents a potential target for therapeutic intervention. J. Hepatol. 2009, 50, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, W.; Doughtie, A.; Cui, G.; Li, X.; Pandit, H.; Yang, Y.; Li, S.; Martin, R. Up-regulation of fibroblast growth factor 19 and its receptor associates with progression from fatty liver to hepatocellular carcinoma. Oncotarget 2016, 7, 52329. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Patani, H.; Bunney, T.D.; Thiyagarajan, N.; Norman, R.A.; Ogg, D.; Breed, J.; Asford, P.; Pottertom, A.; Edwards, M.; Williams, S.V.; et al. Landscape of activating cancer mutations in FGFR kinases and their differential responses to inhibitors in clinical use. Oncotarget 2016, 7, 24252–24268. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.H.; Nelson, K.N.; Meyer, A.N.; Donoghue, D.J. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015, 26, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Lew, E.D.; Furdui, C.M.; Anderson, K.S.; Schlessinger, J. The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci. Signal. 2009, 2, ra6. [Google Scholar] [CrossRef] [PubMed]
- Rand, V.; Huang, J.; Stockwell, T.; Ferriera, S.; Buzko, O.; Levy, S.; Busam, D.; Li, K.; Edwards, J.B.; Eberhart, C.; et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc. Natl. Acad. Sci. USA 2005, 102, 14344–14349. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Gartside, M.G.; Dejeza, L.C.; Powell, M.A.; Mallon, M.A.; Davies, H.; Mohammadi, M.; Futreal, P.A.; Stratton, M.R.; Trent, J.M.; et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 2007, 26, 7158–7162. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Pouessel, D.; Neuzillet, Y.; Mertens, L.S.; van der Heijden, M.S.; de Jong, J.; Sanders, J.; Peters, D.; Leroy, K.; Manceau, A.; Maille, P.; et al. Tumor heterogeneity of fibroblast growth factor receptor 3 (FGFR3) mutations in invasive bladder cancer: Implications for perioperative anti-FGFR3 treatment. Ann. Oncol. 2016, 27, 1311–1316. [Google Scholar] [CrossRef]
- Hafner, C.; van Oers, J.M.; Vogt, T.; Landthaler, M.; Stoehr, R.; Blaszyk, H.; Hofstaedter, F.; Zwarthoff, E.C.; Hartmann, A. Mosaicism of activating FGFR3 mutations in human skin causes epidermal nevi. J. Clin. Investig. 2006, 116, 2201–2207. [Google Scholar] [CrossRef]
- Marks, J.L.; McLellan, M.D.; Zakowski, M.F.; Lash, A.E.; Kasai, Y.; Broderick, S.; Sakaria, I.S.; Pham, D.; Sing, B.; Miner, T.L.; et al. Mutational Analysis of EGFR and Related Signaling Pathway Genes in Lung Adenocarcinomas Identifies a Novel Somatic Kinase Domain Mutation in FGFR4. PLoS ONE 2007, 2, e426. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Hunter, C.; Smith, R.; Stephens, P.; Greenman, C.; Bignell, G.; Teague, J.; Butler, A.; Edkins, S.; Stevens, C.; et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005, 65, 7591–7595. [Google Scholar] [CrossRef]
- Ibrahimi, O.A.; Eliseenkova, A.V.; Plotnikov, A.N.; Yu, K.; Ornitz, D.M.; Mohammadi, M. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc. Natl. Acad. Sci. USA 2001, 98, 7182–7187. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Herr, A.B.; Waksman, G.; Ornitz, D.M. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 14536–14541. [Google Scholar] [CrossRef] [PubMed]
- van Rhijn, B.W.G.; Lurkin, I.; van Rhijn, F.; Kirkels, W.J.; van der Kwast, T.H.; Zwarthoff, E.C. Zwarthoff The Fibroblast Growth Factor Receptor 3 (FGFR3) Mutation Is a Strong Indicator of Superficial Bladder Cancer with Low Recurrence Rate. Cancer Res. 2001, 61, 1265–1268. [Google Scholar] [PubMed]
- Bernard-Pierrot, I.; Brams, A.; Dunois-Lardé, C.; Caillault, A.; Diez de Medina, S.G.; Cappellen, D.; Graff, G.; Thiery, J.P.; Chopin, D.; Ricol, D.; Radvanyi, F. Oncogenic properties of the mutated forms of fibroblast growth factor receptor 3b. Carcinogenesis 2006, 27, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Roidl, A.; Foo, P.; Wong, W.; Mann, C.; Bechtold, S.; Berger, H.J.; Streit, S.; Ruhe, J.E.; Hart, S.; Ullrich, A.; et al. The FGFR4 Y367C mutant is a dominant oncogene in MDA-MB453 breast cancer cells. Oncogene 2010, 29, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.K.; Donoghue, D.J. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J. 1996, 15, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Lievens, P.M.; Mutinelli, C.; Baynes, D.; Liboi, E. The kinase activity of fibroblast growth factor receptor 3 with activation loop mutations affects receptor trafficking and signaling. J. Biol. Chem. 2004, 279, 43254–43260. [Google Scholar] [CrossRef]
- Gartside, M.G.; Chen, H.; Ibrahimi, O.A.; Byron, S.A.; Curtis, A.V.; Wellens, C.L.; Bengston, A.; Yudt, L.M.; Eliseenkova, A.V.; Ma, J.; et al. Loss-of-function fibroblast growth factor receptor-2 mutations in melanoma. Mol. Cancer Res. 2009, 7, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Nalabolu, S.R.; Aster, J.C.; Ma, J.; Abruzzo, L.; Jaffe, E.S.; Stone, R.; Weissman, S.M.; Hudson, T.J.; Fletcher, J.A. FGFR1 is fused with a novel zinc-finger gene, ZNF198, in the t(8;13) leukaemia/lymphoma syndrome. Nat. Genet. 1998, 18, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Sohal, J.; Chase, A.; Mould, S.; Corcoran, M.; Oscier, D.; Iqbal, S.; Parker, S.; Welborn, J.; Harris, R.I.; Martinelli, G.; et al. Identification of four new translocations involving FGFR1 in myeloid disorders. Genes Chromosomes Cancer 2001, 32, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Grand, E.K.; Grand, F.H.; Chase, A.J.; Ross, F.M.; Corcoran, M.M.; Oscier, D.G.; Cross, N.C. Identification of a novel gene, FGFR1OP2, fused to FGFR1 in 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer 2004, 40, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Takahashi, K.; Ishikawa, N.; Takano, A.; Yasui, W.; Inai, K.; Nishimura, H.; Tsuchiya, E.; Nakamura, Y.; Daigo, Y. Fibroblast growth factor receptor 1 oncogene partner as a novel prognostic biomarker and therapeutic target for lung cancer. Cancer Sci. 2007, 98, 1902–1913. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.L.; Goss, V.L.; Reeves, C.; Popova, L.; Nardone, J.; Macneill, J.; Walters, D.K.; Wang, Y.; Rush, J.; Comb, M.J.; et al. Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood 2006, 108, 4202–4204. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef]
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 gene fusions in bladder cancer. Hum. Mol. Genet. 2013, 22, 795–803. [Google Scholar] [CrossRef]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef]
- Jackson, C.C.; Medeiros, L.J.; Miranda, R.N. 8p11 myeloproliferative syndrome: A review. Hum. Pathol. 2010, 41, 461–476. [Google Scholar] [CrossRef]
- Medves, S.; Demoulin, J.B. Tyrosine kinase gene fusions in cancer: Translating mechanisms into targeted therapies. J. Cell. Mol. Med. 2012, 16, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Guasch, G.; Ollendorff, V.; Borg, J.P.; Birnbaum, D.; Pébusque, M.J. 8p12 stem cell myeloproliferative disorder: The FOP-fibroblast growth factor receptor 1 fusion protein of the t(6;8) translocation induces cell survival mediated by mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt/mTOR pathways. Mol. Cell. Biol. 2001, 21, 8129–8142. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Yagasaki, F.; Ishikawa, M.; Takahashi, N.; Bessho, M. Transforming property of TEL-FGFR3 mediated through PI3-K in a T-cell lymphoma that subsequently progressed to AML. Blood 2005, 105, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.N.; Meyer, A.N.; Siari, A.; Campos, A.R.; Motamedchaboki, K.; Donoghue, D.J. Oncogenic Gene Fusion FGFR3-TACC3 Is Regulated by Tyrosine Phosphorylation. Mol. Cancer Res. 2016, 14, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, M.K.; Stachowiak, E.K. Evidence-Based Theory for Integrated Genome Regulation of Ontogeny--An Unprecedented Role of Nuclear FGFR1 Signaling. J. Cell. Physiol. 2016, 231, 1199–1218. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Fan, J.; Chung, T.W.; Lythgoe, K.; Wang, X.; Xie, J.; Ge, Q.; Gu, T.L.; Polakiewicz, R.D.; Roesel, J.L.; et al. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 2011, 44, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.J.; Bruce, C.; Chioni, A.M.; Kocher, H.M.; Grose, R.P. The ins and outs of fibroblast growth factor receptor signalling. Clin. Sci. 2014, 127, 217–231. [Google Scholar] [CrossRef]
- Coleman, S.J.; Chioni, A.M.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481. [Google Scholar] [CrossRef]
- Goh, L.K.; Sorkin, A. Endocytosis of receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a017459. [Google Scholar] [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-independent pathways of endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.P.A.; Boucrot, E. Mechanisms of Carrier Formation during Clathrin-Independent Endocytosis. Trends Cell Biol. 2018, 28, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.B.; Popoff, M.R.; McCluskey, A.; Robinson, P.J.; Meunier, F.A. Targeting membrane trafficking in infection prophylaxis: Dynamin inhibitors. Trends Cell Biol. 2013, 23, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Maib, H.; Smythe, E.; Ayscough, K. Forty years on: Clathrin-coated pits continue to fascinate. Mol. Biol. Cell 2017, 28, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Gesbert, F.; Sauvonnet, N.; Dautry-Varsat, A. Clathrin-lndependent endocytosis and signalling of interleukin 2 receptors IL-2R endocytosis and signalling. Curr. Top. Microbiol. Immunol. 2004, 286, 119–148. [Google Scholar] [PubMed]
- Eyster, C.A.; Higginson, J.D.; Huebner, R.; Porat-Shliom, N.; Weigert, R.; Wu, W.W.; Shen, R.F.; Donaldson, J.G. Discovery of new cargo proteins that enter cells through clathrin-independent endocytosis. Traffic 2009, 10, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Canton, J. Macropinocytosis: New Insights Into Its Underappreciated Role in Innate Immune Cell Surveillance. Front. Immunol. 2018, 9, 2286. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef]
- Abella, J.V.; Park, M. Breakdown of endocytosis in the oncogenic activation of receptor tyrosine kinases. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E973–E984. [Google Scholar] [CrossRef]
- Mellman, I.; Yarden, Y. Endocytosis and cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a016949. [Google Scholar] [CrossRef]
- Sorokin, A.; Mohammadi, M.; Huang, J.; Schlessinger, J. Internalization of fibroblast growth factor receptor is inhibited by a point mutation at tyrosine 766. J. Biol. Chem. 1994, 269, 17056–17061. [Google Scholar] [PubMed]
- Marchese, C.; Mancini, P.; Belleudi, F.; Felici, A.; Gradini, R.; Sansolini, T.; Frati, L.; Torrisi, M.R. Receptor-mediated endocytosis of keratinocyte growth factor. J. Cell Sci. 1998, 111, 3517–3527. [Google Scholar] [PubMed]
- Fannon, M.; Nugent, M.A. Basic fibroblast growth factor binds its receptors, is internalized, and stimulates DNA synthesis in Balb/c3T3 cells in the absence of heparan sulfate. J. Biol. Chem. 1996, 271, 17949–17956. [Google Scholar] [CrossRef] [PubMed]
- Auciello, G.; Cunningham, D.L.; Tatar, T.; Heath, J.K.; Rappoport, J.Z. Regulation of fibroblast growth factor receptor signalling and trafficking by Src and Eps8. J. Cell Sci. 2013, 126, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.; Mikryukov, A.; Tremblay, M.G.; Baril, J.; Guillou, F.; Bellenfant, S.; Moss, T. Extended-synaptotagmin-2 mediates FGF receptor endocytosis and ERK activation in vivo. Dev. Cell 2010, 19, 426–439. [Google Scholar] [CrossRef]
- Tremblay, M.G.; Herdman, C.; Guillou, F.; Mishra, P.K.; Baril, J.; Bellenfant, S.; Moss, T. Extended Synaptotagmin Interaction with the Fibroblast Growth Factor Receptor Depends on Receptor Conformation, Not Catalytic Activity. J. Biol. Chem. 2015, 290, 16142–16156. [Google Scholar] [CrossRef]
- Jean, S.; Tremblay, M.G.; Herdman, C.; Guillou, F.; Moss, T. The endocytic adapter E-Syt2 recruits the p21 GTPase activated kinase PAK1 to mediate actin dynamics and FGF signalling. Biol. Open 2012, 1, 731–738. [Google Scholar] [CrossRef]
- Sandilands, E.; Akbarzadeh, S.; Vecchione, A.; McEwan, D.G.; Frame, M.C.; Heath, J.K. Src kinase modulates the activation, transport and signalling dynamics of fibroblast growth factor receptors. EMBO Rep. 2007, 8, 1162–1169. [Google Scholar] [CrossRef]
- Feng, L.; Liao, W.X.; Luo, Q.; Zhang, H.H.; Wang, W.; Zheng, J.; Chen, D.B. Caveolin-1 orchestrates fibroblast growth factor 2 signaling control of angiogenesis in placental artery endothelial cell caveolae. J. Cell. Physiol. 2012, 227, 2480–2491. [Google Scholar] [CrossRef]
- Sahni, A.; Patel, J.; Narra, H.P.; Schroeder, C.L.C.; Walker, D.H.; Sahni, S.K. Fibroblast growth factor receptor-1 mediates internalization of pathogenic spotted fever rickettsiae into host endothelium. PLoS ONE 2017, 12, e0183181. [Google Scholar] [CrossRef]
- Elfenbein, A.; Lanahan, A.; Zhou, T.X.; Yamasaki, A.; Tkachenko, E.; Matsuda, M.; Simons, M. Syndecan 4 regulates FGFR1 signaling in endothelial cells by directing macropinocytosis. Sci. Signal. 2012, 5, ra36. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.F.; Mizukoshi, E.; Maher, P.A. Ligand dependent and independent internalization and nuclear translocation of fibroblast growth factor (FGF) receptor 1. DNA Cell Biol. 2004, 23, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Haugsten, E.M.; Zakrzewska, M.; Brech, A.; Pust, S.; Olsnes, S.; Sandvig, K.; Wesche, J. Clathrin- and dynamin-independent endocytosis of FGFR3--implications for signalling. PLoS ONE 2011, 6, e21708. [Google Scholar] [CrossRef] [PubMed]
- Citores, L.; Khnykin, D.; Sørensen, V.; Wesche, J.; Klingenberg, O.; Wiedłocha, A.; Olsnes, S. Modulation of intracellular transport of acidic fibroblast growth factor by mutations in the cytoplasmic receptor domain. J. Cell Sci. 2001, 114, 1677–1689. [Google Scholar] [PubMed]
- Francavilla, C.; Cattaneo, P.; Berezin, V.; Bock, E.; Ami, D.; de Marco, A.; Christofori, G.; Cavallaro, U. The binding of NCAM to FGFR1 induces a specific cellular response mediated by receptor trafficking. J. Cell Biol. 2009, 187, 1101–1116. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.M.; Wylie, F.G.; Stow, J.L. Regulation of endocytosis, nuclear translocation, and signaling of fibroblast growth factor receptor 1 by E-cadherin. Mol. Biol. Cell 2005, 16, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Nadratowska-Wesolowska, B.; Haugsten, E.M.; Zakrzewska, M.; Jakimowicz, P.; Zhen, Y.; Pajdzik, D.; Wesche, J.; Wiedlocha, A. RSK2 regulates endocytosis of FGF receptor 1 by phosphorylation on serine 789. Oncogene 2014, 33, 4823–4836. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Sørensen, V.; Brech, A.; Olsnes, S.; Wesche, J. Different intracellular trafficking of FGF1 endocytosed by the four homologous FGF receptors. J. Cell Sci. 2005, 118, 3869–3881. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Malecki, J.; Bjørklund, S.M.; Olsnes, S.; Wesche, J. Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol. Biol. Cell 2008, 19, 3390–3403. [Google Scholar] [CrossRef]
- Belleudi, F.; Leone, L.; Maggio, M.; Torrisi, M.R. Hrs regulates the endocytic sorting of the fibroblast growth factor receptor 2b. Exp. Cell Res. 2009, 315, 2181–2191. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Sørensen, V.; Kunova Bosakova, M.; de Souza, G.A.; Krejci, P.; Wiedlocha, A.; Wesche, J. Proximity Labeling Reveals Molecular Determinants of FGFR4 Endosomal Transport. J. Proteome Res. 2016, 15, 3841–3855. [Google Scholar] [CrossRef] [PubMed]
- Kostas, M.; Haugsten, E.M.; Zhen, Y.; Sørensen, V.; Szybowska, P.; Fiorito, E.; Lorenz, S.; Jones, N.; de Souza, G.A.; Wiedlocha, A.; et al. Protein Tyrosine Phosphatase Receptor Type G (PTPRG) Controls Fibroblast Growth Factor Receptor (FGFR) 1 Activity and Influences Sensitivity to FGFR Kinase Inhibitors. Mol. Cell. Proteomics 2018, 17, 850–870. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Hung, M.C. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. FEBS J. 2015, 282, 3693–3721. [Google Scholar] [CrossRef] [PubMed]
- Merilahti, J.A.M.; Elenius, K. Gamma-secretase-dependent signaling of receptor tyrosine kinases. Oncogene 2018. [Google Scholar] [CrossRef]
- Carpenter, G.; Liao, H.J. Receptor tyrosine kinases in the nucleus. Cold Spring Harb. Perspect. Biol. 2013, 5, a008979. [Google Scholar] [CrossRef]
- Adam, R.M.; Danciu, T.; McLellan, D.L.; Borer, J.G.; Lin, J.; Zurakowski, D.; Weinstein, M.H.; Rajjayabun, P.H.; Mellon, J.K.; Freeman, M.R. A nuclear form of the heparin-binding epidermal growth factor-like growth factor precursor is a feature of aggressive transitional cell carcinoma. Cancer Res. 2003, 63, 484–490. [Google Scholar]
- Li, C.; Iida, M.; Dunn, E.F.; Ghia, A.J.; Wheeler, D.L. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009, 28, 3801–3813. [Google Scholar] [CrossRef]
- Xia, W.; Wei, Y.; Du, Y.; Liu, J.; Chang, B.; Yu, Y.L.; Huo, L.F.; Miller, S.; Hung, M.C. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol. Carcinog. 2009, 48, 610–617. [Google Scholar] [CrossRef]
- Zammit, C.; Barnard, R.; Gomm, J.; Coope, R.; Shousha, S.; Coombes, C.; Johnston, C. Altered intracellular localization of fibroblast growth factor receptor 3 in human breast cancer. J. Pathol. 2001, 194, 27–34. [Google Scholar] [CrossRef]
- May, M.; Mosto, J.; Vazquez, P.M.; Gonzalez, P.; Rojas, P.; Gass, H.; Lanari, C.; Molinolo, A.A. Nuclear staining of fgfr-2/stat-5 and runx-2 in mucinous breast cancer. Exp. Mol. Pathol. 2016, 100, 39–44. [Google Scholar] [CrossRef]
- Reilly, J.F.; Maher, P.A. Importin beta-mediated nuclear import of fibroblast growth factor receptor: Role in cell proliferation. J. Cell Biol. 2001, 152, 1307–1312. [Google Scholar] [CrossRef]
- Song, Q.; Liu, Y.; Jiang, D.; Wang, H.; Huang, J.; Xu, Y.; Sujie, A.; Zeng, H.; Xu, C.; Hou, Y. High amplification of FGFR1 gene is a delayed poor prognostic factor in early stage ESCC patients. Oncotarget 2017, 8, 74539–74553. [Google Scholar] [CrossRef]
- Stachowiak, E.K.; Maher, P.A.; Tucholski, J.; Mordechai, E.; Joy, A.; Moffett, J.; Coons, S.; Stachowiak, M.K. Nuclear accumulation of fibroblast growth factor receptors in human glial cells--association with cell proliferation. Oncogene 1997, 14, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, M.K.; Maher, P.A.; Stachowiak, E.K. Integrative nuclear signaling in cell development--a role for FGF receptor-1. DNA Cell Biol. 2007, 26, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Fang, X.; Dunham, S.M.; Prada, C.; Stachowiak, E.K.; Stachowiak, M.K. 90-kDa ribosomal S6 kinase is a direct target for the nuclear fibroblast growth factor receptor 1 (FGFR1): Role in FGFR1 signaling. J. Biol. Chem. 2004, 279, 29325–29335. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Ems, S.M.; Pudavar, H.E.; Myers, J.M.; Maher, P.A.; Prasad, P.N.; Stachowiak, M.K. Factors controlling fibroblast growth factor receptor-1’s cytoplasmic trafficking and its regulation as revealed by FRAP analysis. Mol. Biol. Cell 2006, 17, 2223–2235. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.A. Nuclear Translocation of fibroblast growth factor (FGF) receptors in response to FGF-2. J. Cell Biol. 1996, 134, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Lord, J.M. Ricin trafficking in cells. Toxins 2015, 7, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Degnin, C.R.; Laedrich, M.B.; Horton, W.A. Ligand activation leads to regulated intramembrane proteolysis of fibroblast growth factor receptor 3. Mol. Biol. Cell 2011, 22, 3861–3873. [Google Scholar] [CrossRef] [PubMed]
- Chioni, A.M.; Grose, R. FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J. Cell Biol. 2012, 197, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef]
- Opalińska, M.; Meisinger, C. Metabolic control via the mitochondrial protein import machinery. Curr. Opin. Cell Biol. 2015, 33, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Salvi, M. Receptor tyrosine kinases take a direct route to mitochondria: An overview. Curr. Protein Pept. Sci. 2013, 14, 635–640. [Google Scholar] [CrossRef]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, J.; Muñoz-Couselo, E.; Soberino, J.; Racca, F.; Cortes, J. Targeting FGFR pathway in breast cancer. Breast 2018, 37, 126–133. [Google Scholar] [CrossRef]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2016, 13, 335–347. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Sochaj, A.M.; Świderska, K.W.; Otlewski, J. Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnol. Adv. 2015, 33, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska-Wedzina, A.; Chodaczek, G.; Chudzian, J.; Borek, A.; Zakrzewska, M.; Otlewski, J. High-Affinity Internalizing Human scFv-Fc Antibody for Targeting FGFR1-Overexpressing Lung Cancer. Mol. Cancer Res. 2017, 15, 1040–1050. [Google Scholar] [CrossRef]
- Opaliński, Ł.; Szymczyk, J.; Szczepara, M.; Kucińska, M.; Krowarsch, D.; Zakrzewska, M.; Otlewski, J. High Affinity Promotes Internalization of Engineered Antibodies Targeting FGFR1. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Opaliński, Ł.; Song, J.; Priesnitz, C.; Wenz, L.S.; Oeljeklaus, S.; Warscheid, B.; Pfanner, N.; Becker, T. Recruitment of Cytosolic J-Proteins by TOM Receptors Promotes Mitochondrial Protein Biogenesis. Cell Rep. 2018, 25, 2036–2043. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Kopitz, C.; Schatz, C.A.; Nising, C.F.; Mahlert, C.; Lerchen, H.G.; Stelte-Ludwig, B.; Hammer, S.; Greven, S.; Schuhmacher, J.; et al. Preclinical Efficacy of the Auristatin-Based Antibody-Drug Conjugate BAY 1187982 for the Treatment of FGFR2-Positive Solid Tumors. Cancer Res. 2016, 76, 6331–6339. [Google Scholar] [CrossRef] [PubMed]
- Borek, A.; Sokolowska-Wedzina, A.; Chodaczek, G.; Otlewski, J. Generation of high-affinity, internalizing anti-FGFR2 single-chain variable antibody fragment fused with Fc for targeting gastrointestinal cancers. PLoS ONE 2018, 13, e0192194. [Google Scholar] [CrossRef] [PubMed]
- Adam Cheuk, Nitya Shivaprasad, Martin Skarzynski, Sivasubramanian Baskar, Peter Azorsa and Javed Khan Abstract 5618: Anti-FGFR4 antibody drug conjugate for immune therapy of rhabdomyosarcoma and hepatocellular carcinoma. Cancer Res. 2018. [CrossRef]
- Szlachcic, A.; Zakrzewska, M.; Lobocki, M.; Jakimowicz, P.; Otlewski, J. Design and characteristics of cytotoxic fibroblast growth factor 1 conjugate for fibroblast growth factor receptor-targeted cancer therapy. Drug Des. Dev. Ther. 2016, 10, 2547–2560. [Google Scholar] [CrossRef] [PubMed]
- Lobocki, M.; Zakrzewska, M.; Szlachcic, A.; Krzyscik, M.A.; Sokolowska-Wedzina, A.; Otlewski, J. High-Yield Site-Specific Conjugation of Fibroblast Growth Factor 1 with Monomethylauristatin E via Cysteine Flanked by Basic Residues. Bioconjug. Chem. 2017, 28, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Krzyscik, M.A.; Zakrzewska, M.; Sørensen, V.; Sokolowska-Wedzina, A.; Lobocki, M.; Swiderska, K.W.; Krowarsch, D.; Wiedlocha, A.; Otlewski, J. Cytotoxic Conjugates of Fibroblast Growth Factor 2 (FGF2) with Monomethyl Auristatin E for Effective Killing of Cells Expressing FGF Receptors. ACS Omega 2017, 2, 3792–3805. [Google Scholar] [CrossRef]
- Swiderska, K.W.; Szlachcic, A.; Czyrek, A.; Zakrzewska, M.; Otlewski, J. Site-specific conjugation of fibroblast growth factor 2 (FGF2) based on incorporation of alkyne-reactive unnatural amino acid. Bioorg. Med. Chem. 2017, 25, 3685–3693. [Google Scholar] [CrossRef]
- Jurek, P.M.; Zabłocki, K.; Waśko, U.; Mazurek, M.P.; Otlewski, J.; Jeleń, F. Anti-FGFR1 aptamer-tagged superparamagnetic conjugates for anticancer hyperthermia therapy. Int. J. Nanomed. 2017, 12, 2941–2950. [Google Scholar] [CrossRef]
- Mahipal, A.; Malafa, M. Importins and exportins as therapeutic targets in cancer. Pharmacol. Ther. 2016, 164, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Papić, D.; Elbaz-Alon, Y.; Koerdt, S.N.; Leopold, K.; Worm, D.; Jung, M.; Schuldiner, M.; Rapaport, D. The role of Djp1 in import of the mitochondrial protein Mim1 demonstrates specificity between a cochaperone and its substrate protein. Mol. Cell. Biol. 2013, 33, 4083–4094. [Google Scholar] [CrossRef] [PubMed]
- Moody, P.R.; Sayers, E.J.; Magnusson, J.P.; Alexander, C.; Borri, P.; Watson, P.; Jones, A.T. Receptor Crosslinking: A General Method to Trigger Internalization and Lysosomal Targeting of Therapeutic Receptor:Ligand Complexes. Mol. Ther. 2015, 23, 1888–1898. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Madshus, I.H.; Stang, E. Cetuximab in combination with anti-human IgG antibodies efficiently down-regulates the EGF receptor by macropinocytosis. Exp. Cell Res. 2012, 318, 2578–2591. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.M.; Rinon, A.; Schechter, B.; Lyass, L.; Lavi, S.; Bacus, S.S.; Sela, M.; Yarden, Y. Synergistic down-regulation of receptor tyrosine kinases by combinations of mAbs: Implications for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2005, 102, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Emde, A.; Pradeep, C.R.; Ferraro, D.A.; Ben-Chetrit, N.; Sela, M.; Ribba, B.; Kam, Z.Y. Combining epitope-distinct antibodies to HER2: Cooperative inhibitory effects on invasive growth. Oncogene 2011, 30, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Resistance to Antibody-Drug Conjugates. Cancer Res. 2018, 78, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Świderska, K.W.; Szlachcic, A.; Opaliński, Ł.; Zakrzewska, M.; Otlewski, J. FGF2 Dual Warhead Conjugate with Monomethyl Auristatin E and α-Amanitin Displays a Cytotoxic Effect towards Cancer Cells Overproducing FGF Receptor 1. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porębska, N.; Latko, M.; Kucińska, M.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Targeting Cellular Trafficking of Fibroblast Growth Factor Receptors as a Strategy for Selective Cancer Treatment. J. Clin. Med. 2019, 8, 7. https://doi.org/10.3390/jcm8010007
Porębska N, Latko M, Kucińska M, Zakrzewska M, Otlewski J, Opaliński Ł. Targeting Cellular Trafficking of Fibroblast Growth Factor Receptors as a Strategy for Selective Cancer Treatment. Journal of Clinical Medicine. 2019; 8(1):7. https://doi.org/10.3390/jcm8010007
Chicago/Turabian StylePorębska, Natalia, Marta Latko, Marika Kucińska, Małgorzata Zakrzewska, Jacek Otlewski, and Łukasz Opaliński. 2019. "Targeting Cellular Trafficking of Fibroblast Growth Factor Receptors as a Strategy for Selective Cancer Treatment" Journal of Clinical Medicine 8, no. 1: 7. https://doi.org/10.3390/jcm8010007
APA StylePorębska, N., Latko, M., Kucińska, M., Zakrzewska, M., Otlewski, J., & Opaliński, Ł. (2019). Targeting Cellular Trafficking of Fibroblast Growth Factor Receptors as a Strategy for Selective Cancer Treatment. Journal of Clinical Medicine, 8(1), 7. https://doi.org/10.3390/jcm8010007