Innate Immune Responses and Viral-Induced Neurologic Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. JHMV-Induced Neurologic Disease
2.1. Secretion of Proinflammatory Cytokines/Chemokines in Response to JHMV Infection of the CNS
2.2. Chemokine Signaling Promotes Immune Cell Infiltration into the CNS
2.3. Microglial Involvement in JHMV Disease Progression
3. JHMV-Induced Demyelination
4. Neutrophils and JHMV-Induced Demyelination
4.1. Neutrophils in MS Patients
4.2. Neutrophil Involvement in Preclinical Models of Demyelination
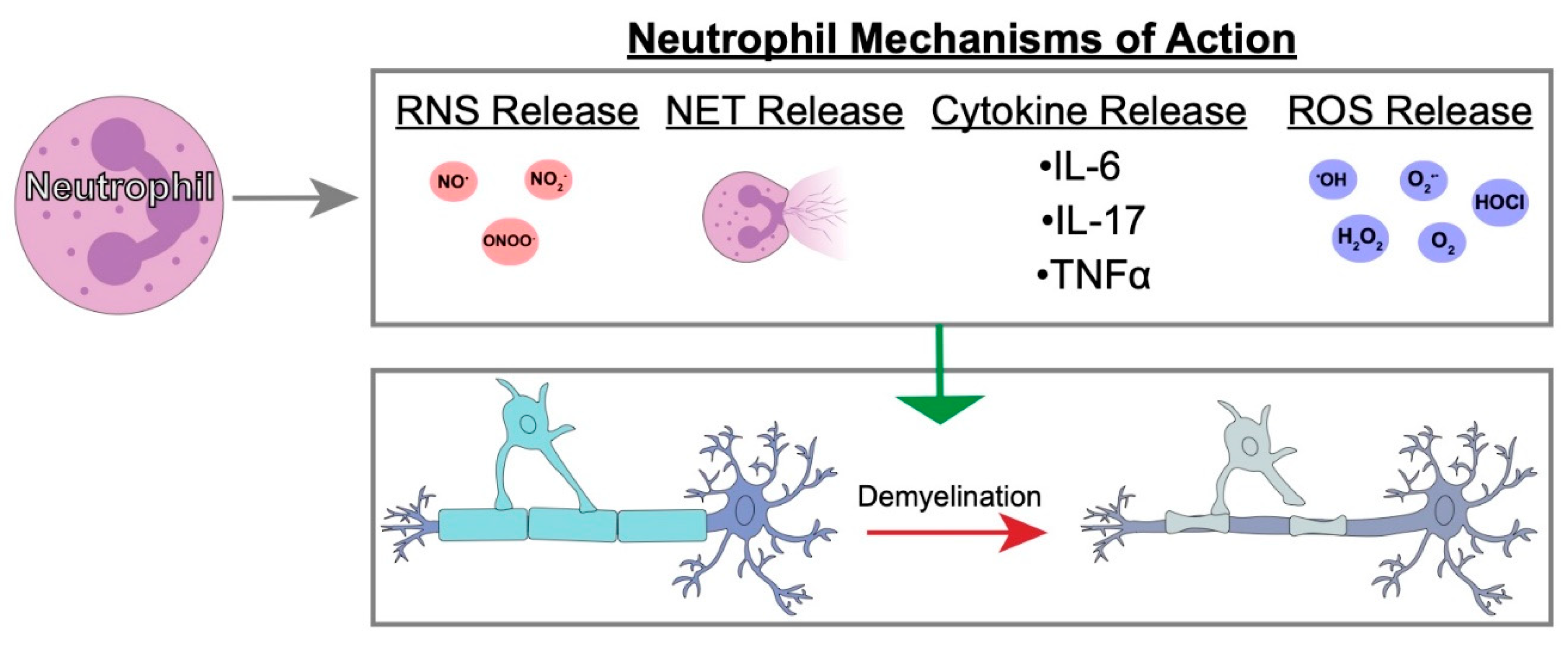
4.3. Possible Neutrophil Mechanisms of Action in Demyelination
5. Perspectives
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Steinman, L. Immunology of Relapse and Remission in Multiple Sclerosis. Annu. Rev. Immunol. 2014, 32, 257–281. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bruck, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Medana, I.M.; Bauer, J.; Lassmann, H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002, 25, 313–319. [Google Scholar] [CrossRef]
- Steinman, L. Multiple sclerosis: A coordinated immunological attack against myelin in the central nervous system. Cell 1996, 85, 299–302. [Google Scholar] [CrossRef]
- Greenfield, A.L.; Hauser, S.L. B-cell Therapy for Multiple Sclerosis: Entering an era. Ann. Neurol. 2018, 83, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Falcao, A.M.; van Bruggen, D.; Marques, S.; Meijer, M.; Jakel, S.; Agirre, E.; Samudyata; Floriddia, E.M.; Vanichkina, D.P.; Ffrench-Constant, C.; et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 2018, 24, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Nishiyama, A.; Peterson, J.; Prineas, J.; Trapp, B.D. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J. Neurosci. 2000, 20, 6404–6412. [Google Scholar] [CrossRef]
- Prineas, J.W.; Kwon, E.E.; Goldenberg, P.Z.; Ilyas, A.A.; Quarles, R.H.; Benjamins, J.A.; Sprinkle, T.J. Multiple sclerosis. Oligodendrocyte proliferation and differentiation in fresh lesions. Lab. Investig. 1989, 61, 489–503. [Google Scholar]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. A study of 113 cases. Brain 1999, 122 Pt 12, 2279–2295. [Google Scholar] [CrossRef]
- Roy, N.S.; Wang, S.; Harrison-Restelli, C.; Benraiss, A.; Fraser, R.A.; Gravel, M.; Braun, P.E.; Goldman, S.A. Identification, isolation, and promoter-defined separation of mitotic oligodendrocyte progenitor cells from the adult human subcortical white matter. J. Neurosci. 1999, 19, 9986–9995. [Google Scholar] [CrossRef]
- Lassmann, H. Comparative neuropathology of chronic experimental allergic encephalomyelitis and multiple sclerosis. Schriftenr. Neurol. 1983, 25, 1–135. [Google Scholar] [PubMed]
- Schlesinger, H. Zur Frage der akuten multiplen Sklerose und der encephalomyelitis disseminata im Kindesalter. Arb. Neurol. Inst. 1909, 17, 410–432. [Google Scholar]
- Halfpenny, C.; Benn, T.; Scolding, N. Cell transplantation, myelin repair, and multiple sclerosis. Lancet Neurol. 2002, 1, 31–40. [Google Scholar] [CrossRef]
- Oksenberg, J.R.; Begovich, A.B.; Erlich, H.A.; Steinman, L. Genetic factors in multiple sclerosis. Jama 1993, 270, 2362–2369. [Google Scholar] [CrossRef] [PubMed]
- Poser, C.M. The epidemiology of multiple sclerosis: A general overview. Ann. Neurol. 1994, 36 (Suppl. 2), S180–S193. [Google Scholar] [CrossRef]
- Raine, C.S. The Dale E. McFarlin Memorial Lecture: The immunology of the multiple sclerosis lesion. Ann. Neurol. 1994, 36 (Suppl. 1), S61–S72. [Google Scholar] [CrossRef]
- Ebers, G.C.; Sadovnick, A.D.; Risch, N.J. A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature 1995, 377, 150–151. [Google Scholar] [CrossRef]
- Friedman, J.E.; Lyons, M.J.; Cu, G.; Ablashl, D.V.; Whitman, J.E.; Edgar, M.; Koskiniemi, M.; Vaheri, A.; Zabriskie, J.B. The association of the human herpesvirus-6 and MS. Mult. Scler. 1999, 5, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.T. The virology of demyelinating diseases. Ann. Neurol. 1994, 36 (Suppl. 1), S54–S60. [Google Scholar] [CrossRef]
- Kennedy, P.G.; Steiner, I. On the possible viral aetiology of multiple sclerosis. QJM 1994, 87, 523–528. [Google Scholar]
- Libbey, J.E.; Fujinami, R.S. Potential triggers of MS. Res. Probl. Cell Differ. 2010, 51, 21–42. [Google Scholar] [CrossRef]
- Lincoln, J.A.; Hankiewicz, K.; Cook, S.D. Could Epstein-Barr virus or canine distemper virus cause multiple sclerosis? Neurol. Clin. 2008, 26, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Lipton, H.L.; Liang, Z.; Hertzler, S.; Son, K.N. A specific viral cause of multiple sclerosis: One virus, one disease. Ann. Neurol. 2007, 61, 514–523. [Google Scholar] [CrossRef]
- McCoy, L.; Tsunoda, I.; Fujinami, R.S. Multiple sclerosis and virus induced immune responses: Autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity 2006, 39, 9–19. [Google Scholar] [CrossRef]
- Olson, J.K.; Ercolini, A.M.; Miller, S.D. A virus-induced molecular mimicry model of multiple sclerosis. Curr. Top. Microbiol. Immunol. 2005, 296, 39–53. [Google Scholar] [PubMed]
- Pugliatti, M.; Harbo, H.F.; Holmoy, T.; Kampman, M.T.; Myhr, K.M.; Riise, T.; Wolfson, C. Environmental risk factors in multiple sclerosis. Acta Neurol. Scand. Suppl. 2008, 188, 34–40. [Google Scholar] [CrossRef]
- Tao, C.; Simpson, S., Jr.; Taylor, B.V.; van der Mei, I. Association between human herpesvirus & human endogenous retrovirus and MS onset & progression. J. Neurol. Sci. 2017, 372, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Fujinami, R.S. Neuropathogenesis of Theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J. Neuroimmune Pharmacol. 2010, 5, 355–369. [Google Scholar] [CrossRef]
- Bergmann, C.C.; Lane, T.E.; Stohlman, S.A. Coronavirus infection of the central nervous system: Host-virus stand-off. Nat. Rev. Microbiol. 2006, 4, 121–132. [Google Scholar] [CrossRef]
- Butchi, N.; Kapil, P.; Puntambekar, S.; Stohlman, S.A.; Hinton, D.R.; Bergmann, C.C. Myd88 Initiates Early Innate Immune Responses and Promotes CD4 T Cells during Coronavirus Encephalomyelitis. J. Virol. 2015, 89, 9299–9312. [Google Scholar] [CrossRef]
- Lane, T.E.; Asensio, V.C.; Yu, N.; Paoletti, A.D.; Campbell, I.L.; Buchmeier, M.J. Dynamic regulation of alpha- and beta-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J. Immunol. 1998, 160, 970–978. [Google Scholar] [PubMed]
- Sun, N.; Grzybicki, D.; Castro, R.F.; Murphy, S.; Perlman, S. Activation of astrocytes in the spinal cord of mice chronically infected with a neurotropic coronavirus. Virology 1995, 213, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Ryman, K.D.; Klimstra, W.B.; Nguyen, K.B.; Biron, C.A.; Johnston, R.E. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J. Virol. 2000, 74, 3366–3378. [Google Scholar] [CrossRef]
- Samuel, M.A.; Diamond, M.S. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef] [PubMed]
- Trottier, M.D., Jr.; Palian, B.M.; Reiss, C.S. VSV replication in neurons is inhibited by type I IFN at multiple stages of infection. Virology 2005, 333, 215–225. [Google Scholar] [CrossRef]
- Ireland, D.D.; Stohlman, S.A.; Hinton, D.R.; Atkinson, R.; Bergmann, C.C. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J. Virol. 2008, 82, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef]
- Vijay, R.; Fehr, A.R.; Janowski, A.M.; Athmer, J.; Wheeler, D.L.; Grunewald, M.; Sompallae, R.; Kurup, S.P.; Meyerholz, D.K.; Sutterwala, F.S.; et al. Virus-induced inflammasome activation is suppressed by prostaglandin D2/DP1 signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E5444–E5453. [Google Scholar] [CrossRef]
- Hosking, M.P.; Liu, L.; Ransohoff, R.M.; Lane, T.E. A protective role for ELR+ chemokines during acute viral encephalomyelitis. PLoS Pathog. 2009, 5, e1000648. [Google Scholar] [CrossRef]
- Vanguri, P.; Farber, J.M. Identification of CRG-2. An interferon-inducible mRNA predicted to encode a murine monokine. J. Biol. Chem. 1990, 265, 15049–15057. [Google Scholar]
- Liu, M.T.; Armstrong, D.; Hamilton, T.A.; Lane, T.E. Expression of Mig (monokine induced by interferon-gamma) is important in T lymphocyte recruitment and host defense following viral infection of the central nervous system. J. Immunol. 2001, 166, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Trifilo, M.J.; Montalto-Morrison, C.; Stiles, L.N.; Hurst, K.R.; Hardison, J.L.; Manning, J.E.; Masters, P.S.; Lane, T.E. CXC chemokine ligand 10 controls viral infection in the central nervous system: Evidence for a role in innate immune response through recruitment and activation of natural killer cells. J. Virol. 2004, 78, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.H.; Dziejman, M.; Liu, M.T.; Leung, J.H.; Lane, T.E.; Luster, A.D. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J. Immunol. 2002, 168, 3195–3204. [Google Scholar] [CrossRef]
- Liu, M.T.; Chen, B.P.; Oertel, P.; Buchmeier, M.J.; Armstrong, D.; Hamilton, T.A.; Lane, T.E. The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J. Immunol. 2000, 165, 2327–2330. [Google Scholar] [CrossRef]
- Stiles, L.N.; Hosking, M.P.; Edwards, R.A.; Strieter, R.M.; Lane, T.E. Differential roles for CXCR3 in CD4+ and CD8+ T cell trafficking following viral infection of the CNS. Eur. J. Immunol. 2006, 36, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Phares, T.W.; Stohlman, S.A.; Hinton, D.R.; Bergmann, C.C. Astrocyte-derived CXCL10 drives accumulation of antibody-secreting cells in the central nervous system during viral encephalomyelitis. J. Virol. 2013, 87, 3382–3392. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.C.; Parra, B.; Hinton, D.R.; Ramakrishna, C.; Dowdell, K.C.; Stohlman, S.A. Perforin and gamma interferon-mediated control of coronavirus central nervous system infection by CD8 T cells in the absence of CD4 T cells. J. Virol. 2004, 78, 1739–1750. [Google Scholar] [CrossRef]
- Parra, B.; Hinton, D.R.; Marten, N.W.; Bergmann, C.C.; Lin, M.T.; Yang, C.S.; Stohlman, S.A. IFN-gamma is required for viral clearance from central nervous system oligodendroglia. J. Immunol. 1999, 162, 1641–1647. [Google Scholar]
- Hwang, M.; Phares, T.W.; Hinton, D.R.; Stohlman, S.A.; Bergmann, C.C.; Min, B. Distinct CD4 T-cell effects on primary versus recall CD8 T-cell responses during viral encephalomyelitis. Immunology 2015, 144, 374–386. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Sariol, A.; Meyerholz, D.K.; Perlman, S. Microglia are required for protection against lethal coronavirus encephalitis in mice. J. Clin. Investig. 2018, 128, 931–943. [Google Scholar] [CrossRef]
- Lane, T.E.; Buchmeier, M.J. Murine coronavirus infection: A paradigm for virus-induced demyelinating disease. Trends Microbiol. 1997, 5, 9–14. [Google Scholar] [CrossRef]
- Hosking, M.P.; Lane, T.E. The Biology of Persistent Infection: Inflammation and Demyelination following Murine Coronavirus Infection of the Central Nervous System. Curr. Immunol. Rev. 2009, 5, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Mangale, V.; McIntyre, L.L.; Walsh, C.M.; Loring, J.F.; Lane, T.E. Promoting remyelination through cell transplantation therapies in a model of viral-induced neurodegenerative disease. Dev. Dyn. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tirotta, E.; Carbajal, K.S.; Schaumburg, C.S.; Whitman, L.; Lane, T.E. Cell replacement therapies to promote remyelination in a viral model of demyelination. J. Neuroimmunol. 2010, 224, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.; Li, F.; Dragomir, I.; Chua, M.M.; Gregory, B.D.; Weiss, S.R. Analysis of the host transcriptome from demyelinating spinal cord of murine coronavirus-infected mice. PLoS ONE 2013, 8, e75346. [Google Scholar] [CrossRef] [PubMed]
- Iacono, K.T.; Kazi, L.; Weiss, S.R. Both spike and background genes contribute to murine coronavirus neurovirulence. J. Virol. 2006, 80, 6834–6843. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.P.; Branigan, P.J.; Del Vecchio, A.M.; Weiss, S.R. Chemokine expression during mouse-hepatitis-virus-induced encephalitis: Contributions of the spike and background genes. J. Neurovirol. 2008, 14, 5–16. [Google Scholar] [CrossRef]
- Glass, W.G.; Lane, T.E. Functional analysis of the CC chemokine receptor 5 (CCR5) on virus-specific CD8+ T cells following coronavirus infection of the central nervous system. Virology 2003, 312, 407–414. [Google Scholar] [CrossRef]
- Glass, W.G.; Lane, T.E. Functional expression of chemokine receptor CCR5 on CD4(+) T cells during virus-induced central nervous system disease. J. Virol. 2003, 77, 191–198. [Google Scholar] [CrossRef]
- Haring, J.S.; Perlman, S. Bystander CD4 T cells do not mediate demyelination in mice infected with a neurotropic coronavirus. J. Neuroimmunol. 2003, 137, 42–50. [Google Scholar] [CrossRef]
- Haring, J.S.; Pewe, L.L.; Perlman, S. High-magnitude, virus-specific CD4 T-cell response in the central nervous system of coronavirus-infected mice. J. Virol. 2001, 75, 3043–3047. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Perlman, S. Viral expression of CCL2 is sufficient to induce demyelination in RAG1-/- mice infected with a neurotropic coronavirus. J. Virol. 2005, 79, 7113–7120. [Google Scholar] [CrossRef] [PubMed]
- Glass, W.G.; Hickey, M.J.; Hardison, J.L.; Liu, M.T.; Manning, J.E.; Lane, T.E. Antibody targeting of the CC chemokine ligand 5 results in diminished leukocyte infiltration into the central nervous system and reduced neurologic disease in a viral model of multiple sclerosis. J. Immunol. 2004, 172, 4018–4025. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.T.; Keirstead, H.S.; Lane, T.E. Neutralization of the chemokine CXCL10 reduces inflammatory cell invasion and demyelination and improves neurological function in a viral model of multiple sclerosis. J. Immunol. 2001, 167, 4091–4097. [Google Scholar] [CrossRef] [PubMed]
- Das Sarma, J. Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J. Neurovirol. 2014, 20, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Biswas, K.; Nag, S.; Ramachandra, S.G.; Das Sarma, J. Microglia play a major role in direct viral-induced demyelination. Clin. Dev. Immunol. 2013, 2013, 510396. [Google Scholar] [CrossRef] [PubMed]
- Aube, B.; Levesque, S.A.; Pare, A.; Chamma, E.; Kebir, H.; Gorina, R.; Lecuyer, M.A.; Alvarez, J.I.; De Koninck, Y.; Engelhardt, B.; et al. Neutrophils mediate blood-spinal cord barrier disruption in demyelinating neuroinflammatory diseases. J. Immunol. 2014, 193, 2438–2454. [Google Scholar] [CrossRef] [PubMed]
- Barthelmes, J.; de Bazo, A.M.; Pewzner-Jung, Y.; Schmitz, K.; Mayer, C.A.; Foerch, C.; Eberle, M.; Tafferner, N.; Ferreiros, N.; Henke, M.; et al. Lack of ceramide synthase 2 suppresses the development of experimental autoimmune encephalomyelitis by impairing the migratory capacity of neutrophils. Brain Behave. Immun. 2015, 46, 280–292. [Google Scholar] [CrossRef]
- Eberle, M.; Ebel, P.; Mayer, C.A.; Barthelmes, J.; Tafferner, N.; Ferreiros, N.; Ulshofer, T.; Henke, M.; Foerch, C.; de Bazo, A.M.; et al. Exacerbation of experimental autoimmune encephalomyelitis in ceramide synthase 6 knockout mice is associated with enhanced activation/migration of neutrophils. Immunol. Cell Boil. 2015, 93, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, K.; Piedavent, M.; Bauer, S.; Neumann, J.T.; Friese, M.A. Neutrophils amplify autoimmune central nervous system infiltrates by maturing local APCs. J. Immunol. 2013, 191, 4531–4539. [Google Scholar] [CrossRef]
- Christy, A.L.; Walker, M.E.; Hessner, M.J.; Brown, M.A. Mast cell activation and neutrophil recruitment promotes early and robust inflammation in the meninges in EAE. J. Autoimmun. 2013, 42, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.L.; Lafrance-Corey, R.G.; Sundsbak, R.S.; Lafrance, S.J. Inflammatory monocytes damage the hippocampus during acute picornavirus infection of the brain. J. Neuroinflamm. 2012, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Sayed, B.A.; Christy, A.L.; Walker, M.E.; Brown, M.A. Meningeal mast cells affect early T cell central nervous system infiltration and blood-brain barrier integrity through TNF: A role for neutrophil recruitment? J. Immunol. 2010, 184, 6891–6900. [Google Scholar] [CrossRef] [PubMed]
- Sewell, D.L.; Nacewicz, B.; Liu, F.; Macvilay, S.; Erdei, A.; Lambris, J.D.; Sandor, M.; Fabry, Z. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: Blocking effects on neutrophil extravasation by C5a receptor antagonist. J. Neuroimmunol. 2004, 155, 55–63. [Google Scholar] [CrossRef]
- Kaplan, M.J. Role of neutrophils in systemic autoimmune diseases. Arthr. Res. Ther. 2013, 15, 219. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef]
- Jarius, S.; Wildemann, B.; Paul, F. Neuromyelitis optica: Clinical features, immunopathogenesis and treatment. Clin. Exp. Immunol. 2014, 176, 149–164. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; Macdonald, C.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Neutrophil Protease Inhibition Reduces Neuromyelitis Optica—Immunoglobulin G—Induced Damage in Mouse Brain. Ann. Neurol. 2012, 71, 323–333. [Google Scholar] [CrossRef]
- Huber, A.K.; Wang, L.; Han, P.; Zhang, X.; Ekholm, S.; Srinivasan, A.; Irani, D.N.; Segal, B.M. Dysregulation of the IL-23/IL-17 axis and myeloid factors in secondary progressive MS. Neurology 2014, 83, 1500–1507. [Google Scholar] [CrossRef]
- Jacob, A.; Saadoun, S.; Kitley, J.; Leite, M.; Palace, J.; Schon, F.; Papadopoulos, M.C. Detrimental role of granulocyte-colony stimulating factor in neuromyelitis optica: Clinical case and histological evidence. Mult. Scler. 2012, 18, 1801–1803. [Google Scholar] [CrossRef]
- Kostic, M.; Dzopalic, T.; Zivanovic, S.; Zivkovic, N.; Cvetanovic, A.; Stojanovic, I.; Vojinovic, S.; Marjanovic, G.; Savic, V.; Colic, M. IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand. J. Immunol. 2014, 79, 181–186. [Google Scholar] [CrossRef]
- Naegele, M.; Tillack, K.; Reinhardt, S.; Schippling, S.; Martin, R.; Sospedra, M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J. Neuroimmunol. 2012, 242, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Openshaw, H.; Stuve, O.; Antel, J.P.; Nash, R.; Lund, B.T.; Weiner, L.P.; Kashyap, A.; McSweeney, P.; Forman, S. Multiple sclerosis flares associated with recombinant granulocyte colony-stimulating factor. Neurology 2000, 54, 2147–2150. [Google Scholar] [CrossRef] [PubMed]
- Rumble, J.M.; Huber, A.K.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-related factors as biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35. [Google Scholar] [CrossRef]
- Liu, L.; Belkadi, A.; Darnall, L.; Hu, T.; Drescher, C.; Cotleur, A.C.; Padovani-Claudio, D.; He, T.; Choi, K.; Lane, T.E.; et al. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: Relevance to multiple sclerosis. Nat. Neurosci. 2010, 13, 319–326. [Google Scholar] [CrossRef]
- Simmons, S.B.; Liggitt, D.; Goverman, J.M. Cytokine-regulated neutrophil recruitment is required for brain but not spinal cord inflammation during experimental autoimmune encephalomyelitis. J. Immunol. 2014, 193, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Stoolman, J.S.; Duncker, P.C.; Huber, A.K.; Segal, B.M. Site-specific chemokine expression regulates central nervous system inflammation and determines clinical phenotype in autoimmune encephalomyelitis. J. Immunol. 2014, 193, 564–570. [Google Scholar] [CrossRef]
- Ishizu, T.; Osoegawa, M.; Mei, F.-J.; Kikuchi, H.; Tanaka, M.; Takakura, Y.; Minohara, M.; Murai, H.; Mihara, F.; Taniwaki, T.; et al. Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain 2005, 128, 988–1002. [Google Scholar] [CrossRef]
- Campbell, S.J.; Meier, U.; Mardiguian, S.; Jiang, Y.; Littleton, E.T.; Bristow, A.; Relton, J.; Connor, T.J.; Anthony, D.C. Sickness behaviour is induced by a peripheral CXC-chemokine also expressed in multiple sclerosis and EAE. Brain behave. Immun. 2010, 24, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Bartosik-Psujek, H.; Stelmasiak, Z. The levels of chemokines CXCL8, CCL2 and CCL5 in multiple sclerosis patients are linked to the activity of the disease. Eur. J. Neurol. 2005, 12, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Carlson, T.; Kroenke, M.; Rao, P.; Lane, T.E.; Segal, B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 2008, 205, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Holdbrooks, A.T.; Meares, G.P.; Buckley, J.A.; Benveniste, E.N.; Qin, H. Preferential Recruitment of Neutrophils into the Cerebellum and Brainstem Contributes to the Atypical Experimental Autoimmune Encephalomyelitis Phenotype. J. Immunol. 2015, 195, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Rubio, N.; Sanz-Rodriguez, F. Induction of the CXCL1 (KC) chemokine in mouse astrocytes by infection with the murine encephalomyelitis virus of Theiler. Virology 2007, 358, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Rubio, N.; Sanz-Rodriguez, F.; Lipton, H.L. Theiler’s virus induces the MIP-2 chemokine (CXCL2) in astrocytes from genetically susceptible but not from resistant mouse strains. Cell. Immunol. 2006, 239, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Hosking, M.P.; Tirotta, E.; Ransohoff, R.M.; Lane, T.E. CXCR2 signaling protects oligodendrocytes and restricts demyelination in a mouse model of viral-induced demyelination. PLoS ONE 2010, 5, e11340. [Google Scholar] [CrossRef]
- Kang, Z.; Wang, C.; Zepp, J.; Wu, L.; Sun, K.; Zhao, J.; Chandrasekharan, U.; DiCorleto, P.E.; Trapp, B.D.; Ransohoff, R.M.; et al. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat. Neurosci. 2013, 16, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Glabinski, A.R.; Krakowski, M.; Han, Y.; Owens, T.; Ransohoff, R.M. Chemokine expression in GKO mice (lacking interferon-gamma) with experimental autoimmune encephalomyelitis. J. Neurovirol. 1999, 5, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Glabinski, A.R.; Tuohy, V.K.; Ransohoff, R.M. Expression of chemokines RANTES, MIP-1alpha and GRO-alpha correlates with inflammation in acute experimental autoimmune encephalomyelitis. Neuroimmunomodulation 1998, 5, 166–171. [Google Scholar] [CrossRef] [PubMed]
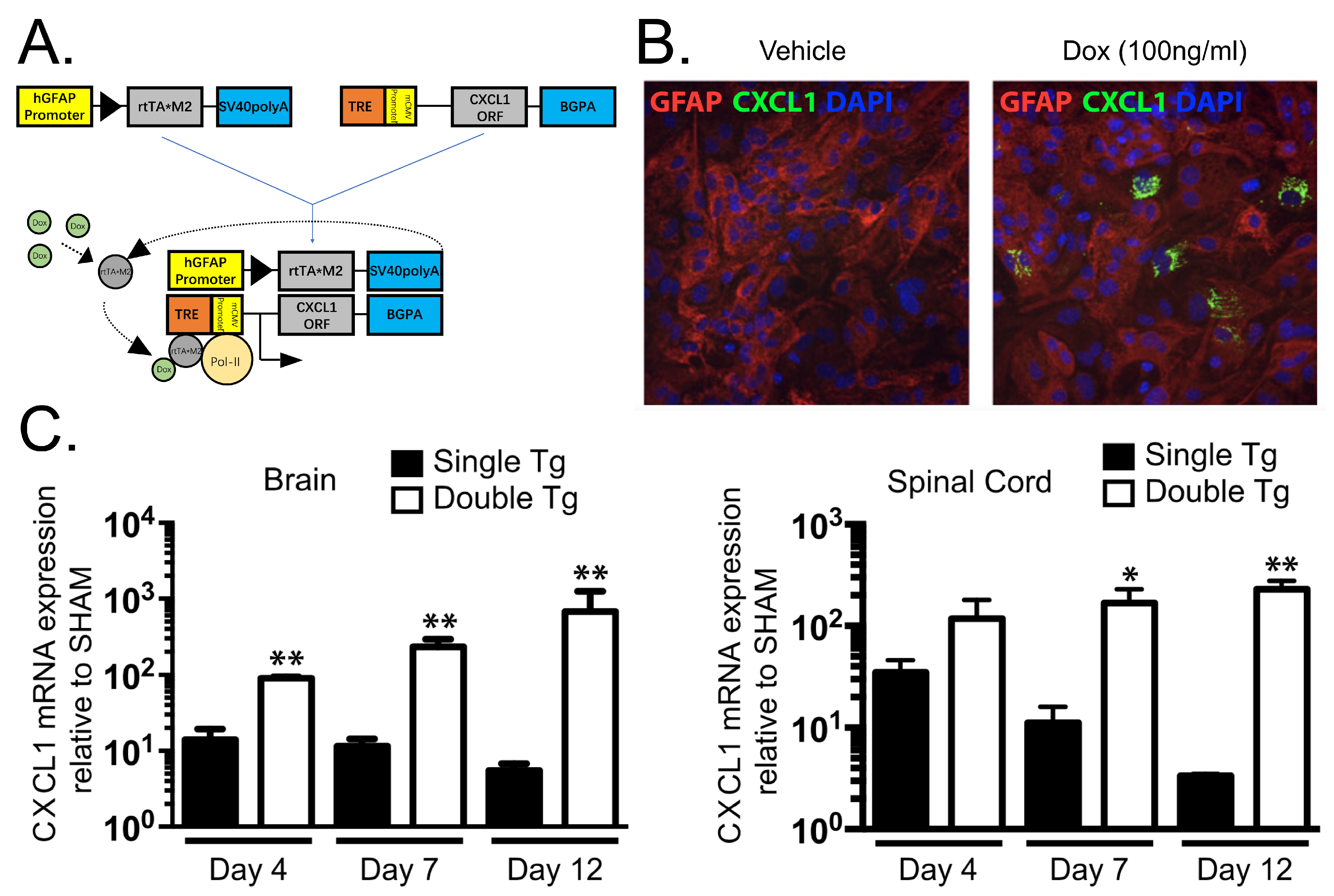
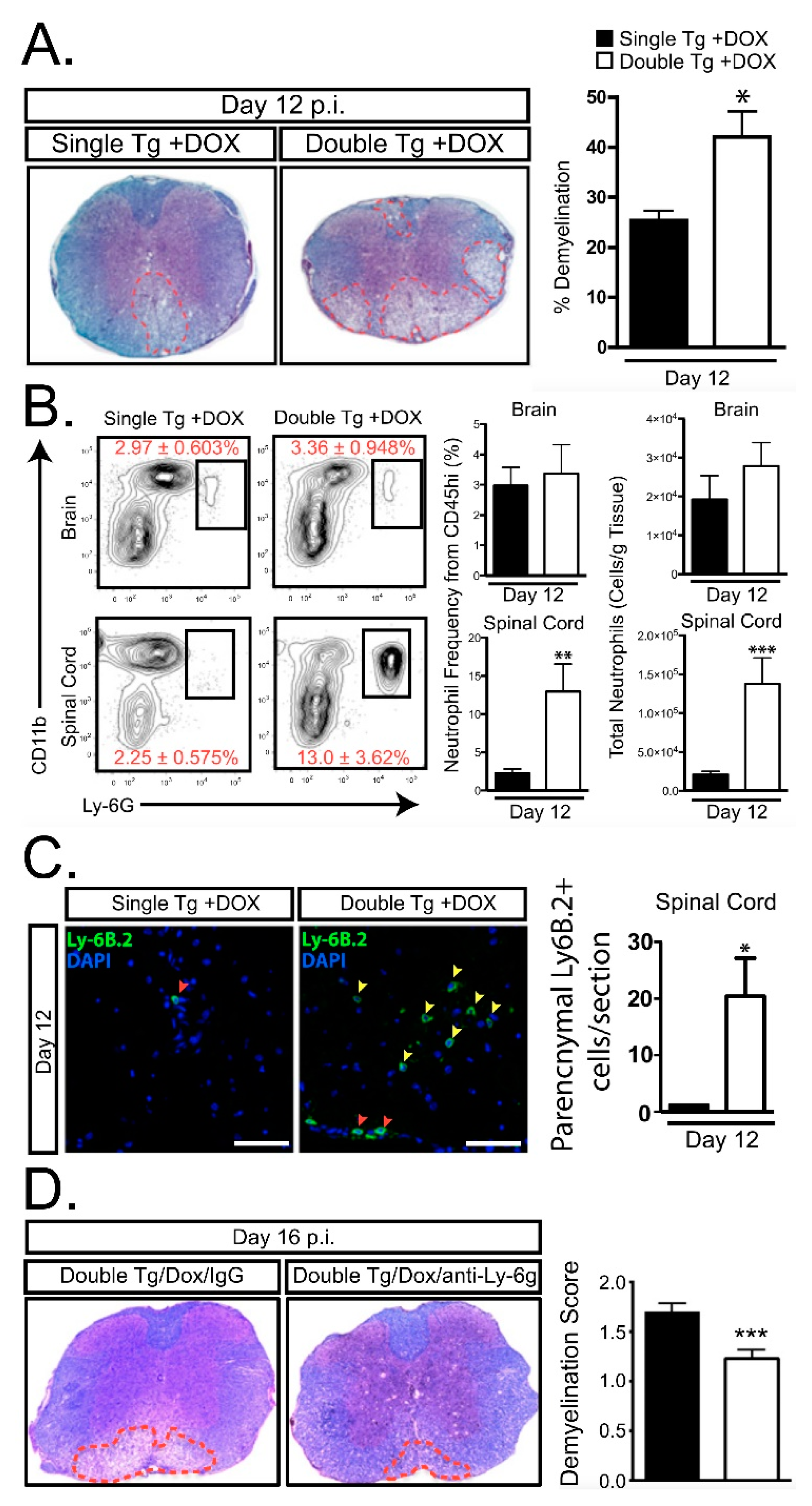
- Marro, B.S.; Grist, J.J.; Lane, T.E. Inducible Expression of CXCL1 within the Central Nervous System Amplifies Viral-Induced Demyelination. J. Immunol. 2016, 196, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Grist, J.J.; Marro, B.S.; Skinner, D.D.; Syage, A.R.; Worne, C.; Doty, D.J.; Fujinami, R.S.; Lane, T.E. Induced CNS expression of CXCL1 augments neurologic disease in a murine model of multiple sclerosis via enhanced neutrophil recruitment. Eur. J. Immunol. 2018, 48, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, L.; Wang, Y.; Downey, G.P. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Boil. Med. 2007, 42, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Tecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-derived cytokines: Facts beyond expression. Front. Immunol. 2014, 5, 508. [Google Scholar] [CrossRef]
- Merrill, J.E.; Ignarro, L.J.; Sherman, M.P.; Melinek, J.; Lane, T.E. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J. Immunol. 1993, 151, 2132–2141. [Google Scholar]
- Mitrovic, B.; Ignarro, L.J.; Montestruque, S.; Smoll, A.; Merrill, J.E. Nitric oxide as a potential pathological mechanism in demyelination: Its differential effects on primary glial cells in vitro. Neuroscience 1994, 61, 575–585. [Google Scholar] [CrossRef]
- Delgado-Rizo, V.; Martinez-Guzman, M.A.; Iniguez-Gutierrez, L.; Garcia-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef]
- Al-Khafaji, A.B.; Tohme, S.; Yazdani, H.O.; Miller, D.; Huang, H.; Tsung, A. Superoxide induces Neutrophil Extracellular Trap Formation in a TLR-4 and NOX-dependent mechanism. Mol. Med. 2016, 22, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed]
- Tillack, K.; Naegele, M.; Haueis, C.; Schippling, S.; Wandinger, K.P.; Martin, R.; Sospedra, M. Gender differences in circulating levels of neutrophil extracellular traps in serum of multiple sclerosis patients. J. Neuroimmunol. 2013, 261, 108–119. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Skinner, D.D.; Lane, T.E. Innate Immune Responses and Viral-Induced Neurologic Disease. J. Clin. Med. 2019, 8, 3. https://doi.org/10.3390/jcm8010003
Cheng Y, Skinner DD, Lane TE. Innate Immune Responses and Viral-Induced Neurologic Disease. Journal of Clinical Medicine. 2019; 8(1):3. https://doi.org/10.3390/jcm8010003
Chicago/Turabian StyleCheng, Yuting, Dominic D. Skinner, and Thomas E. Lane. 2019. "Innate Immune Responses and Viral-Induced Neurologic Disease" Journal of Clinical Medicine 8, no. 1: 3. https://doi.org/10.3390/jcm8010003
APA StyleCheng, Y., Skinner, D. D., & Lane, T. E. (2019). Innate Immune Responses and Viral-Induced Neurologic Disease. Journal of Clinical Medicine, 8(1), 3. https://doi.org/10.3390/jcm8010003