The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations



Abstract
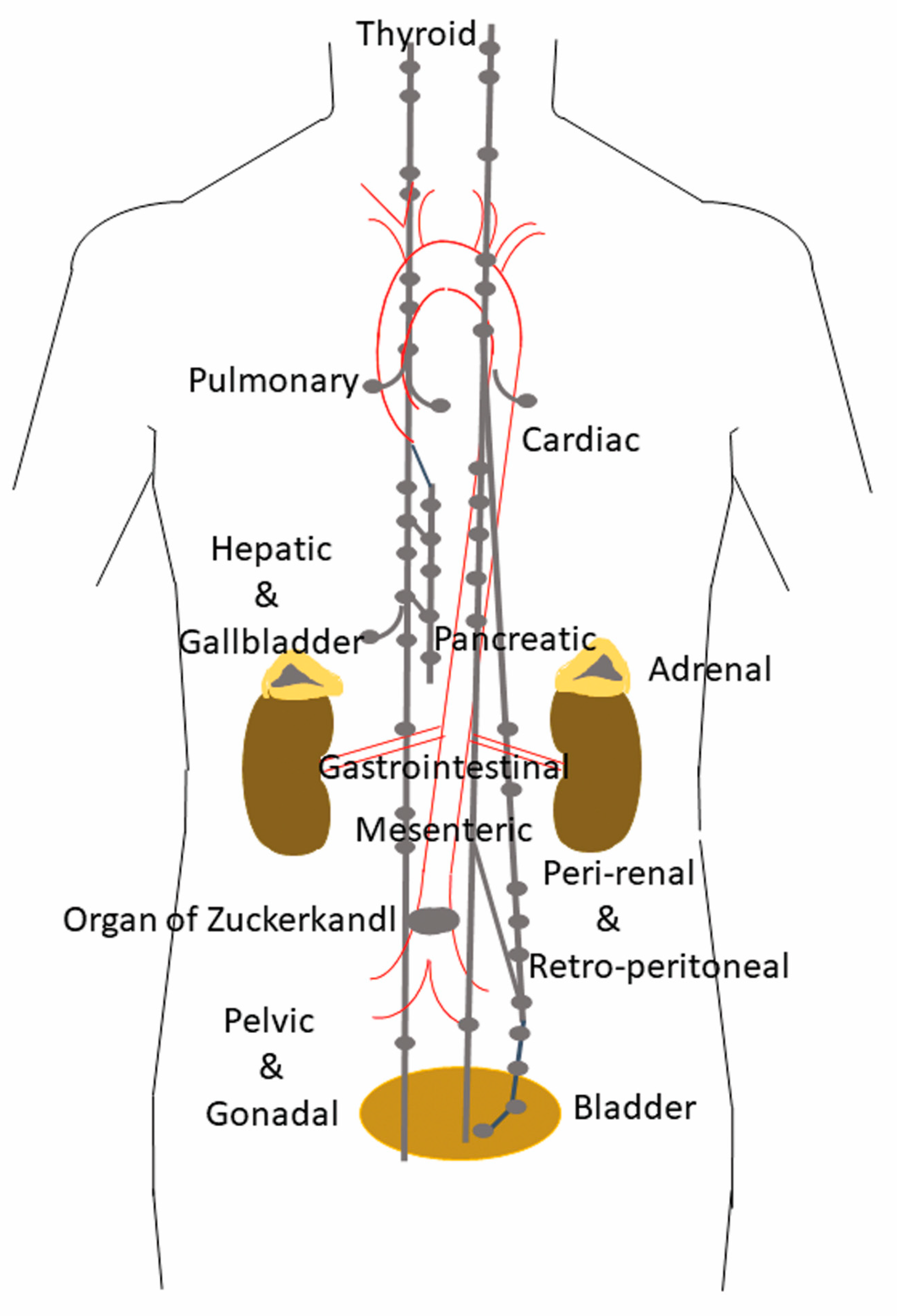
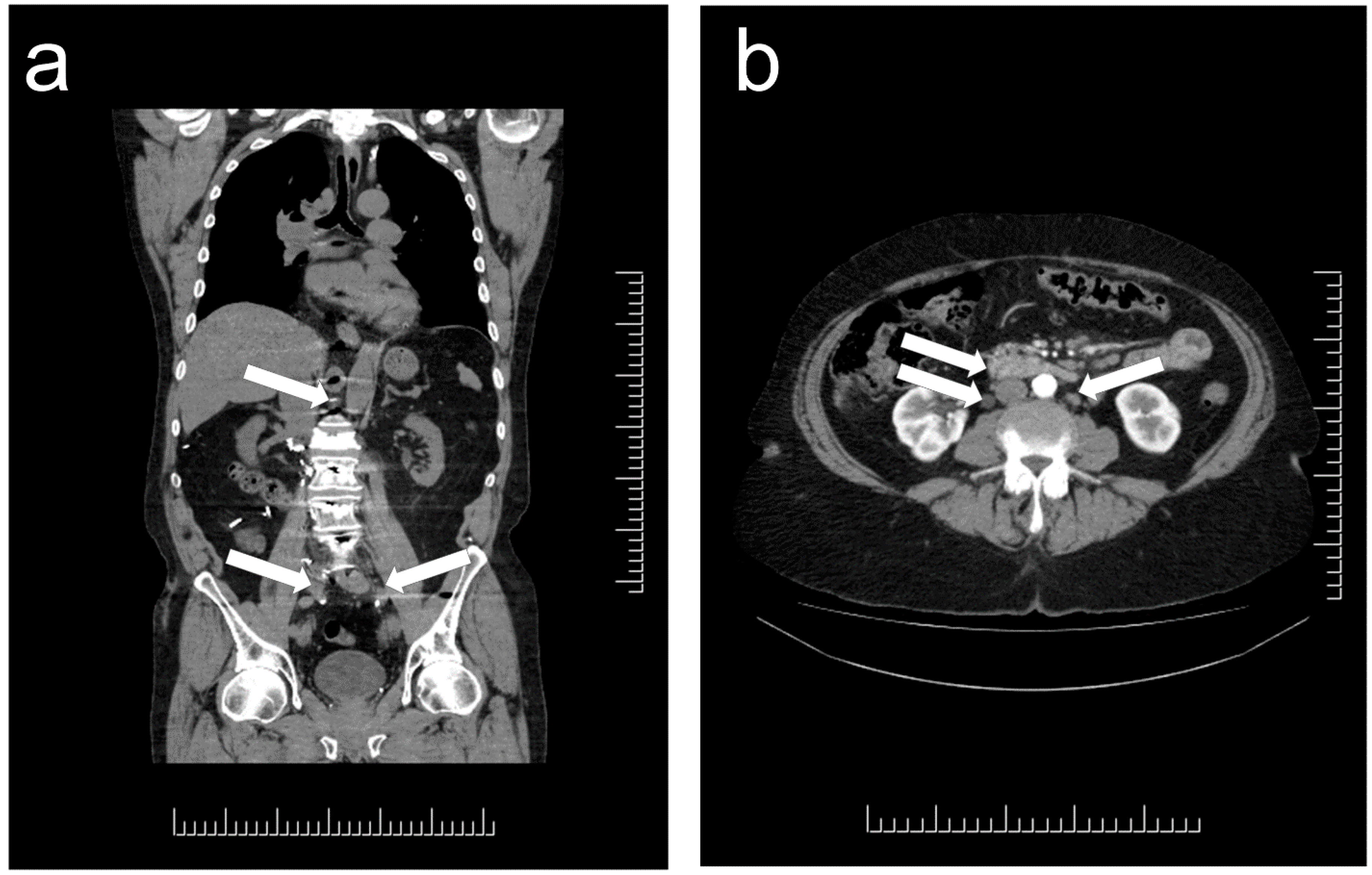
1. Distribution and Localization of Paragangliomas
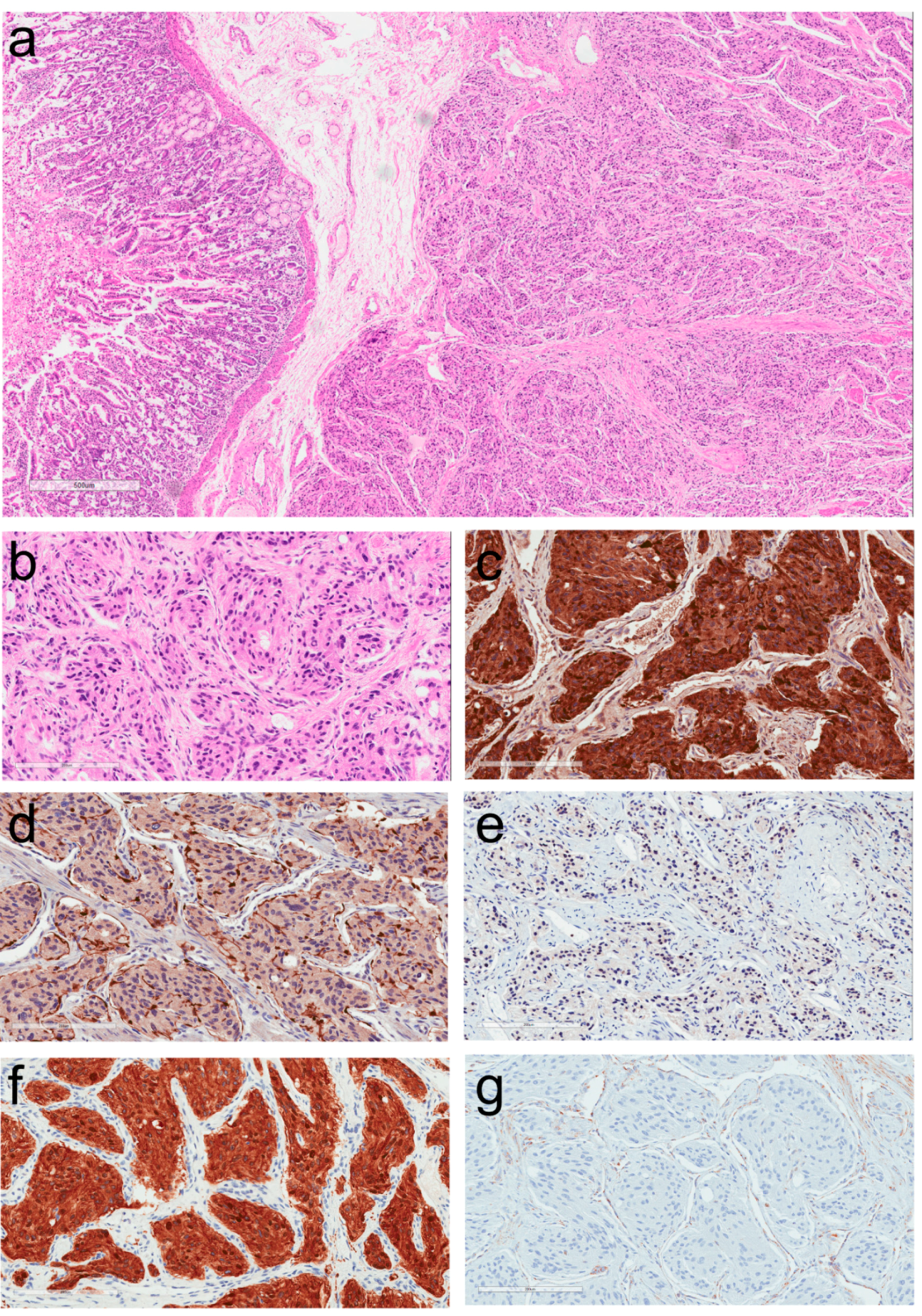
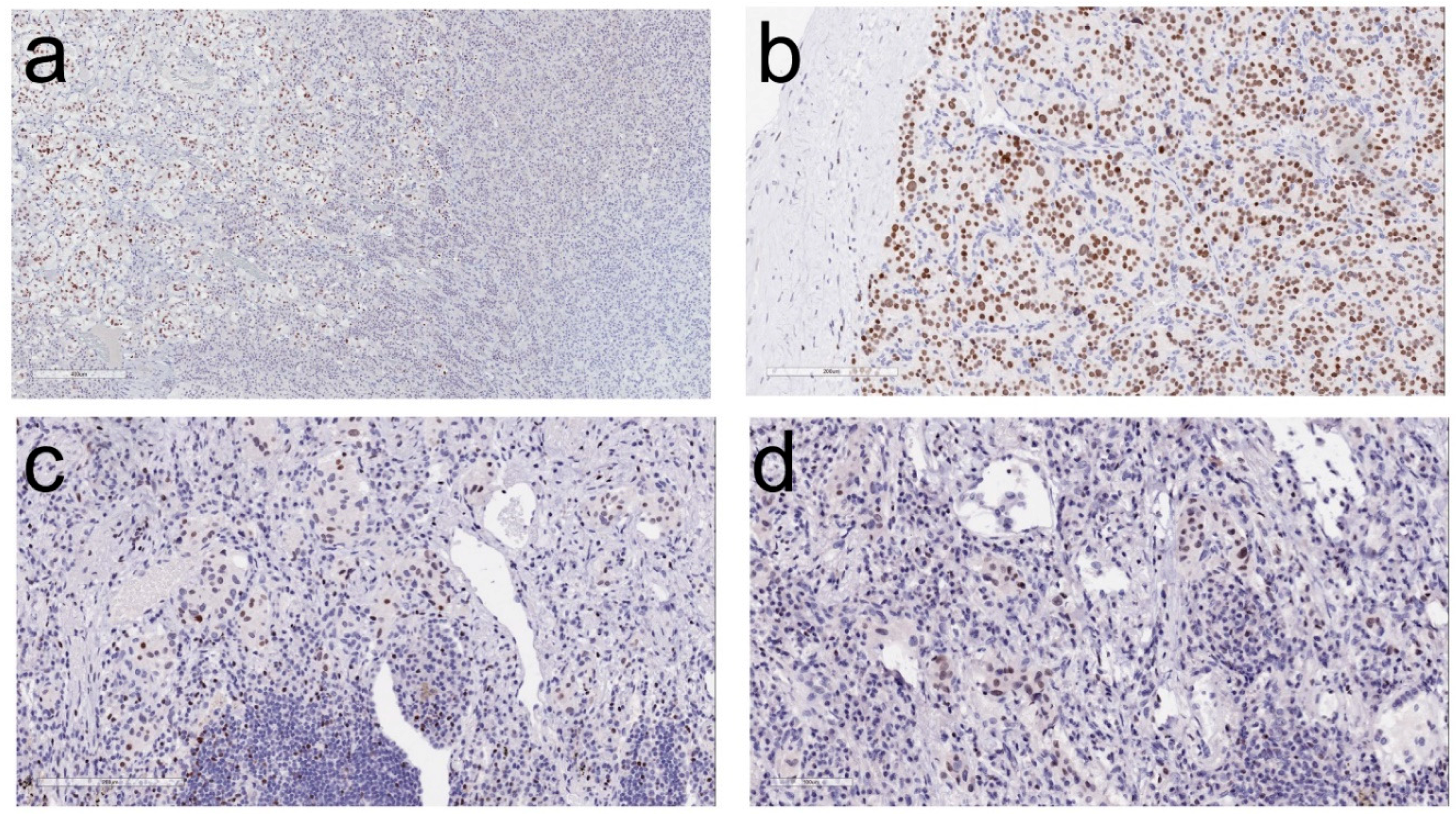
2. Morphologic Diagnosis of Paragangliomas
3. Clinical Implications of the Diagnosis of Paraganglioma
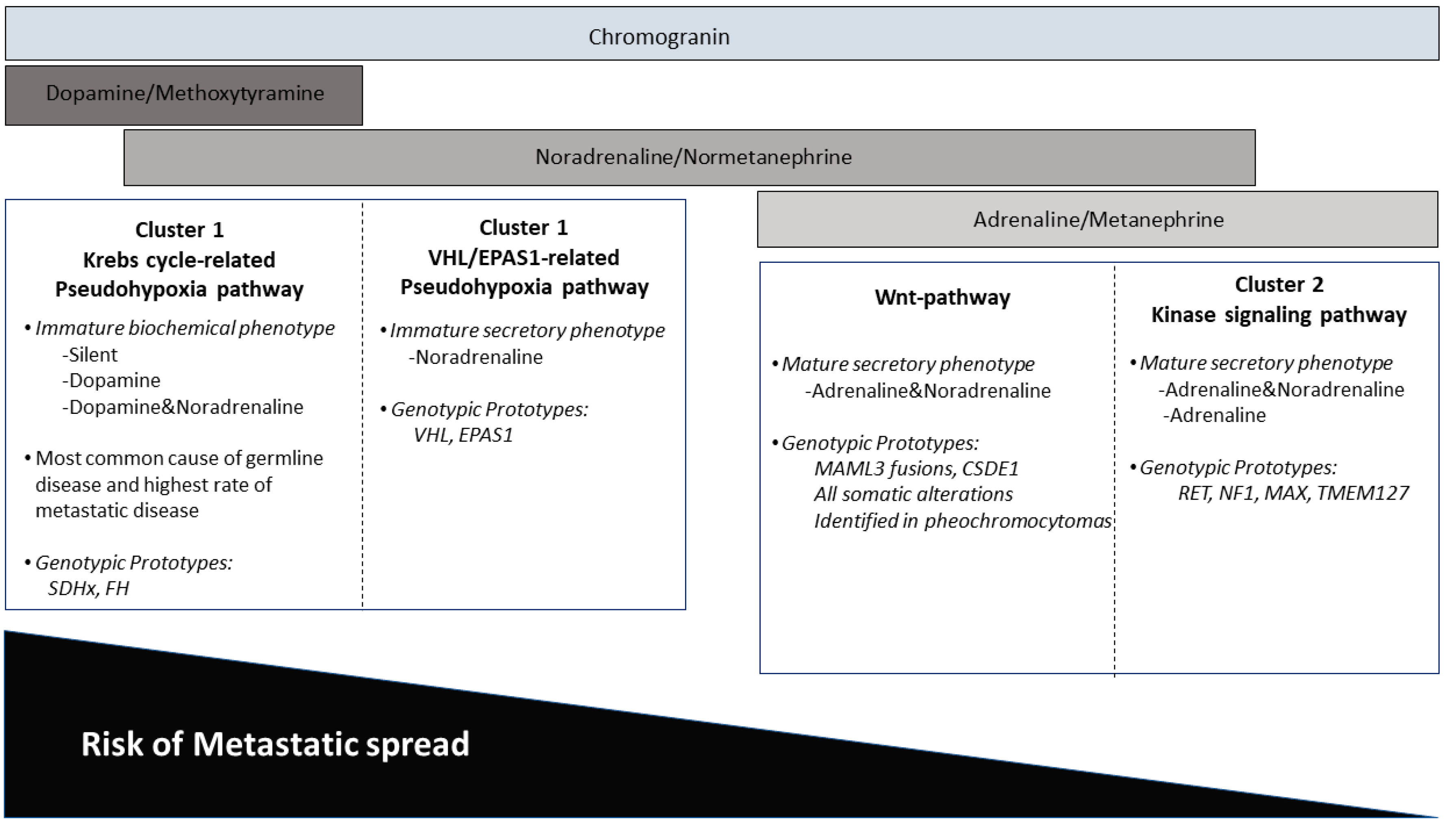
4. Impact of Genotype-Phenotype Correlations in Paraganglioma
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Rindi, G.; Klimstra, D.S.; Abedi-Ardekani, B.; Asa, S.L.; Bosman, F.T.; Brambilla, E.; Busam, K.J.; de Krijger, R.R.; Dietel, M.; El-Naggar, A.K.; et al. A common classification framework for neuroendocrine neoplasms: An international Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lack, E.E. Tumors of the Adrenal Gland and Extraadrenal Paraganglia. Armed Forces Institute of Pathology. In Atlas of Tumor Pathology; Series 4, Faccicle 8; American Registry of Pathology Press: Washington, DC, USA, 2007. [Google Scholar]
- Tischler, A.S. The adrenal medulla and extra-adrenal paraganglia. In Functional Endocrine Pathology; Kovacs, K., Asa, S.L., Eds.; Blackwell Science: Hoboken, NJ, USA, 1998; pp. 550–595. [Google Scholar]
- Oudijk, L.; de Krijger, R.R.; Pacak, K.; Tischler, A.S. Adrenal medulla and extra-adrenal paraganglia. In Endocrine Pathology; Mete, O., Asa, S.L., Eds.; Cambridge University Press: Cambridge, UK, 2016; pp. 628–676. [Google Scholar]
- Hayashi, T.; Mete, O. Head and neck paragangliomas: What does the pathologist need to know? Diagn. Histopathol. 2014, 20, 316–325. [Google Scholar] [CrossRef]
- Tischler, A.S.; Asa, S.L. Paraganglia. In Histology for Pathologists; Mills, S.E., Ed.; Wolters Kluwer: Philadelphia, PA, USA, 2018; in press. [Google Scholar]
- Lack, E.E. Tumors of the Adrenal Gland and Extra-Adrenal Paraganglia. In AFIP Atlas of Tumor Pathology; Series 3, Fascicle 19; Armed Forces Institute of Pathology: Washington, DC, USA, 1997. [Google Scholar]
- Mete, O.; Pakbaz, S.; Cassol, C.; Asa, S.L. The Spectrum and Clinical Features of Paragangliomas. Lab. Investig. 2018, 98, 234. [Google Scholar]
- Ober, W.B. Emil Zuckerkandl and his delightful little organ. Pathol. Annu. 1983, 18 Pt 1, 103–119. [Google Scholar]
- Chow, L.T.C.; Chan, M.H.M.; Wong, S.K.C. Functional Ulnar Nerve Paraganglioma: First Documented Occurrence in the Extremity With Hitherto Undescribed Associated Extensive Glomus Cell Hyperplasia and Tumorlet Formation. Int. J. Surg. Pathol. 2018, 26, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Mehra, S.; Chung-Park, M. Gallbladder paraganglioma: A case report with review of the literature. Arch. Pathol. Lab Med. 2005, 129, 523–526. [Google Scholar] [PubMed]
- Ece, I.; Alptekin, H.; Celik, Z.E.; Sahin, M. Gallbladder paraganglioma. Ulus. Cerrahi. Derg. 2015, 31, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Koh, P.S.; Koong, J.K.; Westerhout, C.J.; Yoong, B.K. Hepatobiliary and pancreatic: A huge liver paraganglioma. J. Gastroenterol. Hepatol. 2013, 28, 1075. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Ding, Z.Y.; Zhang, B.; Chen, L.; Li, G.X.; Wu, J.J.; Zhang, B.X.; Chen, X.P.; Zhu, P. Primary functioning hepatic paraganglioma mimicking hepatocellular carcinoma: A case report and literature review. Medicine 2018, 97, e0293. [Google Scholar] [CrossRef] [PubMed]
- Steel, T.R.; Dailey, A.T.; Born, D.; Berger, M.S.; Mayberg, M.R. Paragangliomas of the sellar region: Report of two cases. Neurosurgery 1993, 32, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Scheithauer, B.W.; Parameswaran, A.; Burdick, B. Intrasellar paraganglioma: Report of a case in a sibship of von Hippel-Lindau disease. Neurosurgery 1996, 38, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Mokry, M.; Kleinert, R.; Clarici, G.; Obermayer-Pietsch, B. Primary paraganglioma simulating pituitary macroadenoma: A case report and review of the literature. Neuroradiology 1998, 40, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Sambaziotis, D.; Kontogeorgos, G.; Kovacs, K.; Horvath, E.; Levedis, A. Intrasellar paraganglioma presenting as nonfunctioning pituitary adenoma. Arch. Pathol. Lab. Med. 1999, 123, 429–432. [Google Scholar] [PubMed]
- Naggara, O.; Varlet, P.; Page, P.; Oppenheim, C.; Meder, J.F. Suprasellar paraganglioma: A case report and review of the literature. Neuroradiology 2005, 47, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Boari, N.; Losa, M.; Mortini, P.; Snider, S.; Terreni, M.R.; Giovanelli, M. Intrasellar paraganglioma: A case report and review of the literature. Acta Neurochir. 2006, 148, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Özüm, Ü.; Egilmez, R.; Yildirim, A. Paraganglioma in pituitary fossa. Neuropathology 2008, 28, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Sharma, M.C.; Sharma, B.S. Malignant paraganglioma of the sellar region mimicking a pituitary macroadenoma. J. Clin. Neurosci. 2008, 15, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Haresh, K.P.; Prabhakar, R.; Anand, R.K.D.; Sharma, D.N.; Julka, P.K.; Rath, G.K. A rare case of paraganglioma of the sella with bone metastases. Pituitary 2009, 12, 276–279. [Google Scholar] [CrossRef] [PubMed]
- LaGuette, J.; Matias-Guiu, X.; Rosai, J. Thyroid paraganglioma: A clinicopathologic and immunohistochemical study of three cases. Am. J. Surg. Pathol. 1997, 21, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Nagahama, M.; Sugino, K.; Ito, K.; Kameyama, K.; Ito, K. Paraganglioma of the thyroid: Report of a male case with ultrasonographic imagings, cytologic, histologic, and immunohistochemical features. Thyroid 2007, 17, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Castelblanco, E.; Gallel, P.; Ros, S.; Gatius, S.; Valls, J.; De-Cubas, A.A.; Maliszewska, A.; Yebra-Pimentel, M.T.; Menarguez, J.; Gamallo, C.; et al. Thyroid paraganglioma. Report of 3 cases and description of an immunohistochemical profile useful in the differential diagnosis with medullary thyroid carcinoma, based on complementary DNA array results. Hum. Pathol. 2012, 43, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Policarpio-Nicolas, M.L. Thyroid Paraganglioma. Arch. Pathol. Lab. Med. 2015, 139, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Taweevisit, M.; Bunyayothin, W.; Thorner, P.S. Thyroid Paraganglioma: “Naked” Nuclei as a Clue to Diagnosis on Imprint Cytology. Endocr. Pathol. 2015, 26, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Navaratne, L.; Mathew, R.G.; Kousparos, G.; McCombe, A. The Management of Locally Invasive Primary Thyroid Paraganglioma: A Case Report and Review of the Literature. Head Neck Pathol. 2017, 11, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.T.; Braun, J.T.; Pennant, M.; Thompson, L.D. Primary paraganglioma of the parathyroid: A case report and clinicopathologic review. Head Neck Pathol. 2010, 4, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nogimura, H.; Kuriki, K.; Kobayashi, R. Superior mediastinal paraganglioma associated with von Hippel-Lindau syndrome: Report of a case. World J. Surg. Oncol. 2014, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Mehta, C.K.; Gillespie, C.T.; Lin, X.; Yeldandi, A.; DeCamp, M.; Bharat, A. Rare Middle Mediastinal Paraganglioma Mimicking Metastatic Neuroendocrine Tumor. Ann. Thorac. Surg. 2015, 100, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Kamitani, T.; Kawanami, S.; Nagao, M.; Yonezawa, M.; Yamasaki, Y.; Baba, S.; Yabuuchi, H.; Hida, T.; Kohashi, K.; et al. Mediastinal paraganglioma. Jpn J. Radiol. 2015, 33, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Millar, A.C.; Mete, O.; Cusimano, R.J.; Fremes, S.E.; Keshavjee, S.; Morgan Sylvia, C.D.; Asa, L.A.; Ezzat, S.; Gilbert, J. Functional Cardiac Paraganglioma Associated with a Rare SDHC Mutation. Endocr. Pathol. 2014, 25, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Gucer, H.; Mete, O. Endobronchial gangliocytic paraganglioma: Not all keratin-positive endobronchial neuroendocrine neoplasms are pulmonary carcinoids. Endocr. Pathol. 2014, 25, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liang, Q.L.; Jiang, L.; Liu, Q.L.; Qu, W.T.; Li, D.H.; Zhang, H.J.; Yuan, G.L. Primary Pulmonary Paraganglioma: A Case Report and Review of Literature. Medicine 2015, 94, e1271. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, G.; Annunziata, A.; de Rosa, N. Primary paraganglioma of the lung: A case report. J. Med. Case Rep. 2015, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Samuel, N.; Ejaz, R.; Silver, J.; Ezzat, S.; Cusimano, R.J.; Kim, R.H. Primary mediastinal paraganglioma associated with a familial variant in the succinate dehydrogenase B subunit gene. J. Surg. Oncol. 2018, 117, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, N.; Santos, P.; Nikfarjam, M. Paraganglioma mimicking a pancreatic neoplasm. J. Pancreas 2011, 12, 259–261. [Google Scholar] [CrossRef]
- Meng, L.; Wang, J.; Fang, S.H. Primary pancreatic paraganglioma: A report of two cases and literature review. World J. Gastroenterol. 2015, 21, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Jacob, N.C.; Howard, M.; Kelly, M.; Hale, P.C. Mesenteric paraganglioma’s: An important differential diagnosis in intra-abdominal tumours. BMJ Case Rep. 2012. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Kamiya, K.; Takahashi, Y.; Miyazaki, S.; Iino, I.; Kikuchi, H.; Hiramatsu, Y.; Ohta, M.; Baba, S.; Konno, H. Mesenteric paraganglioma: Report of a case. World J. Gastrointest. Surg. 2013, 5, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.S.; de Krijger, R.R.; Gill, A.; Kawashima, A.; Kimura, N.; Komminoth, P.; Papathomas, T.G.; Thomposon, L.D.R.; Tissier, F.; Williams, M.D.; et al. Pheochromocytoma. In WHO Classification of Tumours of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloeppel, G., Rosai, J., Eds.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 10, pp. 183–189. [Google Scholar]
- Kimura, N.; Capella, C.; DeLellis, R.A.; Epstein, J.I.; Gill, A.; Kawashima, A.; Koch, C.A.; Komminoth, P.; Lam, A.K.Y.; Merino, M.J.; et al. Extra-adrenal paragangliomas. In WHO Classification of Tumours of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloeppel, G., Rosai, J., Eds.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 10, pp. 190–195. [Google Scholar]
- Assadipour, Y.; Sadowski, S.M.; Alimchandani, M.; Quezado, M.; Steinberg, S.M.; Nilubol, N.; Patel, D.; Prodanov, T.; Pacak, K.; Kebebew, E. SDHB mutation status and tumor size but not tumor grade are important predictors of clinical outcome in pheochromocytoma and abdominal paraganglioma. Surgery 2017, 161, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Feng, L.; Johnson, M.M.; Ejaz, S.; Habra, M.A.; Rich, T.; Busaidy, N.; Cote, G.J.; Perrier, N.; Phan, A.; et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: Primary tumor size and primary tumor location as prognostic indicators. J. Clin. Endocrinol. Metab. 2011, 96, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Othman, N.H.; Ezzat, S.; Kovacs, K.; Horvath, E.; Poulin, E.; Smyth, H.S.; Asa, S.L. Growth hormone-releasing hormone (GHRH) and GHRH receptor (GHRH-R) isoform expression in ectopic acromegaly. Clin. Endocrinol. 2001, 55, 135–140. [Google Scholar] [CrossRef]
- Gosney, J.R.; Denley, H.; Resl, M. Sustentacular cells in pulmonary neuroendocrine tumours. Histopathology 1999, 34, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Asa, S.L.; Dey, C.; Kennecke, H.; Laidley, D.; Law, C.; Asmis, T.; Chan, D.; Ezzat, S.; Goodwin, R.; et al. Diagnosis and management of gastrointestinal neuroendocrine tumors: An evidence-based Canadian consensus. Cancer Treat. Rev. 2016, 47, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Takako, N.; Hamada, M.; Maeda, A.; Fujioka, Y.; Kuroha, T.; Huber, R.E.; Hasegawa, S.L.; Rao, A.; Yamamoto, M.; et al. Gata3 participates in a complex transcriptional feedback network to regulate sympathoadrenal differentiation. Development 2006, 133, 3871–3881. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Shi, J.; Wilkerson, M.L.; Lin, F. Immunohistochemical evaluation of GATA3 expression in tumors and normal tissues: A useful immunomarker for breast and urothelial carcinomas. Am. J. Clin. Pathol. 2012, 138, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, N.G. Value of GATA3 immunostaining in tumor diagnosis: A review. Adv. Anat. Pathol. 2013, 20, 352–360. [Google Scholar] [CrossRef] [PubMed]
- So, J.S.; Epstein, J.I. GATA3 expression in paragangliomas: A pitfall potentially leading to misdiagnosis of urothelial carcinoma. Mod. Pathol. 2013, 26, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- Weissferdt, A.; Kalhor, N.; Liu, H.; Rodriguez, J.; Fujimoto, J.; Tang, X.M.; Wistuba, I.I.; Moran, C.A. Thymic neuroendocrine tumors (paraganglioma and carcinoid tumors): A comparative immunohistochemical study of 46 cases. Hum. Pathol. 2014, 45, 2463–2470. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; McCue, P.A.; Sarlomo-Rikala, M.; Rys, J.; Czapiewski, P.; Wazny, K.; Langfort, R.; Waloszczyk, P.; Biernat, W.; Lasota, J.; et al. GATA3: A multispecific but potentially useful marker in surgical pathology: A systematic analysis of 2500 epithelial and nonepithelial tumors. Am. J. Surg. Pathol. 2014, 38, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Kefeli, M.; Çalişkan, S.; Asa, S.L. GATA-3 Immunoreactivity Expands The Transcription Factor Profile of Pituitary Neuroendocrine Tumors. Mod. Pathol. 2018. submitted. [Google Scholar]
- Hervonen, A.; Pickel, V.M.; Joh, T.H.; Reis, D.J.; Linnoila, I.; Kanerva, L.; Miller, R.J. Immunocytochemical demonstration of the catecholamine-synthesizing enzymes and neuropeptides in the catecholamine-storing cells of human fetal sympathetic nervous system. Adv. Biochem. Psychopharmacol. 1980, 25, 373–378. [Google Scholar] [PubMed]
- Eisenhofer, G.; Tischler, A.S.; de Krijger, R.R. Diagnostic tests and biomarkers for pheochromocytoma and extra-adrenal paraganglioma: From routine laboratory methods to disease stratification. Endocr. Pathol. 2012, 23, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.S. Pheochromocytoma and extra-adrenal paraganglioma: Updates. Arch. Pathol. Lab. Med. 2008, 132, 1272–1284. [Google Scholar] [PubMed]
- Mete, O.; Tischler, A.S.; de Krijger, R.; McNicol, A.M.; Eisenhofer, G.; Pacak, K.; Ezzat, S.; Asa, S.L. Protocol for the examination of specimens from patients with pheochromocytomas and extra-adrenal paragangliomas. Arch. Pathol. Lab. Med. 2014, 138, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Lenders, J.W.; Timmers, H.; Mannelli, M.; Grebe, S.K.; Hofbauer, L.C.; Bornstein, S.R.; Tiebel, O.; Adams, K.; Bratslavsky, G.; et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin. Chem. 2011, 57, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.; Peitzsch, M.; Prejbisz, A.; Hanus, K.; Fassnacht, M.; Beuschlein, F.; Brugger, C.; Fliedner, S.; Langton, K.; Pamporaki, C.; et al. Plasma methoxytyramine: Clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur. J. Endocrinol. 2017, 177, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Osinga, T.E.; van der Horst-Schrivers, A.N.A.; van Faassen, M.; Kerstens, M.N.; Dullaart, R.P.; Peters, M.A.; van der Laan, B.F.A.M.; de Bock, G.H.; Linkset, T.P.; Kema, I.P. Dopamine concentration in blood platelets is elevated in patients with head and neck paragangliomas. Clin. Chem. Lab. Med. 2016, 54, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.A.; Burnichon, N.; Cascon, A.; Benn, D.E.; Bayley, J.P.; Welander, J.; Tops, C.M.; Firth, H.; Dwight, T.; Ercolino, T.; et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat. Rev. Endocrinol. 2017, 13, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Turchini, J.; Cheung, V.K.Y.; Tischler, A.S.; de Krijger, R.R.; Gill, A.J. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology 2018, 72, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Wadt, K.; Choi, J.; Chung, J.Y.; Kiilgaard, J.; Heegaard, S.; Drzewiecki, K.T.; Trent, J.M.; Hewitt, S.M.; Hayward, N.K.; Gerdes, A.M.; et al. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res. 2012, 25, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Welander, J.; Andreasson, A.; Juhlin, C.C.; Wiseman, R.W.; Bäckdahl, M.; Höög, A.; Larsson, C.; Gimm, O.; Söderkvist, P. Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2014, 99, E1352–E1360. [Google Scholar] [CrossRef] [PubMed]
- Juhlin, C.C.; Stenman, A.; Haglund, F.; Clark, V.E.; Brown, T.C.; Baranoski, J.; Bilguvar, K.; Goh, G.; Welander, J.; Svahn, F.; Rubinstein, J.C.; et al. Whole-exome sequencing defines the mutational landscape of pheochromocytoma and identifies KMT2D as a recurrently mutated gene. Gene Chromosome Cancer 2015, 54, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Remacha, L.; Curras-Freixes, M.; Torres-Ruiz, R.; Schiavi, F.; Torres-Pérez, R.; Calsina, B.; Letón, R.; Comino-Méndez, I.; Roldán-Romero, J.M.; Montero-Conde, C.; et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Calsina, B.; Curras-Freixes, M.; Buffet, A.; Pons, T.; Contreras, L.; Letón, R.; Comino-Méndez, I.; Remacha, L.; Calatayud, M.; Obispo, B.; et al. Role of MDH2 pathogenic variant in pheochromocytoma and paraganglioma patients. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Udager, A.M.; Magers, M.J.; Goerke, D.M.; Vinco, M.L.; Siddiqui, J.; Cao, X.H.; Lucas, D.R.; Myers, J.L.; Chinnaiyan, A.M.; McHugh, J.B.; et al. The utility of SDHB and FH immunohistochemistry in patients evaluated for hereditary paraganglioma-pheochromocytoma syndromes. Hum. Pathol. 2018, 71, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Korpershoek, E.; Favier, J.; Gaal, J.; Burnichon, N.; van Gessel, B.; Oudijk, L.; Badoual, C.; Gadessaud, N.; Venisse, A.; Bayley, J.P.; et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 2011, 96, E1472–E1476. [Google Scholar] [CrossRef] [PubMed]
- Papathomas, T.G.; Oudijk, L.; Persu, A.; Gill, A.J.; Van Nederveen, F.; Tischler, A.S.; Tissier, F.; Volante, M.; Matias-Guiu, X.; Smid, M.; et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: A multicenter interobserver variation analysis using virtual microscopy: A Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod. Pathol. 2015, 28, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Menara, M.; Oudijk, L.; Badoual, C.; Bertherat, J.; Lepoutre-Lussey, C.; Amar, L.; Iturrioz, X.; Sibony, M.; Zinzindohoué, F.; de Krijger, R.; et al. SDHD immunohistochemistry: A new tool to validate SDHx mutations in pheochromocytoma/paraganglioma. J. Clin. Endocrinol. Metab. 2015, 100, E287–E291. [Google Scholar] [CrossRef] [PubMed]
- Roszko, K.L.; Blouch, E.; Blake, M.; Powers, J.F.; Tischler, A.S.; Hodin, R.; Sadow, P.; Lawson, E.A. Case Report of a Prolactinoma in a Patient With a Novel MAX Mutation and Bilateral Pheochromocytomas. J. Endocr. Soc. 2017, 1, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Ramachandran, R.; Toussi, S.T.K.; Vergine, M.; Ngo, N.; Sharma, R.; Lloyd, T.; Meeran, K.; Palazzo, F.; Martin, N.; et al. Immunohistochemical markers of the hypoxic response can identify malignancy in phaeochromocytomas and paragangliomas and optimize the detection of tumours with VHL germline mutations. Br. J. Cancer 2013, 108, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Asa, S.L. Precursor lesions of endocrine system neoplasms. Pathology 2013, 45, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Grogan, R.H.; Pacak, K.; Pasche, L.; Huynh, T.T.; Greco, R.S. Bilateral adrenal medullary hyperplasia associated with an SDHB mutation. J. Clin. Oncol. 2011, 29, e200–e202. [Google Scholar] [CrossRef] [PubMed]
- Toledo, S.P.A.; Lourenco, D.M., Jr.; Sekiya, T.; Lucon, A.M.; Baena, M.E.; Castro, C.C.; Bortolotto, L.A.; Zerbini, M.C.N.; Siqueira, S.A.C.; Toledo, R.A.; et al. Penetrance and clinical features of pheochromocytoma in a six-generation family carrying a germline TMEM127 mutation. J. Clin. Endocrinol. Metab. 2015, 100, E308–E318. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, K.G.; Ezzat, S.; Morel, C.F.; Swallow, C.; Otremba, M.; Dickson, B.C.; Asa, S.L.; Mete, O. Familial pheochromocytoma and renal cell carcinoma syndrome: TMEM127 as a novel candidate gene for the association. Virchows Arch. 2015, 466, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Romanet, P.; Guerin, C.; Pedini, P.; Essamet, W.; Castinetti, F.; Sebag, F.; Roche, P.; Cascon, A.; Tischler, A.S.; Pacak, K.; et al. Pathological and Genetic Characterization of Bilateral Adrenomedullary Hyperplasia in a Patient with Germline MAX Mutation. Endocr. Pathol. 2017, 28, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Crona, J.; Taieb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef] [PubMed]
- Janssen, I.; Blanchet, E.M.; Adams, K.; Chen, C.C.; Millo, C.; Herscovitch, P.; Taïeb, D.; Kebebew, E.; Lehnert, H.; Fojo, A.T.; et al. Superiority of [68Ga]-DOTATATE PET/CT to Other Functional Imaging Modalities in the Localization of SDHB-Associated Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2015, 21, 3888–3895. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Ling, A.; Millo, C.; Gupta, G.; Viana, B.; Lin, F.I.; Herscovitch, P.; Adams, K.P.; Taieb, D.; Metwalli, A.R.; et al. Superiority of (68)Ga-DOTATATE over (18)F-FDG and anatomic imaging in the detection of succinate dehydrogenase mutation (SDHx)-related pheochromocytoma and paraganglioma in the pediatric population. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.D.R. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am. J. Surg. Pathol. 2002, 26, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Takayanagi, R.; Takizawa, N.; Itagaki, E.; Katabami, T.; Kakoi, N.; Rakugi, H.; Ikeda, Y.; Tanabe, A.; Nigawara, T.; et al. Phaechromocytoma Study Group in Japan. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr. Relat. Cancer 2014, 21, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, O.; Young, W.F., Jr.; Gruber, L.; Smestad, J.; Yan, Q.; Ponce, O.J.; Prokop, L.; Murad, M.H.; Bancos, I. Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: A systematic review and meta-analysis. Clin. Endocrinol. 2017, 87, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Niemeijer, N.D.; Papathomas, T.G.; Korpershoek, E.; de Krijger, R.R.; Oudijk, L.; Morreau, H.; Bayley, J.P.; Hes, F.J.; Jansen, J.C.; Dinjens, W.N.M.; et al. Succinate Dehydrogenase (SDH)-Deficient Pancreatic Neuroendocrine Tumor Expands the SDH-Related Tumor Spectrum. J. Clin. Endocrinol. Metab. 2015, 100, E1386–E1393. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; de Jesus, A.C.; Kerr, D.; Mete, O.; Nose, V.; Papotti, M.; Volante, M. Thyroid. In Endocrine Pathology; Mete, O., Asa, S.L., Eds.; Cambridge University Press: Cambridge, UK, 2016; pp. 398–572. [Google Scholar]
- Duan, K.; Mete, O. Hereditary endocrine tumor syndromes: The clinical and predictive role of molecular hoistopathology. AJSP Rev. Rep. 2017, 22, 246–268. [Google Scholar] [CrossRef]
- Gill, A.J. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018, 72, 106–116. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Transcription Factors Frequently Expressed |
|---|---|
| Pituitary | Pit-1, Tpit, SF-1, ER-alpha, GATA-3, GATA-2 |
| Thyroid | PAX-8 *, TTF-1 |
| Parathyroid | GATA-3, GCM-2 |
| Lung | TTF-1 |
| Stomach | CDX-2 |
| Duodenum | ISL-1, PDX-1, CDX-2 |
| Pancreas | PDX-1, ISL-1, CDX-2 |
| Jejunum/Ileum | CDX-2 |
| Appendix | CDX-2 |
| Colon/Rectum | CDX-2 |
| Paragangliomas | GATA-3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asa, S.L.; Ezzat, S.; Mete, O. The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. J. Clin. Med. 2018, 7, 280. https://doi.org/10.3390/jcm7090280
Asa SL, Ezzat S, Mete O. The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. Journal of Clinical Medicine. 2018; 7(9):280. https://doi.org/10.3390/jcm7090280
Chicago/Turabian StyleAsa, Sylvia L., Shereen Ezzat, and Ozgur Mete. 2018. "The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations" Journal of Clinical Medicine 7, no. 9: 280. https://doi.org/10.3390/jcm7090280
APA StyleAsa, S. L., Ezzat, S., & Mete, O. (2018). The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. Journal of Clinical Medicine, 7(9), 280. https://doi.org/10.3390/jcm7090280