Antimycobacterial, Enzyme Inhibition, and Molecular Interaction Studies of Psoromic Acid in Mycobacterium tuberculosis: Efficacy and Safety Investigations

 ,
, 
Abstract
1. Introduction
2. Experimental Section
2.1. Antimicrobial Activity
2.1.1. Bacterial Strains, Chemicals, Medium, and Cultures
2.1.2. In Vitro Antimycobacterial Efficacy Evaluation
2.2. Determination of Cytotoxicity
2.3. Determination of Lipophilicity
2.4. Anti-UDP-Galactopyranose Mutase (UGM) Activity
2.4.1. Enzyme Expression and Purification
2.4.2. Anti-UGM Activity
2.5. Anti-arylamine-N-acetyltransferase Activity (TBNAT)
2.5.1. Enzyme Production and Purification
2.5.2. Anti-TBNAT Activity
2.6. Molecular Docking Analysis
3. Results and Discussion
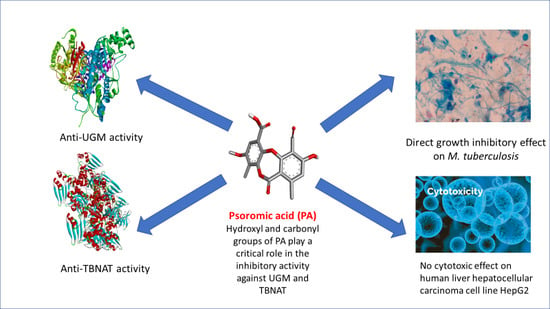
3.1. Antimycobacterial Properties
3.2. Cytotoxicity and Lipophilicity Studies
3.3. Anti-UGM Evaluation
3.4. Anti-TBNAT Evaluation
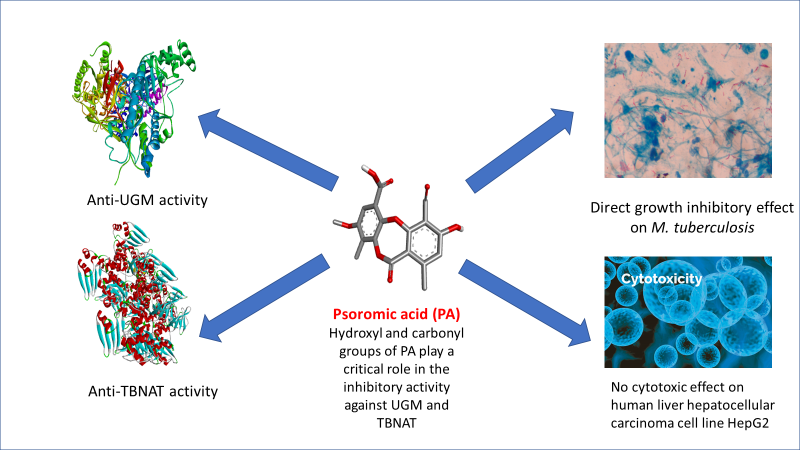
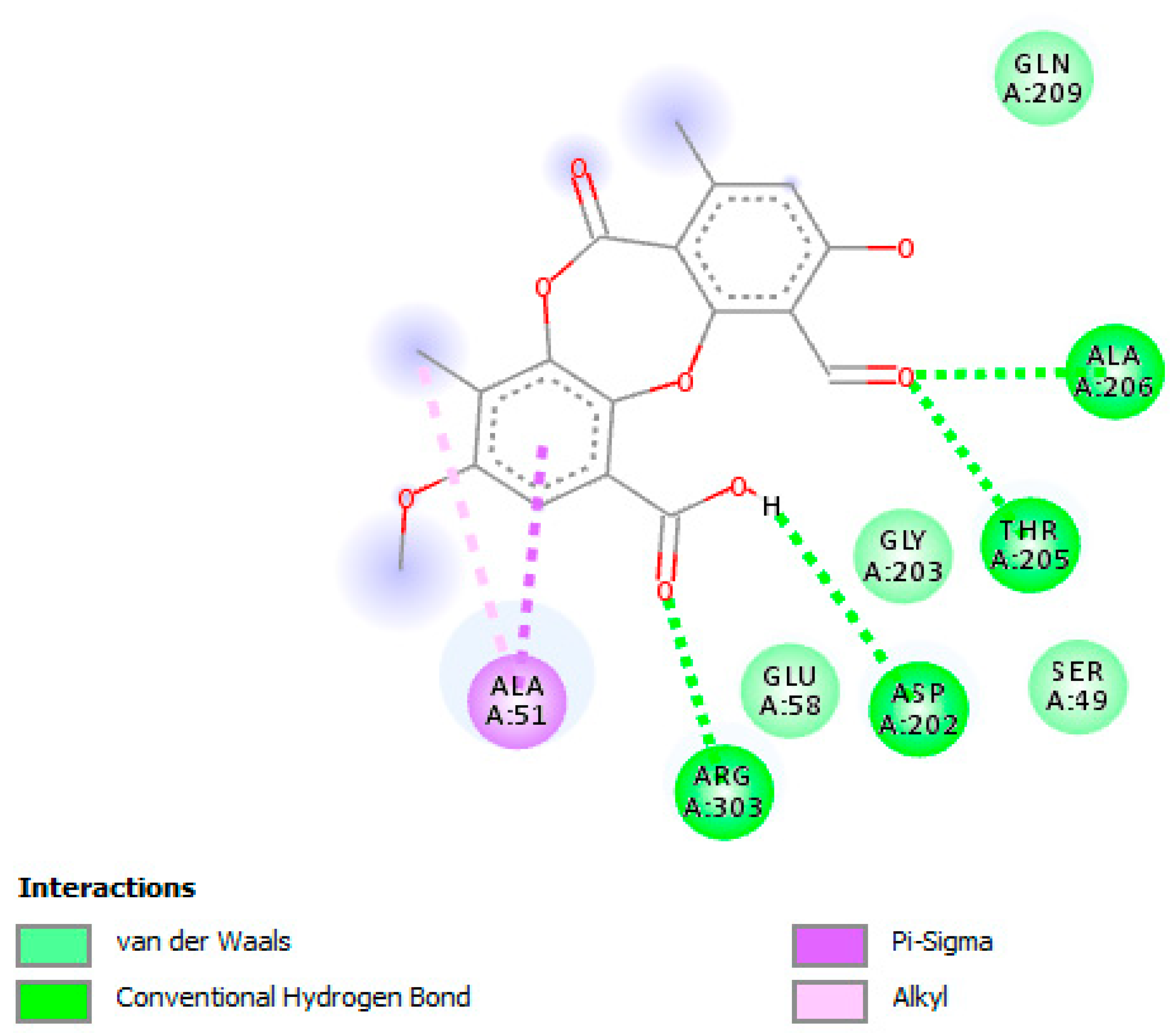
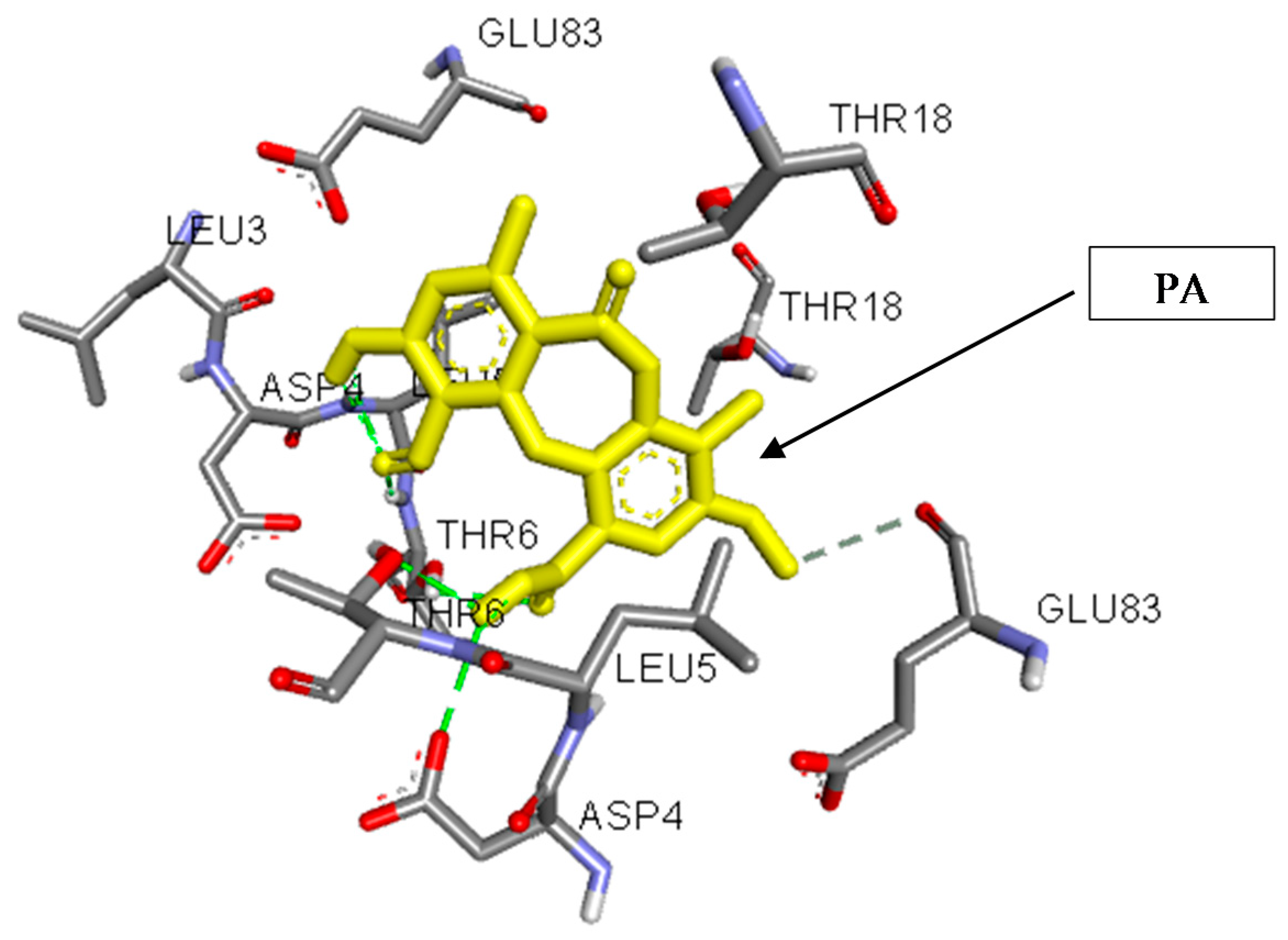
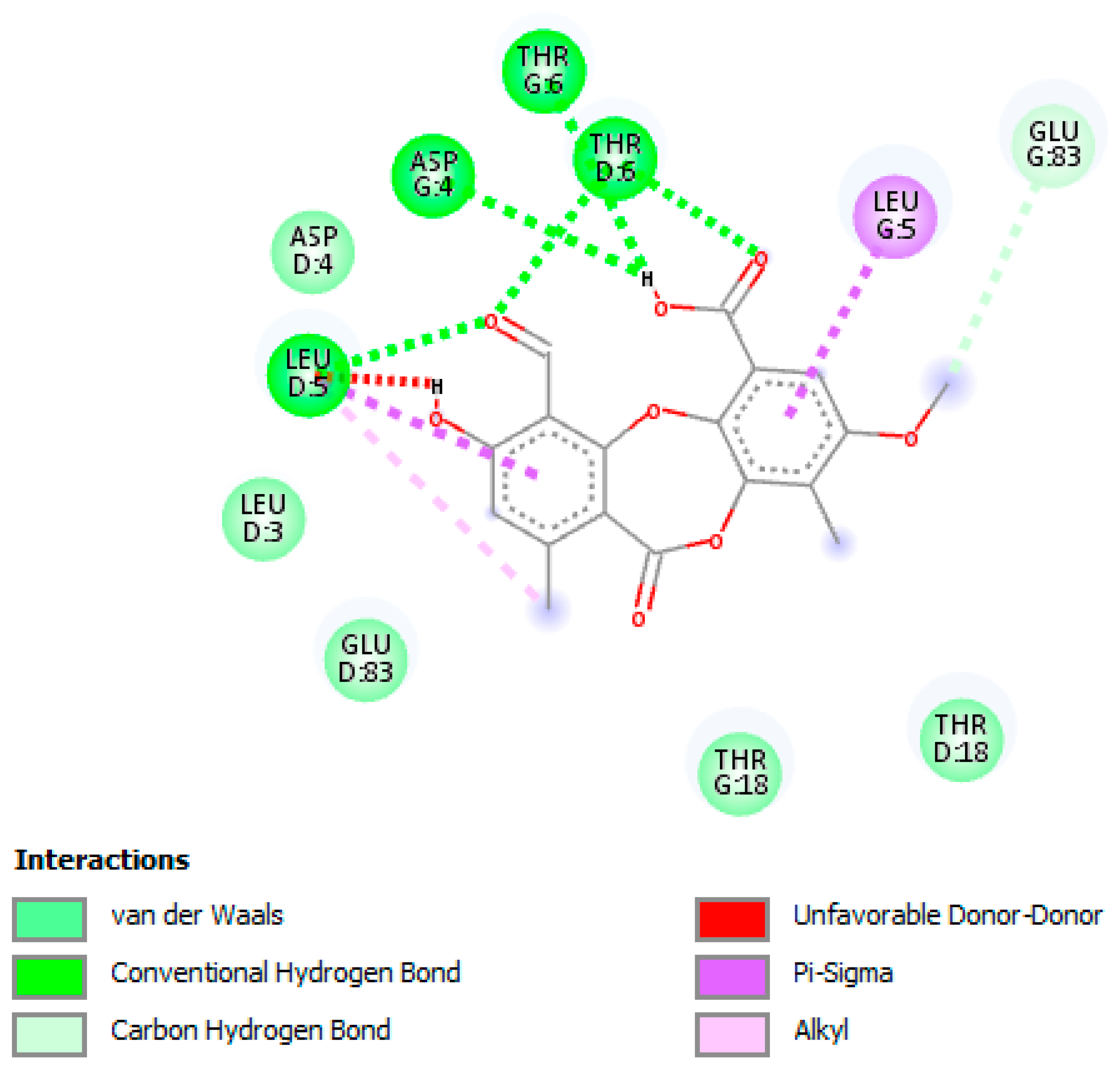
3.5. In Silico Studies and Molecular Interactions
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Raj, R.; Biot, C.; Carrère-Kremer, S.; Kremer, L.; Guérardel, Y.; Gut, J.; Rosenthal, P.J.; Forge, D.; Kumar, V. 7-chloroquinoline–isatin conjugates: Antimalarial, antitubercular, and cytotoxic evaluation. Chem. Biol. Drug Des. 2014, 83, 622–629. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Fact Sheet on Tuberculosis (Updated January 2018). Available online: http://www.who.int/en/news-room/fact-sheets/detail/tuberculosis (accessed on 30 January 2018).
- WHO. Drug-Resistant TB: Global Situation. 2018. Available online: http://www.who.int/tb/areas-of-work/drug-resistant-tb/global-situation/en/ (accessed on 30 January 2018).
- Herrmann, J.; Rybniker, J.; Müller, R. Novel and revisited approaches in antituberculosis drug discovery. Curr. Opin. Biotechnol. 2017, 48, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Pfeiffer, B.; Altmann, K.-H. Recent developments in natural product-based drug discovery for tuberculosis. Drug Discov. Today 2017, 22, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Young, D.B.; Perkins, M.D.; Duncan, K.; Barry, C.E. Confronting the scientific obstacles to global control of tuberculosis. J Clin. Investig. 2008, 118, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, S.; Zandberg, W.F.; Mohan, S.; Ko, M.; Martinez-Gutierrez, F.; Partha, S.K.; Sanders, D.A.; Av-Gay, Y.; Pinto, B.M. Antimycobacterial activity of UDP-galactopyranose mutase inhibitors. Int. J. Antimicrob. Agents 2010, 36, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.S.M.; Eckshtain-Levi, M.; Vogelaar, N.J.; Sobrado, P. Identification of Aspergillus fumigatus UDP-Galactopyranose mutase inhibitors. Sci. Rep. 2017, 7, 10836. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.J.; Boechi, L.; McCammon, J.A.; Sobrado, P. Structure, mechanism, and dynamics of udp-galactopyranose mutase. Arch. Biochem. Biophys. 2014, 544, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Fu, H.; Dieu, M.; Halloum, I.; Kremer, L.; Xia, Y.; Pan, W.; Vincent, S.P. Identification of inhibitors targeting Mycobacterium tuberculosis cell wall biosynthesis via dynamic combinatorial chemistry. Chem. Commun. 2017, 53, 10632–10635. [Google Scholar] [CrossRef] [PubMed]
- Villaume, S.A.; Fu, J.; N’Go, I.; Liang, H.; Lou, H.; Kremer, L.; Pan, W.; Vincent, S.P. Natural and synthetic flavonoids as potent Mycobacterium tuberculosis UGM inhibitors. Chem. Eur. J. 2017, 23, 10423–10429. [Google Scholar] [CrossRef] [PubMed]
- Partha, S.K.; Sadeghi-Khomami, A.; Slowski, K.; Kotake, T.; Thomas, N.R.; Jakeman, D.L.; Sanders, D.A. Chemoenzymatic synthesis, inhibition studies, and x-ray crystallographic analysis of the phosphono analog of UDP-galp as an inhibitor and mechanistic probe for UDP-galactopyranose mutase. J. Mol. Biol. 2010, 403, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Westwood, I.M.; Bhakta, S.; Russell, A.J.; Fullam, E.; Anderton, M.C.; Kawamura, A.; Mulvaney, A.W.; Vickers, R.J.; Bhowruth, V.; Besra, G.S. Identification of arylamine N-acetyltransferase inhibitors as an approach towards novel anti-tuberculars. Protein Cell 2010, 1, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Butcher, N.J.; Tiang, J.; Minchin, R.F. Regulation of arylamine N-acetyltransferases. Curr. Drug Metab. 2008, 9, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Madikane, V.E.; Bhakta, S.; Russell, A.J.; Campbell, W.E.; Claridge, T.D.; Elisha, B.G.; Davies, S.G.; Smith, P.; Sim, E. Inhibition of mycobacterial arylamine N-acetyltransferase contributes to anti-mycobacterial activity of Warburgia salutaris. Bioorg. Med. Chem. 2007, 15, 3579–3586. [Google Scholar] [CrossRef] [PubMed]
- Sim, E.; Sandy, J.; Evangelopoulos, D.; Fullam, E.; Bhakta, S.; Westwood, I.; Krylova, A.; Lack, N.; Noble, M. Arylamine N-acetyltransferases in mycobacteria. Curr. Drug Metab. 2008, 9, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Sim, E.; Pinter, K.; Mushtaq, A.; Upton, A.; Sandy, J.; Bhakta, S.; Noble, M. Arylamine N-acetyltransferases: A pharmacogenomic approach to drug metabolism and endogenous function. Biochem. Soc. Trans. 2003, 31, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Sweidan, A.; Chollet-Krugler, M.; Sauvager, A.; Van de Weghe, P.; Chokr, A.; Bonnaure-Mallet, M.; Tomasi, S.; Bousarghin, L. Antibacterial activities of natural lichen compounds against Streptococcus gordonii and Porphyromonas gingivalis. Fitoterapia 2017, 121, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Vartia, K.O. Antibiotics in lichens. In The Lichens; Academic Press, Inc.: New York, NY, USA, 1973; pp. 547–561. [Google Scholar]
- Shibata, S. Especial compounds of lichens. In Der stoffwechsel sekundärer pflanzenstoffe/the Metabolism of Secondary Plant Products; Springer: Berlin, Heidelberg, Germany, 1958; pp. 560–623. [Google Scholar]
- Emsen, B.; Aslan, A.; Togar, B.; Turkez, H. In vitro antitumor activities of the lichen compounds olivetoric, physodic and psoromic acid in rat neuron and glioblastoma cells. Pharm. Biol. 2016, 54, 1748–1762. [Google Scholar] [CrossRef] [PubMed]
- Da Rosa Guterres, Z.; Honda, N.K.; Coelho, R.G.; Alcantara, G.B.; Micheletti, A.C. Antigenotoxicity of depsidones isolated from Brazilian lichens. Orbital. Electron. J. Chem. 2017, 9, 50–54. [Google Scholar] [CrossRef]
- Brandão, L.F.G.; Alcantara, G.B.; Matos, M.D.F.C.; Bogo, D.; dos Santos Freitas, D.; Oyama, N.M.; Honda, N.K. Cytotoxic evaluation of phenolic compounds from lichens against melanoma cells. Chem. Pharm. Bull. 2013, 61, 176–183. [Google Scholar] [PubMed]
- Behera, B.C.; Mahadik, N.; Morey, M. Antioxidative and cardiovascular-protective activities of metabolite usnic acid and psoromic acid produced by lichen species Usnea complanata under submerged fermentation. Pharm. Biol. 2012, 50, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Deraeve, C.L.; Guo, Z.; Bon, R.S.; Blankenfeldt, W.; DiLucrezia, R.; Wolf, A.; Menninger, S.; Stigter, E.A.; Wetzel, S.; Choidas, A. Psoromic acid is a selective and covalent rab-prenylation inhibitor targeting autoinhibited rabggtase. J. Am. Chem. Soc. 2012, 134, 7384–7391. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute. Laboratory Detection and Identification of Mycobacteria, 1st ed.; Approved Guideline; CLSI Document M48-A; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008. [Google Scholar]
- Clinical and Laboratory Standards Institute. Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes, 2nd ed.; Approved Standard M24-A2; CLSI: Wayne, PA, USA, 2011. [Google Scholar]
- Semelková, L.; Janošcová, P.; Fernandes, C.; Bouz, G.; Janďourek, O.; Konečná, K.; Paterová, P.; Navrátilová, L.; Kuneš, J.; Doležal, M. Design, synthesis, antimycobacterial evaluation, and in silico studies of 3-(phenylcarbamoyl)-pyrazine-2-carboxylic acids. Molecules 2017, 22, 1491. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, H.W. Studies of UDP-galactopyranose mutase from Escherichia coli: An unusual role of reduced fad in its catalysis. J. Am. Chem. Soc. 2000, 122, 9065–9070. [Google Scholar] [CrossRef]
- Veerapen, N.; Yuan, Y.; Sanders, D.A.; Pinto, B.M. Synthesis of novel ammonium and selenonium ions and their evaluation as inhibitors of udp-galactopyranose mutase. Carbohydr. Res. 2004, 339, 2205–2217. [Google Scholar] [CrossRef] [PubMed]
- Abuhammad, A.; Lack, N.; Schweichler, J.; Staunton, D.; Sim, R.B.; Sim, E. Improvement of the expression and purification of Mycobacterium tuberculosis arylamine N-acetyltransferase (TBNAT) a potential target for novel anti-tubercular agents. Protein Expr. Purif. 2011, 80, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Brooke, E.W.; Davies, S.G.; Mulvaney, A.W.; Pompeo, F.; Sim, E.; Vickers, R.J. An approach to identifying novel substrates of bacterial arylamine N-acetyltransferases. Bioorg. Med. chem. 2003, 11, 1227–1234. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.T.S.; Švajdlenka, E. Biological evaluation and molecular docking of protocatechuic acid from Hibiscus sabdariffa L. As a potent urease inhibitor by an esi-ms based method. Molecules 2017, 22, 1696. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, D.S. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Ramis, I.B.; Vianna, J.S.; Reis, A.J.; von Groll, A.; Ramos, D.F.; Viveiros, M.; da Silva, P.E.A. Antimicrobial and efflux inhibitor activity of usnic acid against Mycobacterium abscessus. Planta Med. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Fadipe, V.O.; Mongalo, N.I.; Opoku, A.R.; Dikhoba, P.M.; Makhafola, T.J. Isolation of anti-mycobacterial compounds from Curtisia dentata (burm. F.) ca sm (curtisiaceae). BMC Complement. Altern. Med. 2017, 17, 306. [Google Scholar] [CrossRef] [PubMed]
- Ganihigama, D.U.; Sureram, S.; Sangher, S.; Hongmanee, P.; Aree, T.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Antimycobacterial activity of natural products and synthetic agents: Pyrrolodiquinolines and vermelhotin as anti-tubercular leads against clinical multidrug resistant isolates of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2015, 89, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, D.; Franzblau, S.G. In vitro antituberculotic activity of several lichen metabolites. Planta Med. 2007, 73, 174. [Google Scholar] [CrossRef]
- Zhang, Y.; Permar, S.; Sun, Z.H. Conditions that may affect the results of susceptibility testing of Mycobacterium tuberculosis to pyrazinamide. J. Med. Microbiol. 2002, 51, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Portaels, F.; Pattyn, S.R. Growth of mycobacteria in relation to the pH of the medium. Ann. Microbiol. (Paris) 1982, 133, 213–221. [Google Scholar] [PubMed]
- Yew, W.W.; Leung, C.C. Antituberculosis drugs and hepatotoxicity. Respirology 2006, 11, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.; Benet, L.Z. Evaluation of the relevance of dili predictive hypotheses in early drug development: Review of in vitro methodologies vs. BDDCS classification. Toxicol. Res. 2018, 7, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Dykhuizen, E.C.; May, J.F.; Tongpenyai, A.; Kiessling, L.L. Inhibitors of UDP-galactopyranose mutase thwart mycobacterial growth. J. Am. Chem. Soc. 2008, 130, 6706–6707. [Google Scholar] [CrossRef] [PubMed]
- Castagnolo, D.; de Logu, A.; Radi, M.; Bechi, B.; Manetti, F.; Magnani, M.; Supino, S.; Meleddu, R.; Chisu, L.; Botta, M. Synthesis, biological evaluation and sar study of novel pyrazole analogues as inhibitors of Mycobacterium tuberculosis. Bioorg. Med. Chem. 2008, 16, 8587–8591. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-H.; Chung, J. The effects of plant phenolics, caffeic acid, chlorogenic acid and ferulic acid on arylamine N-acetyltransferase activities in human gastrointestinal microflora. Anticancer Res. 1999, 19, 133–139. [Google Scholar] [PubMed]
- Kukongviriyapan, V.; Phromsopha, N.; Tassaneeyakul, W.; Kukongviriyapan, U.; Sripa, B.; Hahnvajanawong, V.; Bhudhisawasdi, V. Inhibitory effects of polyphenolic compounds on human arylamine N-acetyltransferase 1 and 2. Xenobiotica 2006, 36, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Chung, J.G.; Ho, C.C.; Wu, L.T.; Chang, S.H. Aloe-emodin effects on arylamine N-acetyltransferase activity in the bacterium Helicobacter pylori. Planta Med. 1998, 64, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, A.; Graham, J.; Mushtaq, A.; Tsiftsoglou, S.A.; Vath, G.M.; Hanna, P.E.; Wagner, C.R.; Sim, E. Eukaryotic arylamine N-acetyltransferase: Investigation of substrate specificity by high-throughput screening. Biochem. Pharm. 2005, 69, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Van Straaten, K.E.; Kuttiyatveetil, J.R.; Sevrain, C.M.; Villaume, S.A.; Jiménez-Barbero, J.S.; Linclau, B.; Vincent, S.P.P.; Sanders, D.A. Structural basis of ligand binding to UDP-galactopyranose mutase from Mycobacterium tuberculosis using substrate and tetrafluorinated substrate analogues. J. Am. Chem. Soc. 2015, 137, 1230–1244. [Google Scholar] [CrossRef] [PubMed]
- Fullam, E.; Talbot, J.; Abuhammed, A.; Westwood, I.; Davies, S.G.; Russell, A.J.; Sim, E. Design, synthesis and structure—Activity relationships of 3,5-diaryl-1H-pyrazoles as inhibitors of arylamine N-acetyltransferase. Bioorg. Med. Chem. Lett. 2013, 23, 2759–2764. [Google Scholar] [CrossRef] [PubMed]
- Abuhammad, A.; Fullam, E.; Lowe, E.D.; Staunton, D.; Kawamura, A.; Westwood, I.M.; Bhakta, S.; Garner, A.C.; Wilson, D.L.; Seden, P.T. Piperidinols that show anti-tubercular activity as inhibitors of arylamine N-acetyltransferase: An essential enzyme for mycobacterial survival inside macrophages. PLoS ONE 2012, 7, e52790. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.D.; Borrok, M.J.; Westler, W.M.; Forest, K.T.; Kiessling, L.L. Ligand binding and substrate discrimination by UDP-galactopyranose mutase. J. Mol. Biol. 2009, 391, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Rahim, F.; Zaman, K.; Ullah, H.; Taha, M.; Wadood, A.; Javed, M.T.; Rehman, W.; Ashraf, M.; Uddin, R.; Uddin, I. Synthesis of 4-thiazolidinone analogs as potent in vitro anti-urease agents. Bioorg. Chem. 2015, 63, 123–131. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M.tb Strains | MIC (µM) | Cytotoxicity (IC50) (µM) for PA and INH | Selectivity Index (SI) | ||
|---|---|---|---|---|---|
| PA | INH | PA | INH | ||
| M.tb H37Rv CNCTC My 331-88 (ATCC 27294) | 3.2 | 5.8 | >75 | >23.4 | >13.0 |
| M.tb-01 * | 3.9 | 5.5 | >75 | >19.2 | >13.6 |
| M.tb-02 * | 3.6 | 5.6 | >75 | >20.8 | >13.4 |
| M.tb-03 * | 3.8 | 5.4 | >75 | >19.7 | >13.9 |
| M.tb-04 * | 4.1 | 5.8 | >75 | >18.3 | >13.0 |
| M.tb-05 * | 3.2 | 5.7 | >75 | >23.4 | >13.2 |
| M.tb-06 * | 3.5 | 5.7 | >75 | >21.4 | >13.2 |
| M.tb-07 * | 3.9 | 5.5 | >75 | >19.2 | >13.6 |
| M.tb-08 * | 4.0 | 5.7 | >75 | >18.6 | >13.2 |
| Compound | Lipophilicity (Log P) Value |
|---|---|
| PA | 2.8 |
| INH | –0.7 |
| Compounds | Turnover a (%) | Inhibition b (%) |
|---|---|---|
| PA | 8.3 ± 2.6 | 85.8 |
| UDP | 0.4 ± 2.3 | 99.3 |
| No inhibition | 58.4 ± 3.4 | Nd |
| Compounds | Inhibition (%) | IC50 (µM) |
|---|---|---|
| PA | 77.4 ± 0.33 | 8.7 ± 1.44 |
| INH | 96.3 ± 1.3 | 6.2 ± 1.22 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, S.T.S.; Šudomová, M.; Berchová-Bímová, K.; Gowrishankar, S.; Rengasamy, K.R.R. Antimycobacterial, Enzyme Inhibition, and Molecular Interaction Studies of Psoromic Acid in Mycobacterium tuberculosis: Efficacy and Safety Investigations. J. Clin. Med. 2018, 7, 226. https://doi.org/10.3390/jcm7080226
Hassan STS, Šudomová M, Berchová-Bímová K, Gowrishankar S, Rengasamy KRR. Antimycobacterial, Enzyme Inhibition, and Molecular Interaction Studies of Psoromic Acid in Mycobacterium tuberculosis: Efficacy and Safety Investigations. Journal of Clinical Medicine. 2018; 7(8):226. https://doi.org/10.3390/jcm7080226
Chicago/Turabian StyleHassan, Sherif T. S., Miroslava Šudomová, Kateřina Berchová-Bímová, Shanmugaraj Gowrishankar, and Kannan R. R. Rengasamy. 2018. "Antimycobacterial, Enzyme Inhibition, and Molecular Interaction Studies of Psoromic Acid in Mycobacterium tuberculosis: Efficacy and Safety Investigations" Journal of Clinical Medicine 7, no. 8: 226. https://doi.org/10.3390/jcm7080226
APA StyleHassan, S. T. S., Šudomová, M., Berchová-Bímová, K., Gowrishankar, S., & Rengasamy, K. R. R. (2018). Antimycobacterial, Enzyme Inhibition, and Molecular Interaction Studies of Psoromic Acid in Mycobacterium tuberculosis: Efficacy and Safety Investigations. Journal of Clinical Medicine, 7(8), 226. https://doi.org/10.3390/jcm7080226