The Emerging Role of Pathogenesis of IgA Nephropathy

 ,
,  ,
, 
Abstract
1. Introduction
2. Epidemiologic and Clinical Characteristics of IgAN
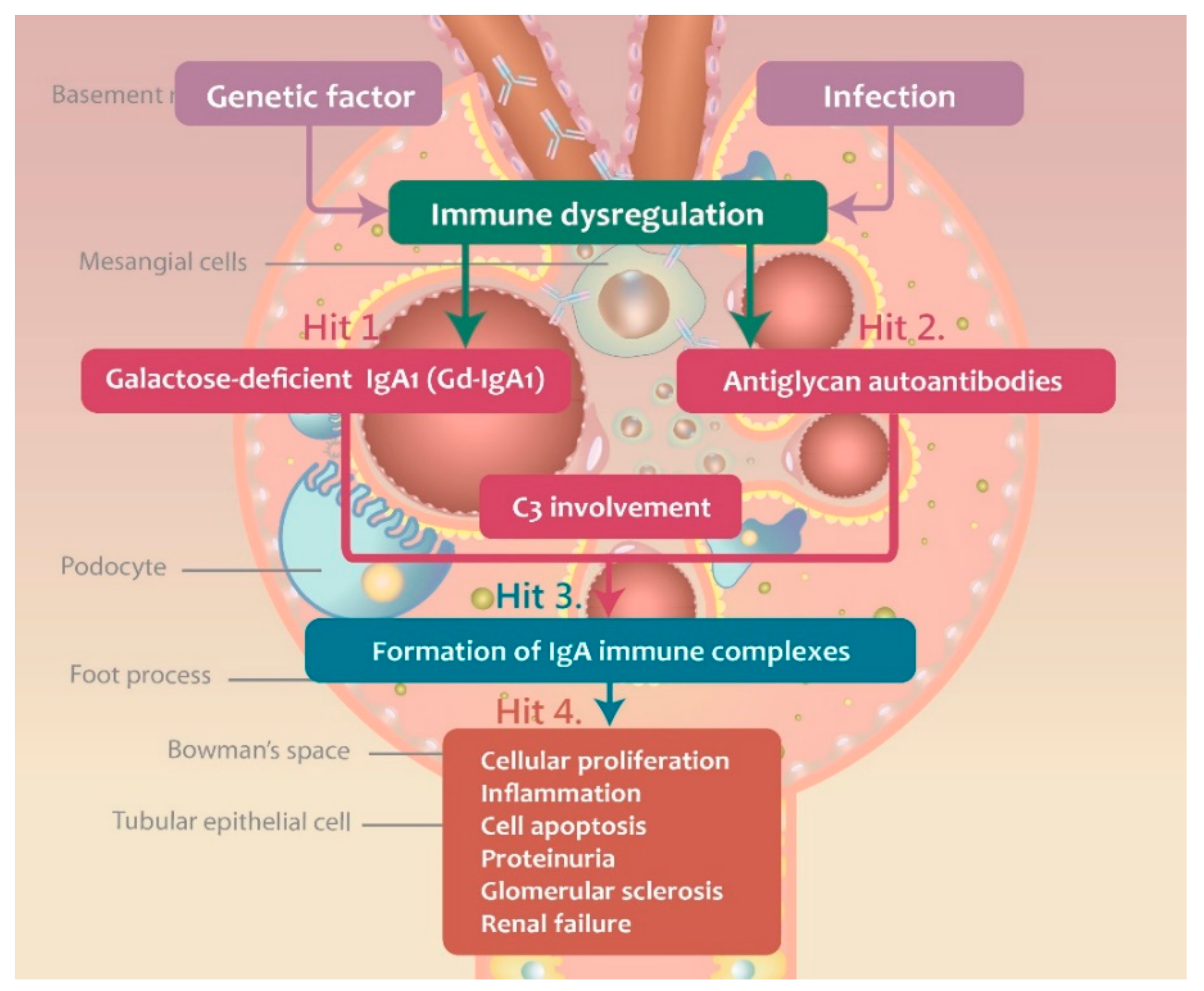
3. Pathogenesis of IgAN
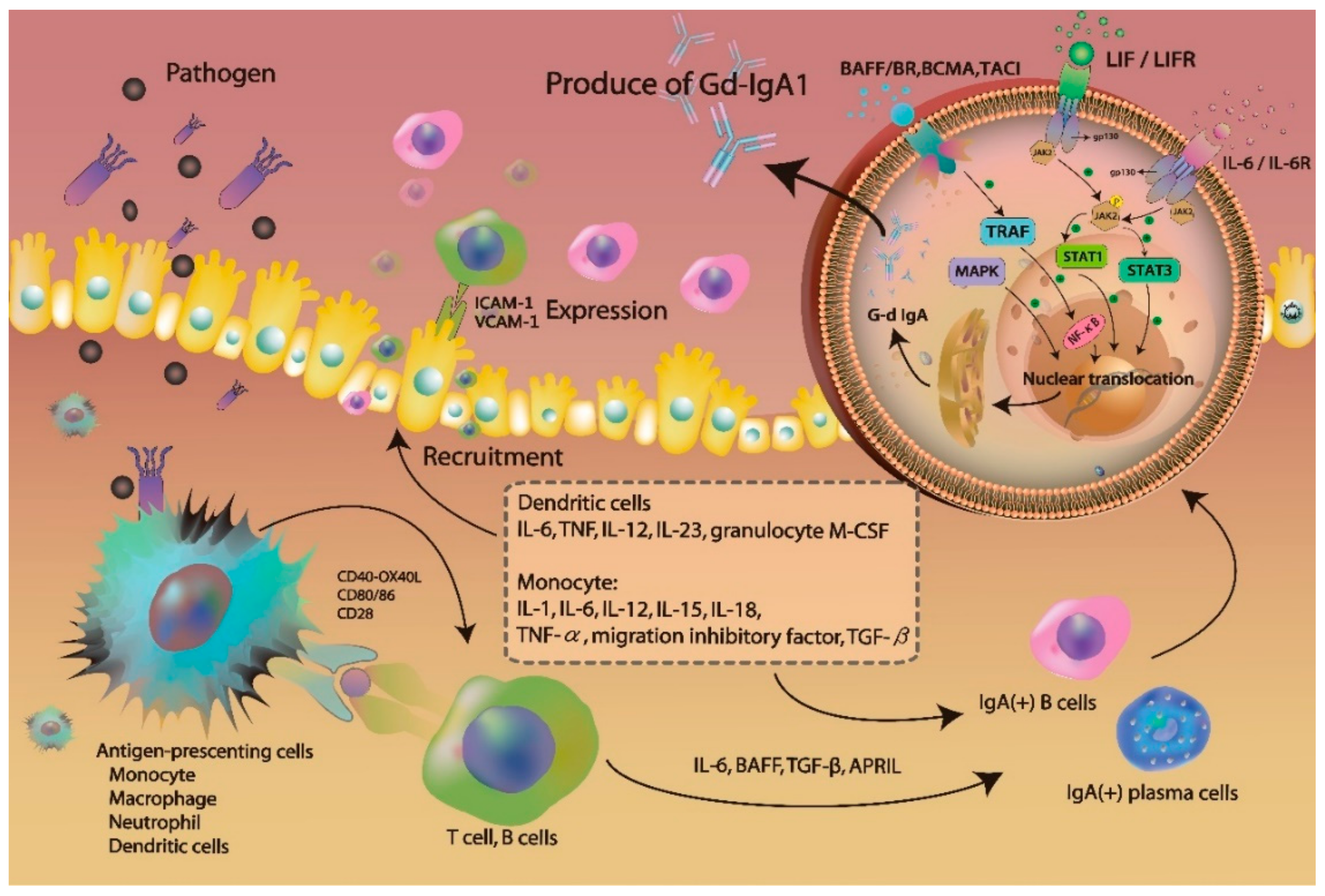
3.1. Gd-IgA1 Signaling and Production
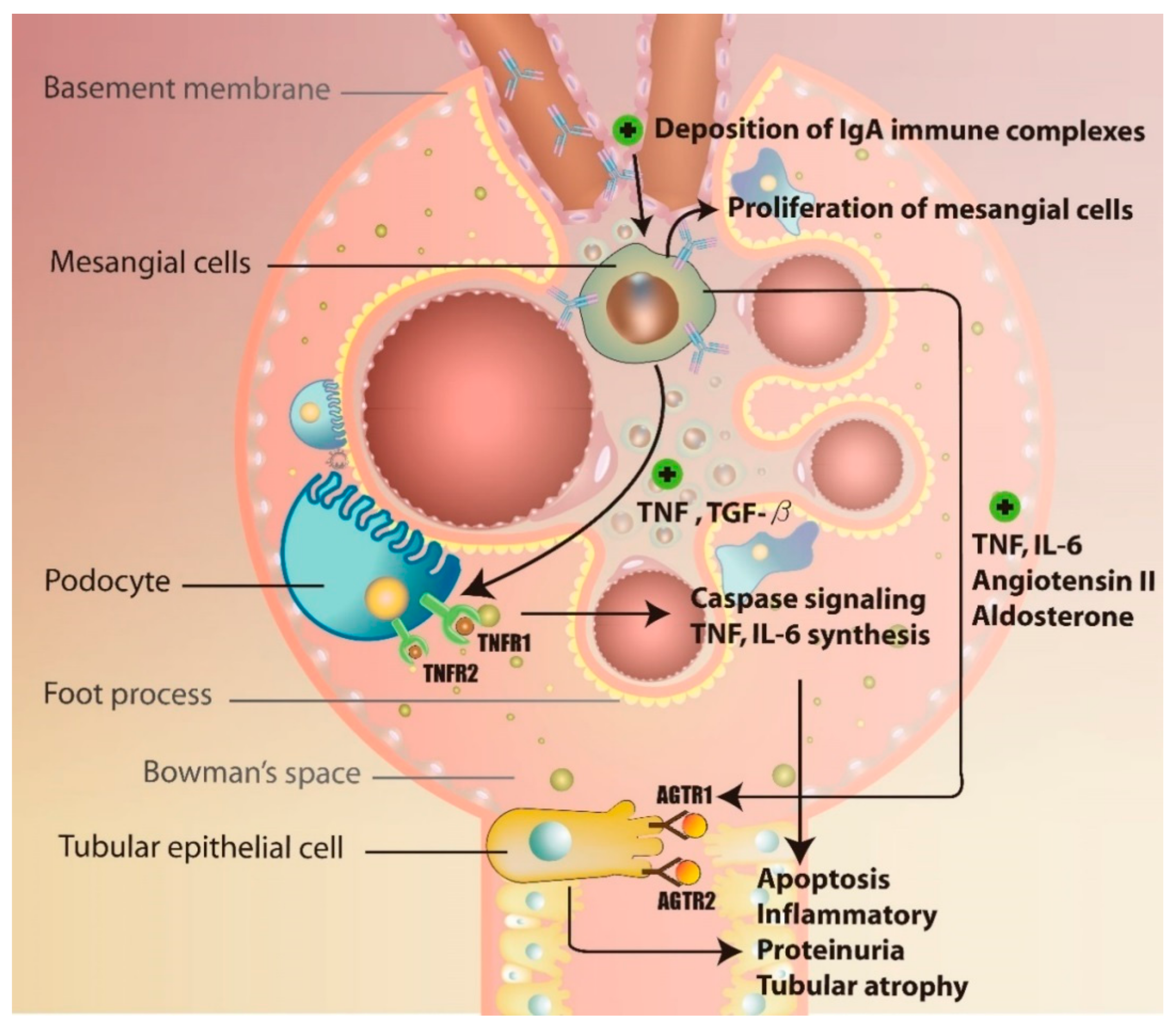
3.2. Immune Complex Deposition-Associated Induction of Inflammation and Complement Activation
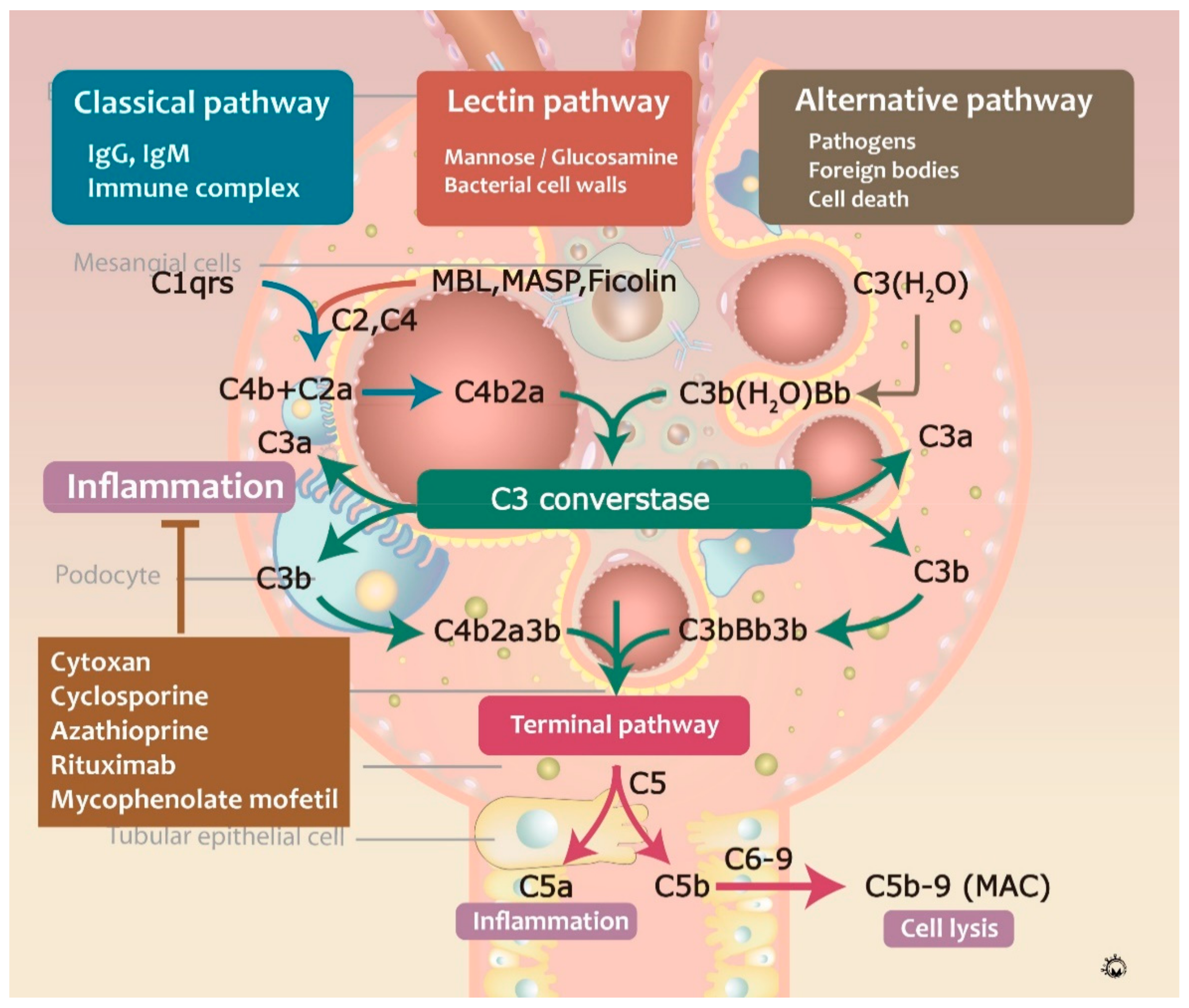
3.3. Complement Activation during IgAN Pathogenesis
4. Therapeutic Strategies for the Treatment of IgAN
4.1. Non-Immunosuppressive Therapies
4.2. Corticosteroids
4.3. Mycophenolate Mofetil
4.4. B-cell Depletion/Inhibition
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Geddes, C.C. A Tricontinental View of IgA Nephropathy. Nephrol. Dial. Transplant. 2003, 18, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.G. Disappearance of glomerular mesangial IgA deposits after renal allograft transplantation. Transplantation 1982, 33, 241–246. [Google Scholar] [PubMed]
- Suzuki, H. The Pathophysiology of IgA Nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- McGrogan, A.; Franssen, C.F.; De Vries, C.S. The incidence of primary glomerulonephritis worldwide: A systematic review of the literature. Nephrol. Dial. Transplant. 2011, 26, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Barbour, S.J. Individuals of Pacific Asian origin with IgA nephropathy have an increased risk of progression to end-stage renal disease. Kidney Int. 2013, 84, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Barbour, S.J. The MEST score provides earlier risk prediction in lgA nephropathy. Kidney Int. 2016, 89, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.J. Epidemiology of IgA nephropathy in central and eastern Kentucky for the period 1975 through 1994 Central Kentucky Region of the Southeastern United States IgA Nephropathy DATABANK Project. J. Am. Soc. Nephrol. 1998, 9, 853–858. [Google Scholar] [PubMed]
- Lai, K.N. An overlapping syndrome of IgA nephropathy and lipoid nephrosis. Am. J. Clin. Pathol. 1986, 86, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W. Long-term outcome of IgA nephropathy with minimal change disease: A comparison between patients with and without minimal change disease. J. Nephrol. 2016, 29, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.S. The Oxford classification of IgA nephropathy: Pathology definitions, correlations, and reproducibility. Kidney Int. 2009, 76, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Herzenberg, A.M. Validation of the Oxford classification of IgA nephropathy. Kidney Int. 2011, 80, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R. Validation of the Oxford classification of IgA nephropathy in cohorts with different presentations and treatments. Kidney Int. 2014, 86, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Abe, T. Participation of extracapillary lesions (ECL) in progression of IgA nephropathy. Clin. Nephrol. 1986, 25, 37–41. [Google Scholar] [PubMed]
- Lv, J. Evaluation of the Oxford Classification of IgA nephropathy: A systematic review and meta-analysis. Am. J. Kidney Dis. 2013, 62, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N. IgA nephropathy. Nat. Rev. Dis. Prim. 2016, 2, 16001. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J. Clin. Investig. 2009, 119, 1668–1677. [Google Scholar] [CrossRef] [PubMed]
- Mestecky, J. Defective galactosylation and clearance of IgA1 molecules as a possible etiopathogenic factor in IgA nephropathy. Contrib. Nephrol. 1993, 104, 172–182. [Google Scholar] [PubMed]
- Aryal, R.P.; Ju, T.; Cummings, R.D. The endoplasmic reticulum chaperone Cosmc directly promotes in vitro folding of T.-synthase. J. Biol. Chem. 2010, 285, 2456–2462. [Google Scholar] [CrossRef] [PubMed]
- Qin, W. External suppression causes the low expression of the Cosmc gene in IgA nephropathy. Nephrol. Dial. Transpl. 2008, 23, 1608–1614. [Google Scholar] [CrossRef] [PubMed]
- Szeto, C.C; Li, P.K. MicroRNAs in IgA nephropathy. Nat. Rev. Nephrol. 2014, 10, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.; Cummings, R.D. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc. Natl. Acad. Sci. USA 2002, 99, 16613–16618. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.C. Charge-dependent binding of polymeric IgA1 to human mesangial cells in IgA nephropathy. Kidney Int. 2001, 59, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.G. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J. Am. Soc. Nephrol. 2008, 19, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Expression and correlation analysis of IL-4, IFN-gamma and FcalphaRI in tonsillar mononuclear cells in patients with IgA nephropathy. Cell Immunol. 2014, 289, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Akira, S. Mammalian Toll-like receptors. Curr. Opin. Immunol. 2003, 15, 5–11. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, I. Chapter Five—Leukocytes Crossing the Endothelium: A Matter of Communication. In International Review of Cell and Molecular Biology; Academic Press: Cambridge, MA, USA, 2016; Volume 322, pp. 281–329. [Google Scholar]
- Reily, C. Cellular Signaling and Production of Galactose-Deficient IgA1 in IgA Nephropathy, an Autoimmune Disease. J. Immunol. Res. 2014, 2014, 197548. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H. Cytokines Alter IgA1 O-Glycosylation by Dysregulating C1GalT1 and ST6GalNAc-II Enzymes. J. Biol. Chem. 2014, 289, 5330–5339. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.J.; Julian, B.A. IgA Nephropathy. New Eng. J. Med. 2013, 368, 2402–2414. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.G. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J. Immunol. 2004, 173, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Browning, J.L. BAFF: A fundamental survival factor for B cells. Nat. Rev. Immunol. 2002, 2, 465. [Google Scholar] [CrossRef] [PubMed]
- Scapini, P.; Bazzoni, F.; Cassatella, M.A. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol. Lett. 2008, 116, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.L. Increased APRIL Expression Induces IgA1 Aberrant Glycosylation in IgA Nephropathy. Medicine (Baltimore) 2016, 95, e3099. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G. Pathogenic Role of a Proliferation-Inducing Ligand (APRIL) in Murine IgA Nephropathy. PLoS ONE 2015, 10, e0137044. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.G. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 43, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Xin, G. Serum BAFF is elevated in patients with IgA nephropathy and associated with clinical and histopathological features. J. Nephrol. 2013, 26, 683–690. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.D. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J. Clin. Investig. 2011, 121, 3991–4002. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K. Aberrant glycosylation of IgA1 is inherited in both pediatric IgA nephropathy and Henoch-Schonlein purpura nephritis. Kidney Int. 2011, 80, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Tomana, M. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J. Clin. Investig. 1999, 104, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.C. Abnormal IgA glycosylation in Henoch-Schonlein purpura restricted to patients with clinical nephritis. Nephrol. Dial. Transplant. 1998, 13, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Levinsky, R.J.; Barratt, T.M. IgA immune complexes in Henoch-Schonlein purpura. Lancet 1979, 2, 1100–1103. [Google Scholar] [CrossRef]
- Lau, K.K. Serum levels of galactose-deficient IgA in children with IgA nephropathy and Henoch-Schonlein purpura. Pediatr. Nephrol. 2007, 22, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H. IgA nephropathy: Characterization of IgG antibodies specific for galactose-deficient IgA1. Contrib. Nephrol. 2007, 157, 129–133. [Google Scholar] [PubMed]
- Tomana, M. Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney Int. 1997, 52, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J. Clin. Investig. 2008, 118, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N. Pathogenesis of IgA nephropathy. Nat. Rev. Nephrol. 2012, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Berthoux, F. Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Novak, J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin. Nephrol. 2008, 28, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Feehally, J. Sequential study of the IgA system in relapsing IgA nephropathy. Kidney Int. 1986, 30, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N. Activation of podocytes by mesangial-derived TNF-alpha: Glomerulo-podocytic communication in IgA nephropathy. Am. J. Physiol. Renal. Physiol. 2008, 294, F945–F955. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.Y. Activation of tubular epithelial cells by mesangial-derived TNF-alpha: Glomerulotubular communication in IgA nephropathy. Kidney Int. 2005, 67, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Tam, K.Y. Macromolecular IgA1 taken from patients with familial IgA nephropathy or their asymptomatic relatives have higher reactivity to mesangial cells in vitro. Kidney Int. 2009, 75, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N. Podocyte injury induced by mesangial-derived cytokines in IgA nephropathy. Nephrol. Dial. Transplant. 2009, 24, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N. Mesangial expression of angiotensin II receptor in IgA nephropathy and its regulation by polymeric IgA1. Kidney Int. 2004, 66, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Flannery, P.J.; Spurney, R.F. Characterization of angiotensin II-receptor subtypes in podocytes. J. Lab. Clin. Med. 2003, 142, 313–321. [Google Scholar] [CrossRef]
- Chan, L.Y. Tubular expression of angiotensin II receptors and their regulation in IgA nephropathy. J. Am. Soc. Nephrol. 2005, 16, 2306–2317. [Google Scholar] [CrossRef] [PubMed]
- Van Kooten, C.; Daha, M.R.; Van Es, L.A. Tubular epithelial cells: A critical cell type in the regulation of renal inflammatory processes. Exp. Nephrol. 1999, 7, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Wünsche, C. Transforming growth factor β2 (TGF-β2)-induced connective tissue growth factor (CTGF) expression requires sphingosine 1-phosphate receptor 5 (S1P5) in human mesangial cells. BBA—Mol. Cell Biol. Lipids 2015, 2015, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Castro, N.E. Transforming growth factor beta1 (TGF-beta1) enhances expression of profibrotic genes through a novel signaling cascade and microRNAs in renal mesangial cells. J. Biol. Chem. 2014, 289, 29001–29013. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.C.; Haas, M.; Reich, H.N. IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 357, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C. The immunohistology of IgA nephropathy. Am. J. Kidney Dis. 1988, 12, 348–352. [Google Scholar] [CrossRef]
- Cooper, N.R. The classical complement pathway: Activation and regulation of the first complement component. Adv. Immunol. 1985, 37, 151–216. [Google Scholar] [PubMed]
- Ferreira, V. The Classical Activation Pathway of the Human Complement System Is Specifically Inhibited by Calreticulin from Trypanosoma cruzi. J. Immunol. 2004, 172, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Fishelson, Z.; Pangburn, M.K.; Muller-Eberhard, H.J. Characterization of the initial C3 convertase of the alternative pathway of human complement. J. Immunol. 1984, 132, 1430–1434. [Google Scholar] [PubMed]
- Petersen, S.V.; Thiel, S.; Jensenius, J.C. The mannan-binding lectin pathway of complement activation: Biology and disease association. Mol. Immunol. 2001, 38, 133–149. [Google Scholar] [CrossRef]
- Sahu, A.; Lambris, J.D. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 2001, 180, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 2014, 46, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L. Variants in Complement Factor H and Complement Factor H-Related Protein Genes, CFHR3 and CFHR1, Affect Complement Activation in IgA Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Rauterberg, E.W. Complement membrane attack (MAC) in idiopathic IgA-glomerulonephritis. Kidney Int. 1987, 31, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, H. Immunohistochemical study of the membrane attack complex of complement in IgA nephropathy. Virchows Arch. A. Pathol. Anat. Histopathol. 1988, 413, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Onda, K. Excretion of complement proteins and its activation marker C5b-9 in IgA nephropathy in relation to renal function. BMC Nephrol. 2011, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Shankland, S.J.; Couser, W.G. Cellular response to injury in membranous nephropathy. J. Am. Soc. Nephrol. 2005, 16, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, A.V. Complement-induced phospholipase A2 activation in experimental membranous nephropathy. Kidney Int. 2000, 57, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W. Sublytic C5b-9 complexes induce proliferative changes of glomerular mesangial cells in rat Thy-1 nephritis through TRAF6-mediated PI3K-dependent Akt1 activation. J. Pathol. 2012, 226, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W. Sublytic C5b-9 triggers glomerular mesangial cell apoptosis via XAF1 gene activation mediated by p300-dependent IRF-1 acetylation. Cell Death Dis. 2014, 5, e1176. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H. Relationship between serum IgA/C3 ratio and progression of IgA nephropathy. Intern. Med. 2004, 43, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Serum immunoglobulin A/C3 ratio predicts progression of immunoglobulin A nephropathy. Nephrology (Carlton) 2013, 18, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Yang, P. Comparative Efficacy and Safety of Therapies in IgA Nephropathy: A Network Meta-analysis of Randomized Controlled Trials. Kidney Int. Rep. 2018, 3, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Maneiro, L.; Puente-Garcia, A. Renin-Angiotensin-Aldosterone System Blockade in Diabetic Nephropathy. Present Evidences. J. Clin. Med. 2015, 4, 1908–1937. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y. New Insights into the Immune Molecular Regulation of the Pathogenesis of Acute Respiratory Distress Syndrome. Intern. J. Mol. Sci. 2018, 19, 588. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J. ACEI/ARB therapy for IgA nephropathy: A meta analysis of randomised controlled trials. Int. J. Clin. Pract. 2009, 63, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, C.; Paradis, P.; Schiffrin, E.L. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol. Sci. 2008, 29, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Han, C. Angiotensin II induces C-reactive protein expression through ERK1/2 and JNK signaling in human aortic endothelial cells. Atherosclerosis 2010, 212, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. Omega-3 fatty acids in inflammation and autoimmune diseases. J. Am. Coll. Nutr. 2002, 21, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Donadio, J.V.; Grande, J.P. The role of fish oil/omega-3 fatty acids in the treatment of IgA nephropathy. Semin. Nephrol. 2004, 24, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Reid, S. Non-immunosuppressive treatment for IgA nephropathy. Cochrane Database Syst. Rev. 2011, 3, Cd003962. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.W. Anti-inflammatory action of corticosteroids. Postgrad Med. J. 1976, 52, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory Action of Glucocorticoids—New Mechanisms for Old Drugs. New Eng. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R. IgA Nephropathy: A European Perspective in the Corticosteroid Treatment. Kidney Dis. (Basel) 2018, 4, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, C. Corticosteroid effectiveness in IgA nephropathy: Long-term results of a randomized, controlled trial. J. Am. Soc. Nephrol. 2004, 15, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Lv, J. Combination therapy of prednisone and ACE inhibitor versus ACE-inhibitor therapy alone in patients with IgA nephropathy: A randomized controlled trial. Am. J. Kidney Dis. 2009, 53, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Manno, C. Randomized controlled clinical trial of corticosteroids plus ACE-inhibitors with long-term follow-up in proteinuric IgA nephropathy. Nephrol. Dial. Transplant. 2009, 24, 3694–3701. [Google Scholar] [CrossRef] [PubMed]
- Ballardie, F.W.; Roberts, I.S. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J. Am. Soc. Nephrol. 2002, 13, 142–148. [Google Scholar] [PubMed]
- Pozzi, C. Addition of azathioprine to corticosteroids does not benefit patients with IgA nephropathy. J. Am. Soc. Nephrol. 2010, 21, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, C. IgA nephropathy with severe chronic renal failure: A randomized controlled trial of corticosteroids and azathioprine. J. Nephrol. 2013, 26, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Smerud, H.K. New treatment for IgA nephropathy: Enteric budesonide targeted to the ileocecal region ameliorates proteinuria. Nephrol. Dial. Transplant. 2011, 26, 3237–3242. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Eugui, E.M. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000, 47, 85–118. [Google Scholar] [CrossRef]
- Kang, Z. Mycophenolate mofetil therapy for steroid-resistant IgA nephropathy with the nephrotic syndrome in children. Pediatr. Nephrol. 2015, 30, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H. Mycophenolate mofetil in the treatment of IgA nephropathy: A systematic review. Singap. Med. J. 2008, 49, 780–785. [Google Scholar]
- Kim, M.J. Spleen tyrosine kinase is important in the production of proinflammatory cytokines and cell proliferation in human mesangial cells following stimulation with IgA1 isolated from IgA nephropathy patients. J. Immunol. 2012, 189, 3751–3758. [Google Scholar] [CrossRef] [PubMed]
- Du, B. Efficacy and safety of mycophenolate mofetil in patients with IgA nephropathy: An update meta-analysis. BMC Nephrol. 2017, 18, 245. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C. Long-term study of mycophenolate mofetil treatment in IgA nephropathy. Kidney Int. 2010, 77, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Tang, S. Mycophenolate mofetil alleviates persistent proteinuria in IgA nephropathy. Kidney Int. 2005, 68, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Appel, G.B. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J. Am. Soc. Nephrol. 2009, 20, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Hogg, R.J. Randomized controlled trial of mycophenolate mofetil in children, adolescents, and adults with IgA nephropathy. Am. J. Kidney Dis. 2015, 66, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Beck, L. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for glomerulonephritis. Am. J. Kidney Dis. 2013, 62, 403–441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Cutting Edge: A Role for B Lymphocyte Stimulator in Systemic Lupus Erythematosus. J. Immunol. 2001, 166, 6–10. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Science. Safety and Efficacy Study of Fostamatinib to Treat Immunoglobin A (IgA) Nephropathy; ClinicalTrials.gov: Bethesda, MD, USA, 2018.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histological Finding | Score |
|---|---|
| Mesangial hypercellularity | M0: Presence of mesangial hypercellularity in <50% glomeruli M1: Presence of mesangial hypercellularity in >50% glomeruli |
| Endocapillary hypercellularity | E0: No endocapillary hypercellularity E1: Presence of any endocapillary hypercellularity |
| Segmental glomerulosclerosis | S0: No segmental glomerulosclerosis S1: Presence of any segmental glomerulosclerosis |
| Tubular atrophy and interstitial fibrosis | T0: 0–25% tubular atrophy/interstitial fibrosis in cortical area T1: 26–50% tubular atrophy/interstitial fibrosis in cortical area T2: >50% tubular atrophy/interstitial fibrosis in cortical area |
| Cellular or fibrocellular crescents | C0: no cellular or fibrocellular crescents C1: Presence of cellular/fibrocellular crescents in <25% glomeruli C2: Presence of cellular/fibrocellular crescents in >25% glomeruli |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.-Y.; Chen, C.-S.; Yiang, G.-T.; Cheng, P.-W.; Chen, Y.-L.; Chiu, H.-C.; Liu, K.-H.; Lee, W.-C.; Li, C.-J. The Emerging Role of Pathogenesis of IgA Nephropathy. J. Clin. Med. 2018, 7, 225. https://doi.org/10.3390/jcm7080225
Wu M-Y, Chen C-S, Yiang G-T, Cheng P-W, Chen Y-L, Chiu H-C, Liu K-H, Lee W-C, Li C-J. The Emerging Role of Pathogenesis of IgA Nephropathy. Journal of Clinical Medicine. 2018; 7(8):225. https://doi.org/10.3390/jcm7080225
Chicago/Turabian StyleWu, Meng-Yu, Chien-Sheng Chen, Giou-Teng Yiang, Pei-Wen Cheng, Yu-Long Chen, Hsiao-Chen Chiu, Kuan-Hung Liu, Wen-Chin Lee, and Chia-Jung Li. 2018. "The Emerging Role of Pathogenesis of IgA Nephropathy" Journal of Clinical Medicine 7, no. 8: 225. https://doi.org/10.3390/jcm7080225
APA StyleWu, M.-Y., Chen, C.-S., Yiang, G.-T., Cheng, P.-W., Chen, Y.-L., Chiu, H.-C., Liu, K.-H., Lee, W.-C., & Li, C.-J. (2018). The Emerging Role of Pathogenesis of IgA Nephropathy. Journal of Clinical Medicine, 7(8), 225. https://doi.org/10.3390/jcm7080225