Current and Future Molecular Testing in NSCLC, What Can We Expect from New Sequencing Technologies?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
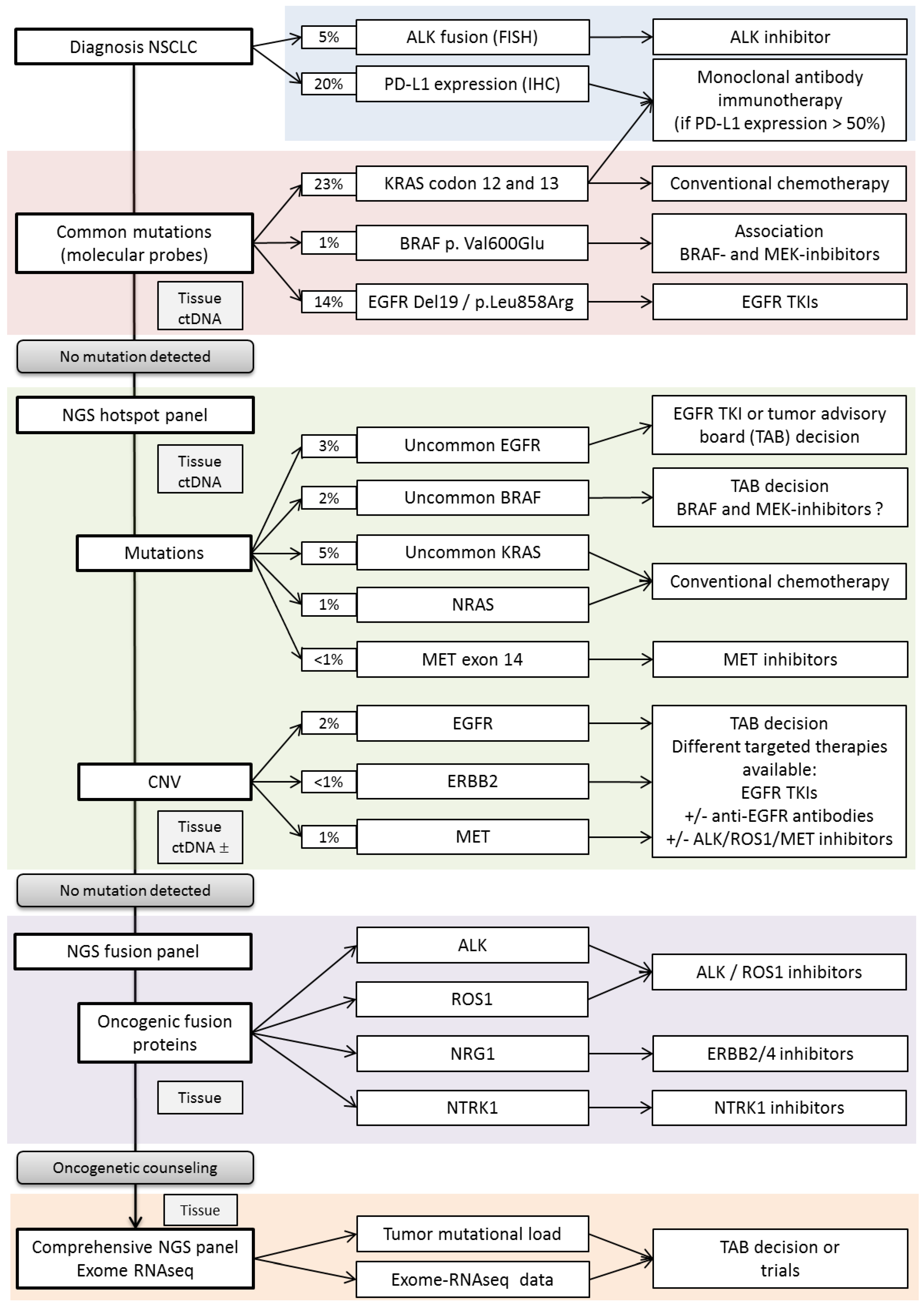
2. 1-Lung Cancer Molecular Screenings, Update on Validated Markers and Emerging Ones
2.1. -Mutation Testing
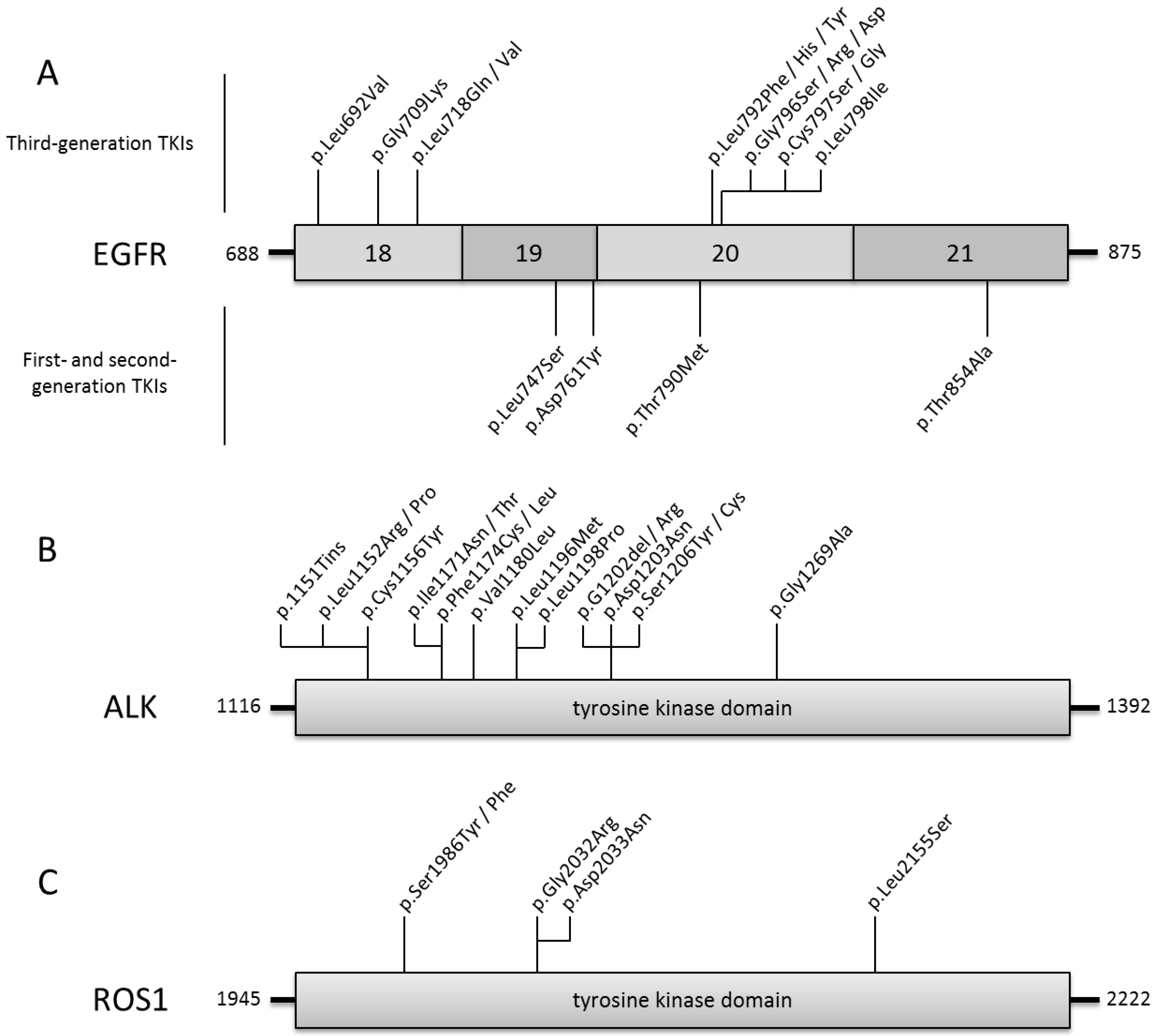
2.1.1. EGFR
First- and Second-Generation EGFR-TKIs
EGFR-TKIs Treatment for Patients with Uncommon EGFR Mutated Tumors
Third-Generation EGFR-TKI
Resistance to Third-Generation EGFR-TKI
Third-Generation EGFR-TKI as First-Line Treatment of EGFR Mutated NSCLC
Allosteric Inhibitors of EGFR
2.1.2. BRAF
2.1.3. MET
2.1.4. KRAS
2.1.5. PI3KCA
2.2. Fusion Testing
2.2.1. ALK
2.2.2. Resistance to ALK-Inhibitors
2.2.3. ROS1
2.2.4. RET
2.2.5. NTRK
2.2.6. NRG1
2.2.7. Gene Fusion Detection
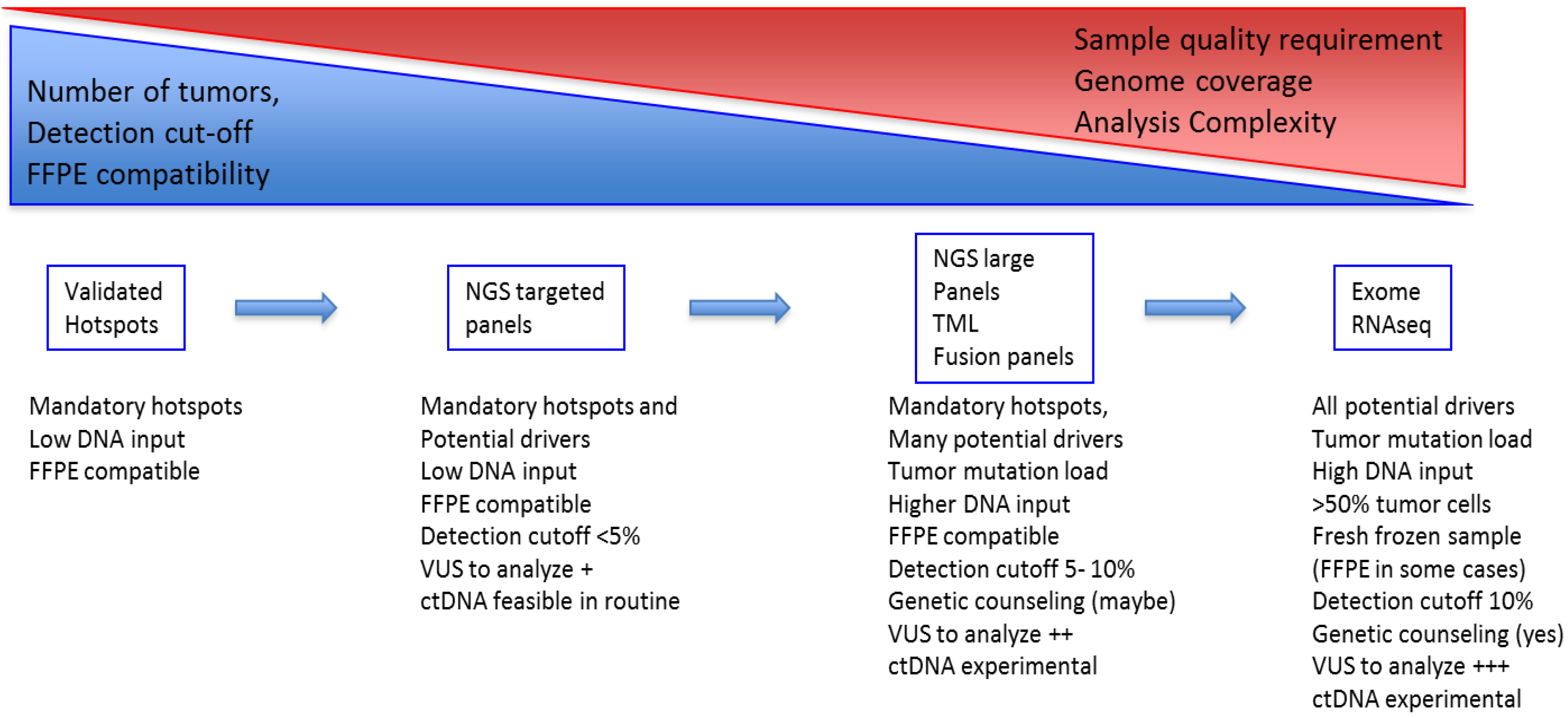
2.3. Technical Evolution in Clinical Molecular Testing
2.3.1. From Single Gene to Multi-Gene Testing/Panels
2.3.2. From Tissue Testing to Circulating DNA
2.3.3. Predictive Markers of Response to Immune Checkpoint Inhibitors, Focus on Genetic Determinants
2.3.4. Driver Mutations as Predictive Markers
2.3.5. Tumor Mutational Load (TML) as a Predictive Marker
2.3.6. Quantification of Tumor Mutational Burden
3. Discussion and Conclusions
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA. Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.G.; Ebert, P.J.; Ting, J.C.; Lim, E.; Wong, S.-S.; Teo, A.S.M.; Yue, Y.G.; Chua, H.-H.; Ma, X.; Loh, G.S.L.; et al. Whole-genome sequencing of Asian lung cancers: Second-hand smoke unlikely to be responsible for higher incidence of lung cancer among Asian never-smokers. Cancer Res. 2014, 74, 6071–6081. [Google Scholar] [CrossRef] [PubMed]
- Deeb, K.K.; Hohman, C.M.; Risch, N.F.; Metzger, D.J.; Starostik, P. Routine Clinical Mutation Profiling of Non-Small Cell Lung Cancer Using Next-Generation Sequencing. Arch. Pathol. Lab. Med. 2015, 139, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Ou, S.-H.I.; Bang, Y.-J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Mayekar, M.K.; Bivona, T.G. Current Landscape of Targeted Therapy in Lung Cancer. Clin. Pharmacol. Ther. 2017, 102, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Balducci, L.; Ciardiello, F.; Di Maio, M.; Felip, E.; Langer, C.; Lilenbaum, R.C.; Perrone, F.; Senan, S.; de Marinis, F. Treatment of Elderly Patients with Non-Small-Cell Lung Cancer: Results of an International Expert Panel Meeting of the Italian Association of Thoracic Oncology. Clin. Lung Cancer 2015, 16, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Girard, N. Optimizing outcomes in EGFR mutation-positive NSCLC: Which tyrosine kinase inhibitor and when? Future Oncol. Lond. Engl. 2018. [Google Scholar] [CrossRef] [PubMed]
- De Leng, W.W.J.; Gadellaa-van Hooijdonk, C.G.; Barendregt-Smouter, F.A.S.; Koudijs, M.J.; Nijman, I.; Hinrichs, J.W.J.; Cuppen, E.; van Lieshout, S.; Loberg, R.D.; de Jonge, M.; et al. Targeted Next Generation Sequencing as a Reliable Diagnostic Assay for the Detection of Somatic Mutations in Tumours Using Minimal DNA Amounts from Formalin Fixed Paraffin Embedded Material. PLoS ONE 2016, 11, e0149405. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R. Intratumoral heterogeneity and subclonal diversification of early breast cancer. Breast 2017, 34 (Suppl. 1), S36–S42. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Jordan, E.J.; Kim, H.R.; Arcila, M.E.; Barron, D.; Chakravarty, D.; Gao, J.; Chang, M.T.; Ni, A.; Kundra, R.; Jonsson, P.; et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017, 7, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Legras, A.; Barritault, M.; Tallet, A.; Fabre, E.; Guyard, A.; Rance, B.; Digan, W.; Pecuchet, N.; Giroux-Leprieur, E.; Julie, C.; et al. Validity of targeted next-generation sequencing in routine care for identifying clinically relevant molecular profiles in non–small-cell lung cancer: Results of a 2-year experience on 1343 samples. J. Mol. Diagn. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yatabe, Y.; Kerr, K.M.; Utomo, A.; Rajadurai, P.; Tran, V.K.; Du, X.; Chou, T.-Y.; Enriquez, M.L.D.; Lee, G.K.; Iqbal, J.; et al. EGFR mutation testing practices within the Asia Pacific region: Results of a multicenter diagnostic survey. J. Thorac. Oncol. 2015, 10, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Barlesi, F.; Mazieres, J.; Merlio, J.-P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Biomarkers France contributors Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet Lond. Engl. 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H.; Chiang, C.-J.; Tseng, J.-S.; Yang, T.-Y.; Hsu, K.-H.; Chen, K.-C.; Wang, C.-L.; Chen, C.-Y.; Yen, S.-H.; Tsai, C.-M.; et al. EGFR mutation, smoking, and gender in advanced lung adenocarcinoma. Oncotarget 2017, 8, 98384–98393. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016, 107, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Galli, G.; Corrao, G.; Imbimbo, M.; Proto, C.; Signorelli, D.; Ganzinelli, M.; Zilembo, N.; Vitali, M.; de Braud, F.; Garassino, M.C.; et al. Uncommon mutations in epidermal growth factor receptor and response to first and second generation tyrosine kinase inhibitors: A case series and literature review. Lung Cancer 2018, 115, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Grigoriu, B.; Berghmans, T.; Meert, A.-P. Management of EGFR mutated nonsmall cell lung carcinoma patients. Eur. Respir. J. 2015, 45, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.-Y.; Ke, E.-E.; Yang, J.-J.; Sun, Y.-L.; Yan, H.-H.; Zheng, M.-Y.; Bai, X.-Y.; Wang, Z.; Su, J.; Chen, Z.-H.; et al. A comprehensive review of uncommon EGFR mutations in patients with non-small cell lung cancer. Lung Cancer 2017, 114, 96–102. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.M.; Bradbury, P.A.; Feld, R.; Leighl, N.B.; Liu, G.; Pisters, K.-M.; Kamel-Reid, S.; Tsao, M.S.; Shepherd, F.A. Uncommon EGFR mutations in advanced non-small cell lung cancer. Lung Cancer 2017, 109, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Beau-Faller, M.; Prim, N.; Ruppert, A.-M.; Nanni-Metéllus, I.; Lacave, R.; Lacroix, L.; Escande, F.; Lizard, S.; Pretet, J.-L.; Rouquette, I.; et al. Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: A multicentre observational study by the French ERMETIC-IFCT network. Ann. Oncol. 2014, 25, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Eck, M.J.; Yun, C.-H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta 2010, 1804, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Bronte, G.; Rolfo, C.; Giovannetti, E.; Cicero, G.; Pauwels, P.; Passiglia, F.; Castiglia, M.; Rizzo, S.; Vullo, F.L.; Fiorentino, E.; et al. Are erlotinib and gefitinib interchangeable, opposite or complementary for non-small cell lung cancer treatment? Biological, pharmacological and clinical aspects. Crit. Rev. Oncol. Hematol. 2014, 89, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Afatinib: A review of its use in the treatment of advanced non-small cell lung cancer. Drugs 2014, 74, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Zhou, C.; Hu, C.-P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Wu, Y.-L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.-P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.-H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.-H.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Tan, E.-H.; O’Byrne, K.; Zhang, L.; Hirsh, V.; Boyer, M.; Yang, J.C.-H.; Mok, T.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: Overall survival data from the phase IIb LUX-Lung 7 trial. Ann. Oncol. 2017, 28, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-H.; Yang, C.-T.; Shih, J.-Y.; Huang, M.-S.; Su, W.-C.; Lai, R.-S.; Wang, C.-C.; Hsiao, S.-H.; Lin, Y.-C.; Ho, C.-L.; et al. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Treatment Response in Advanced Lung Adenocarcinomas with G719X/L861Q/S768I Mutations. J. Thorac. Oncol. 2015, 10, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Hao, X.; Hu, X.; Wang, H.; Wang, Y.; Ying, J. Clinical characteristics and response to tyrosine kinase inhibitors of patients with non-small cell lung cancer harboring uncommon epidermal growth factor receptor mutations. Chin. J. Cancer Res. 2017, 29, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-Y.; Yu, C.-J.; Chang, Y.-C.; Yang, C.-H.; Shih, J.-Y.; Yang, P.-C. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin. Cancer Res. 2011, 17, 3812–3821. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Minegishi, Y.; Yoshizawa, H.; Maemondo, M.; Inoue, A.; Sugawara, S.; Isobe, H.; Harada, M.; Ishii, Y.; Gemma, A.; et al. Effectiveness of gefitinib against non-small-cell lung cancer with the uncommon EGFR mutations G719X and L861Q. J. Thorac. Oncol. 2014, 9, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.; Chirieac, L.R.; D’Amico, T.A.; DeCamp, M.M.; Dilling, T.J.; Dobelbower, M.; et al. Non-Small Cell Lung Cancer, Version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. JNCCN 2017, 15, 504–535. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Sequist, L.V.; Geater, S.L.; Tsai, C.-M.; Mok, T.S.K.; Schuler, M.; Yamamoto, N.; Yu, C.-J.; Ou, S.-H.I.; Zhou, C.; et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: A combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015, 16, 830–838. [Google Scholar] [CrossRef]
- Finlay, M.R.V.; Anderton, M.; Ashton, S.; Ballard, P.; Bethel, P.A.; Box, M.R.; Bradbury, R.H.; Brown, S.J.; Butterworth, S.; Campbell, A.; et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014, 57, 8249–8267. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. AURA3 Investigators Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Tian, S.K.; Drilon, A.E.; Borsu, L.; Riely, G.J.; Arcila, M.E.; Ladanyi, M. Acquired Resistance of EGFR-Mutant Lung Cancer to a T790M-Specific EGFR Inhibitor: Emergence of a Third Mutation (C797S) in the EGFR Tyrosine Kinase Domain. JAMA Oncol. 2015, 1, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.-H.; Lu, J.-J. Osimertinib resistance in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Lett. 2018, 420, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Minari, R.; Bordi, P.; Tiseo, M. Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: Review on emerged mechanisms of resistance. Transl. Lung Cancer Res. 2016, 5, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, M.; Liguori, A.; Karachaliou, N.; Gonzalez-Cao, M.; Daffinà, M.G.; D’Aveni, A.; Marabello, G.; Altavilla, G.; Rosell, R. Osimertinib in the treatment of non-small-cell lung cancer: Design, development and place in therapy. Lung Cancer (Auckl.) 2017, 8, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-S.; Kumarakulasinghe, N.B.; Huang, Y.-Q.; Ang, Y.L.E.; Choo, J.R.-E.; Goh, B.-C.; Soo, R.A. Third generation EGFR TKIs: Current data and future directions. Mol. Cancer 2018, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Minari, R.; Bordi, P.; Del Re, M.; Facchinetti, F.; Mazzoni, F.; Barbieri, F.; Camerini, A.; Comin, C.E.; Gnetti, L.; Azzoni, C.; et al. Primary resistance to osimertinib due to SCLC transformation: Issue of T790M determination on liquid re-biopsy. Lung Cancer 2018, 115, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Tan, D.S.W.; Chiari, R.; Wu, Y.-L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.-J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet Lond. Engl. 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Lin, L.; Asthana, S.; Chan, E.; Bandyopadhyay, S.; Martins, M.M.; Olivas, V.; Yan, J.J.; Pham, L.; Wang, M.M.; Bollag, G.; et al. Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E748–E757. [Google Scholar] [CrossRef] [PubMed]
- Baik, C.S.; Myall, N.J.; Wakelee, H.A. Targeting BRAF-Mutant Non-Small Cell Lung Cancer: From Molecular Profiling to Rationally Designed Therapy. Oncologist 2017, 22, 786–796. [Google Scholar] [CrossRef] [PubMed]
- De Langen, A.J.; Smit, E.F. Therapeutic approach to treating patients with BRAF-mutant lung cancer: Latest evidence and clinical implications. Ther. Adv. Med. Oncol. 2017, 9, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Gautschi, O.; Milia, J.; Cabarrou, B.; Bluthgen, M.-V.; Besse, B.; Smit, E.F.; Wolf, J.; Peters, S.; Früh, M.; Koeberle, D.; et al. Targeted Therapy for Patients with BRAF-Mutant Lung Cancer: Results from the European EURAF Cohort. J. Thorac. Oncol. 2015, 10, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Odogwu, L.; Mathieu, L.; Blumenthal, G.; Larkins, E.; Goldberg, K.B.; Griffin, N.; Bijwaard, K.; Lee, E.Y.; Philip, R.; Jiang, X.; et al. FDA Approval Summary: Dabrafenib and Trametinib for the Treatment of Metastatic Non-Small Cell Lung Cancers Harboring BRAF V600E Mutations. Oncologist 2018. [Google Scholar] [CrossRef] [PubMed]
- Noeparast, A.; Teugels, E.; Giron, P.; Verschelden, G.; De Brakeleer, S.; Decoster, L.; De Grève, J. Non-V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of Trametinib and Dabrafenib. Oncotarget 2017, 8, 60094–60108. [Google Scholar] [CrossRef] [PubMed]
- Buzyn, A.; Blay, J.-Y.; Hoog-Labouret, N.; Jimenez, M.; Nowak, F.; Deley, M.-C.L.; Pérol, D.; Cailliot, C.; Raynaud, J.; Vassal, G. Equal access to innovative therapies and precision cancer care. Nat. Rev. Clin. Oncol. 2016, 13, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.-N.; Liu, P. Targeting MET in cancer therapy. Chronic Dis. Transl. Med. 2017, 3, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Pécuchet, N.; Vieira, T.; Rabbe, N.; Antoine, M.; Blons, H.; Cadranel, J.; Laurent-Puig, P.; Wislez, M. Molecular classification of pulmonary sarcomatoid carcinomas suggests new therapeutic opportunities. Ann. Oncol. 2017, 28, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Maroun, C.R.; Rowlands, T. The Met receptor tyrosine kinase: A key player in oncogenesis and drug resistance. Pharmacol. Ther. 2014, 142, 316–338. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.W.; Dahlberg, S.E.; Wakelee, H.A.; Aisner, S.C.; Bowden, M.; Huang, Y.; Carbone, D.P.; Gerstner, G.J.; Lerner, R.E.; Rubin, J.L.; et al. ECOG-ACRIN 1512 Investigators Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second-line or third-line treatment of patients with EGFR wild-type advanced non-small-cell lung cancer (ECOG-ACRIN 1512): A randomised, controlled, open-label, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Ervin, T.J.; Ramlau, R.A.; Daniel, D.B.; Goldschmidt, J.H.; Blumenschein, G.R.; Krzakowski, M.J.; Robinet, G.; Godbert, B.; Barlesi, F.; et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 4105–4114. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Drilon, A.; Fan, P.-D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Golding, B.; Luu, A.; Jones, R.; Viloria-Petit, A.M. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol. Cancer 2018, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. PROFILE 1014 Investigators First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, T.M.; Crinò, L.; Gridelli, C.; Kiura, K.; Liu, G.; Novello, S.; Bearz, A.; Gautschi, O.; Mok, T.; et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 874–886. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. ALEX Trial Investigators Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [PubMed]
- Bergethon, K.; Shaw, A.T.; Ou, S.-H.I.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Tseng, D.; Yoda, S.; Dagogo-Jack, I.; Friboulet, L.; Lin, J.J.; Hubbeling, H.G.; Dardaei, L.; Farago, A.F.; Schultz, K.R.; et al. Patterns of Metastatic Spread and Mechanisms of Resistance to Crizotinib in ROS1-Positive Non-Small-Cell Lung Cancer. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Yang, J.C.-H.; Kim, D.-W.; Lu, S.; Zhou, J.; Seto, T.; Yang, J.-J.; Yamamoto, N.; Ahn, M.-J.; Takahashi, T.; et al. Phase II Study of Crizotinib in East Asian Patients With ROS1-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, F.; Caramella, C.; Auger, N.; Planchard, D.; Adam, J.; Lacroix, L.; Remon, J.; Massard, C.; Soria, J.-C.; Friboulet, L.; et al. Crizotinib primary resistance overcome by ceritinib in a patient with ALK-rearranged non-small cell lung cancer. Tumori 2016, 102. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Wang, S.; Xie, W.; Chang, J.; Gan, Y. The RET fusion gene and its correlation with demographic and clinicopathological features of non-small cell lung cancer: A meta-analysis. Cancer Biol. Ther. 2015, 16, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Nakaoku, T.; Kohno, T.; Araki, M.; Niho, S.; Chauhan, R.; Knowles, P.P.; Tsuchihara, K.; Matsumoto, S.; Shimada, Y.; Mimaki, S.; et al. A secondary RET mutation in the activation loop conferring resistance to vandetanib. Nat. Commun. 2018, 9, 625. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Kennedy, E.; Sequist, L.V.; Brastianos, P.K.; Goodwin, K.E.; Stevens, S.; Wanat, A.C.; Stober, L.L.; Digumarthy, S.R.; Engelman, J.A.; et al. Clinical Activity of Alectinib in Advanced RET-Rearranged Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11, 2027–2032. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, R.; Auger, N.; Auclin, E.; Besse, B. Clinical and Translational Implications of RET Rearrangements in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and drug sensitive NTRK1 rearrangements in lung cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Farago, A.F.; Le, L.P.; Zheng, Z.; Muzikansky, A.; Drilon, A.; Patel, M.; Bauer, T.M.; Liu, S.V.; Ou, S.-H.I.; Jackman, D.; et al. Durable Clinical Response to Entrectinib in NTRK1-Rearranged Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2015, 10, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Brambilla, M.; Metro, G.; Baglivo, S.; Matocci, R.; Pirro, M.; Chiari, R. Targeting NTRK fusion in non-small cell lung cancer: Rationale and clinical evidence. Med. Oncol. 2017, 34, 105. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Nakaoku, T.; Tsuta, K.; Tsuchihara, K.; Matsumoto, S.; Yoh, K.; Goto, K. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl. Lung Cancer Res. 2015, 4, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.D.; Wang, Y.; Beadling, C.; Warrick, A.; Neff, T.; Corless, C.L.; Tolba, K. Durable Response to Afatinib in Lung Adenocarcinoma Harboring NRG1 Gene Fusions. J. Thorac. Oncol. 2017, 12, e107–e110. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Somwar, R.; Mangatt, B.P.; Edgren, H.; Desmeules, P.; Ruusulehto, A.; Smith, R.S.; Delasos, L.; Vojnic, M.; Plodkowski, A.J.; et al. Response to ERBB3-Directed Targeted Therapy in NRG1-Rearranged Cancers. Cancer Discov. 2018. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, J.A.; Taviaux, S.; Béganton, B.; Godreuil, S.; Audran, P.; Grand, D.; Clermont, E.; Serre, I.; Szablewski, V.; Coopman, P.; et al. Detection of known and novel ALK fusion transcripts in lung cancer patients using next-generation sequencing approaches. Sci. Rep. 2017, 7, 12510. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Tayebwa, J. Evolving techniques for gene fusion detection in soft tissue tumours. Histopathology 2014, 64, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-C.; Zhou, Y.-F.; Wang, W.-X.; Xu, C.-W.; Zhuang, W.; Du, K.-Q.; Chen, G. CEP72-ROS1: A novel ROS1 oncogenic fusion variant in lung adenocarcinoma identified by next-generation sequencing. Thorac. Cancer 2018, 9, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Velizheva, N.P.; Rechsteiner, M.P.; Valtcheva, N.; Freiberger, S.N.; Wong, C.E.; Vrugt, B.; Zhong, Q.; Wagner, U.; Moch, H.; Hillinger, S.; et al. Targeted next-generation-sequencing for reliable detection of targetable rearrangements in lung adenocarcinoma-a single center retrospective study. Pathol. Res. Pract. 2018, 214, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Le Mercier, M.; De Nève, N.; Blanchard, O.; Remmelink, M.; Weynand, B.; Salmon, I.; D’Haene, N. Clinical application of targeted next generation sequencing for lung cancer patients. Belgian J. Med. Oncol. 2015, 27, 2–8. [Google Scholar]
- Lih, C.-J.; Sims, D.J.; Harrington, R.D.; Polley, E.C.; Zhao, Y.; Mehaffey, M.G.; Forbes, T.D.; Das, B.; Walsh, W.D.; Datta, V.; et al. Analytical Validation and Application of a Targeted Next-Generation Sequencing Mutation-Detection Assay for Use in Treatment Assignment in the NCI-MPACT Trial. J. Mol. Diagn. JMD 2016, 18, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Lin, Y.; Liu, J.; Wan, L.; Liu, Z.; Cheng, S.; Fei, L.; Deng, R.; Wang, J.; Chen, X.; et al. A targeted next-generation sequencing method for identifying clinically relevant mutation profiles in lung adenocarcinoma. Sci. Rep. 2016, 6, 22338. [Google Scholar] [CrossRef] [PubMed]
- Bennett, N.C.; Farah, C.S. Next-generation sequencing in clinical oncology: Next steps towards clinical validation. Cancers 2014, 6, 2296–2312. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cespedes, M.; Parrella, P.; Esteller, M.; Nomoto, S.; Trink, B.; Engles, J.M.; Westra, W.H.; Herman, J.G.; Sidransky, D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62, 3659–3662. [Google Scholar] [PubMed]
- Fang, R.; Zheng, C.; Sun, Y.; Han, X.; Gao, B.; Li, C.; Liu, H.; Wong, K.-K.; Liu, X.-Y.; Chen, H.; et al. Integrative genomic analysis reveals a high frequency of LKB1 genetic alteration in Chinese lung adenocarcinomas. J. Thorac. Oncol. 2014, 9, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-X.; Xu, Z.-X. Targeting the LKB1 tumor suppressor. Curr. Drug Targets 2014, 15, 32–52. [Google Scholar] [CrossRef] [PubMed]
- Moghadasi, S.; Eccles, D.M.; Devilee, P.; Vreeswijk, M.P.G.; van Asperen, C.J. Classification and Clinical Management of Variants of Uncertain Significance in High Penetrance Cancer Predisposition Genes. Hum. Mutat. 2016, 37, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, S.; Nagano, M.; Ueno, T.; Suehara, Y.; Hayashi, T.; Shimada, N.; Takahashi, K.; Suzuki, K.; Takamochi, K.; Takahashi, F.; et al. A method of high-throughput functional evaluation ofEGFRgene variants of unknown significance in cancer. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Mandel, P.; Metais, P. [Les acides nucleiques du plasma sanguin chez l’homme]. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Pécuchet, N.; Rozenholc, Y.; Zonta, E.; Pietrasz, D.; Didelot, A.; Combe, P.; Gibault, L.; Bachet, J.-B.; Taly, V.; Fabre, E.; et al. Analysis of Base-Position Error Rate of Next-Generation Sequencing to Detect Tumor Mutations in Circulating DNA. Clin. Chem. 2016, 62, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rizaldos, E.; Grimm, D.G.; Tadigotla, V.; Hurley, J.; Healy, J.; Neal, P.L.; Sher, M.; Venkatesan, R.; Karlovich, C.; Raponi, M.; et al. Exosome-based Detection of EGFR T790M in Plasma from Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Cheng, Z.; Qin, W.; Jiang, L. Exosomes as a liquid biopsy for lung cancer. Lung Cancer 2018, 116, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Couraud, S.; Vaca-Paniagua, F.; Villar, S.; Oliver, J.; Schuster, T.; Blanché, H.; Girard, N.; Trédaniel, J.; Guilleminault, L.; Gervais, R.; et al. BioCAST/IFCT-1002 investigators Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: A proof-of-concept study from BioCAST/IFCT-1002. Clin. Cancer Res. 2014, 20, 4613–4624. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Denis, M.G.; Thress, K.S.; Ratcliffe, M.; Reck, M. Guide to detecting epidermal growth factor receptor (EGFR) mutations in ctDNA of patients with advanced non-small-cell lung cancer. Oncotarget 2017, 8, 12501–12516. [Google Scholar] [CrossRef] [PubMed]
- Uchida, J.; Kato, K.; Kukita, Y.; Kumagai, T.; Nishino, K.; Daga, H.; Nagatomo, I.; Inoue, T.; Kimura, M.; Oba, S.; et al. Diagnostic Accuracy of Noninvasive Genotyping of EGFR in Lung Cancer Patients by Deep Sequencing of Plasma Cell-Free DNA. Clin. Chem. 2015, 61, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Plagnol, V.; Woodhouse, S.; Howarth, K.; Lensing, S.; Smith, M.; Epstein, M.; Madi, M.; Smalley, S.; Leroy, C.; Hinton, J.; et al. Analytical validation of a next generation sequencing liquid biopsy assay for high sensitivity broad molecular profiling. PLoS ONE 2018, 13, e0193802. [Google Scholar] [CrossRef] [PubMed]
- Mambetsariev, I.; Vora, L.; Yu, K.W.; Salgia, R. Effective osimertinib treatment in a patient with discordant T790 M mutation detection between liquid biopsy and tissue biopsy. BMC Cancer 2018, 18, 314. [Google Scholar] [CrossRef] [PubMed]
- McCoach, C.E.; Blakely, C.M.; Banks, K.C.; Levy, B.; Chue, B.M.; Raymond, V.M.; Le, A.T.; Lee, C.E.; Diaz, J.; Waqar, S.N.; et al. Clinical Utility of Cell-Free DNA for the Detection of ALK Fusions and Genomic Mechanisms of ALK Inhibitor Resistance in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Provencio, M.; Torrente, M.; Calvo, V.; Pérez-Callejo, D.; Gutiérrez, L.; Franco, F.; Pérez-Barrios, C.; Barquín, M.; Royuela, A.; García-García, F.; et al. Prognostic value of quantitative ctDNA levels in non small cell lung cancer patients. Oncotarget 2018, 9, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Hyun, M.H.; Sung, J.S.; Kang, E.J.; Choi, Y.J.; Park, K.H.; Shin, S.W.; Lee, S.Y.; Kim, Y.H. Quantification of circulating cell-free DNA to predict patient survival in non-small-cell lung cancer. Oncotarget 2017, 8, 94417–94430. [Google Scholar] [CrossRef] [PubMed]
- Pécuchet, N.; Zonta, E.; Didelot, A.; Combe, P.; Thibault, C.; Gibault, L.; Lours, C.; Rozenholc, Y.; Taly, V.; Laurent-Puig, P.; et al. Base-Position Error Rate Analysis of Next-Generation Sequencing Applied to Circulating Tumor DNA in Non-Small Cell Lung Cancer: A Prospective Study. PLoS Med. 2016, 13, e1002199. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Yoneshima, Y.; Ijichi, K.; Anai, S.; Ota, K.; Otsubo, K.; Iwama, E.; Tanaka, K.; Oda, Y.; Nakanishi, Y.; Okamoto, I. PD-L1 expression in lung adenocarcinoma harboring EGFR mutations or ALK rearrangements. Lung Cancer 2018, 118, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Azuma, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Tanizaki, J.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; et al. Induction of PD-L1 Expression by the EML4-ALK Oncoprotein and Downstream Signaling Pathways in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 4014–4021. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Johnson, D.; Temin, S.; Baker, S.; Brahmer, J.; Ellis, P.M.; Giaccone, G.; Hesketh, P.J.; Jaiyesimi, I.; Leighl, N.B.; et al. Systemic Therapy for Stage IV Non-Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 3484–3515. [Google Scholar] [CrossRef] [PubMed]
- Haratani, K.; Hayashi, H.; Tanaka, T.; Kaneda, H.; Togashi, Y.; Sakai, K.; Hayashi, K.; Tomida, S.; Chiba, Y.; Yonesaka, K.; et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann. Oncol. 2017, 28, 1532–1539. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Man, J.; Lord, S.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C.-H. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non-Small Cell Lung Cancer-A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Hellmann, M.D.; Awad, M.M.; Rizvi, H.; Carter, B.W.; Denning, W.; Elamin, Y.; Zhang, J.; Leonardi, G.C.; Halpenny, D.; et al. STK11/LKB1 co-mutations to predict for de novo resistance to PD-1/PD-L1 axis blockade in KRAS-mutant lung adenocarcinoma. J. Clin. Oncol. 2017, 35, 9016. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Mansuet-Lupo, A.; Alifano, M.; Pécuchet, N.; Biton, J.; Becht, E.; Goc, J.; Germain, C.; Ouakrim, H.; Régnard, J.-F.; Cremer, I.; et al. Intratumoral Immune Cell Densities Are Associated with Lung Adenocarcinoma Gene Alterations. Am. J. Respir. Crit. Care Med. 2016, 194, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Warren, R.L.; Gibb, E.A.; Martin, S.D.; Spinelli, J.J.; Nelson, B.H.; Holt, R.A. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014, 24, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients with Non-Small-Cell Lung Cancer Profiled with Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Steuer, C.E.; Ramalingam, S.S. Tumor Mutation Burden: Leading Immunotherapy to the Era of Precision Medicine? J. Clin. Oncol. 2018, 36, 631–632. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garinet, S.; Laurent-Puig, P.; Blons, H.; Oudart, J.-B. Current and Future Molecular Testing in NSCLC, What Can We Expect from New Sequencing Technologies? J. Clin. Med. 2018, 7, 144. https://doi.org/10.3390/jcm7060144
Garinet S, Laurent-Puig P, Blons H, Oudart J-B. Current and Future Molecular Testing in NSCLC, What Can We Expect from New Sequencing Technologies? Journal of Clinical Medicine. 2018; 7(6):144. https://doi.org/10.3390/jcm7060144
Chicago/Turabian StyleGarinet, Simon, Pierre Laurent-Puig, Hélène Blons, and Jean-Baptiste Oudart. 2018. "Current and Future Molecular Testing in NSCLC, What Can We Expect from New Sequencing Technologies?" Journal of Clinical Medicine 7, no. 6: 144. https://doi.org/10.3390/jcm7060144
APA StyleGarinet, S., Laurent-Puig, P., Blons, H., & Oudart, J.-B. (2018). Current and Future Molecular Testing in NSCLC, What Can We Expect from New Sequencing Technologies? Journal of Clinical Medicine, 7(6), 144. https://doi.org/10.3390/jcm7060144