Androgen Receptor Splice Variant 7 Drives the Growth of Castration Resistant Prostate Cancer without Being Involved in the Efficacy of Taxane Chemotherapy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Cell Proliferation Assay
2.3. Western Blot Analysis
2.4. Real-Time Quantitative Reverse Transcription PCR (Real-Time RT-qPCR)
2.5. AR-Driven PSA(6.1kb)-Luciferase Reporter Gene Assay
2.6. Knockdown of AR-V7
2.7. Statistical Analysis
3. Results
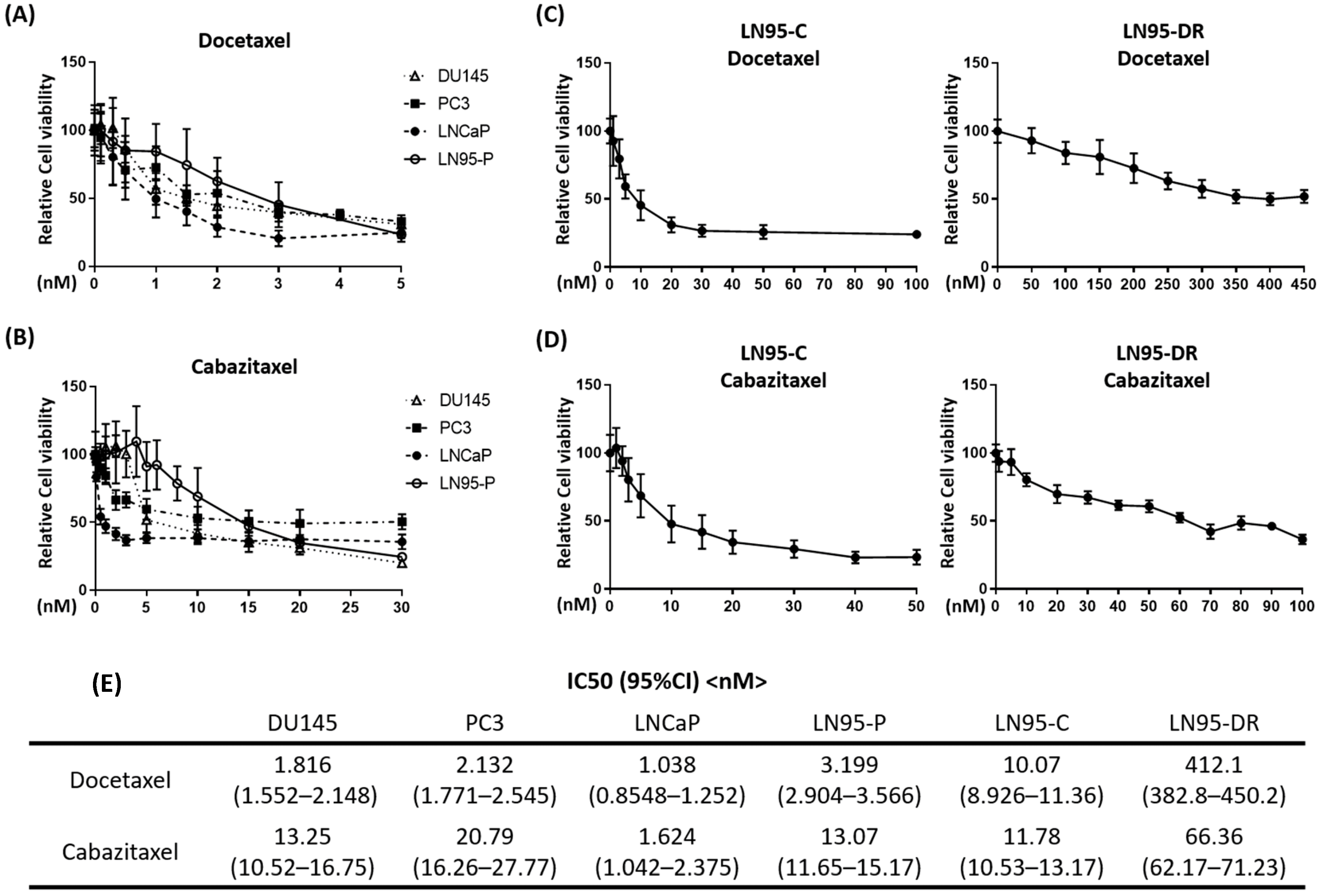
3.1. LNCaP95-DR Cells Were Cross-Resistant to Cabazitaxel
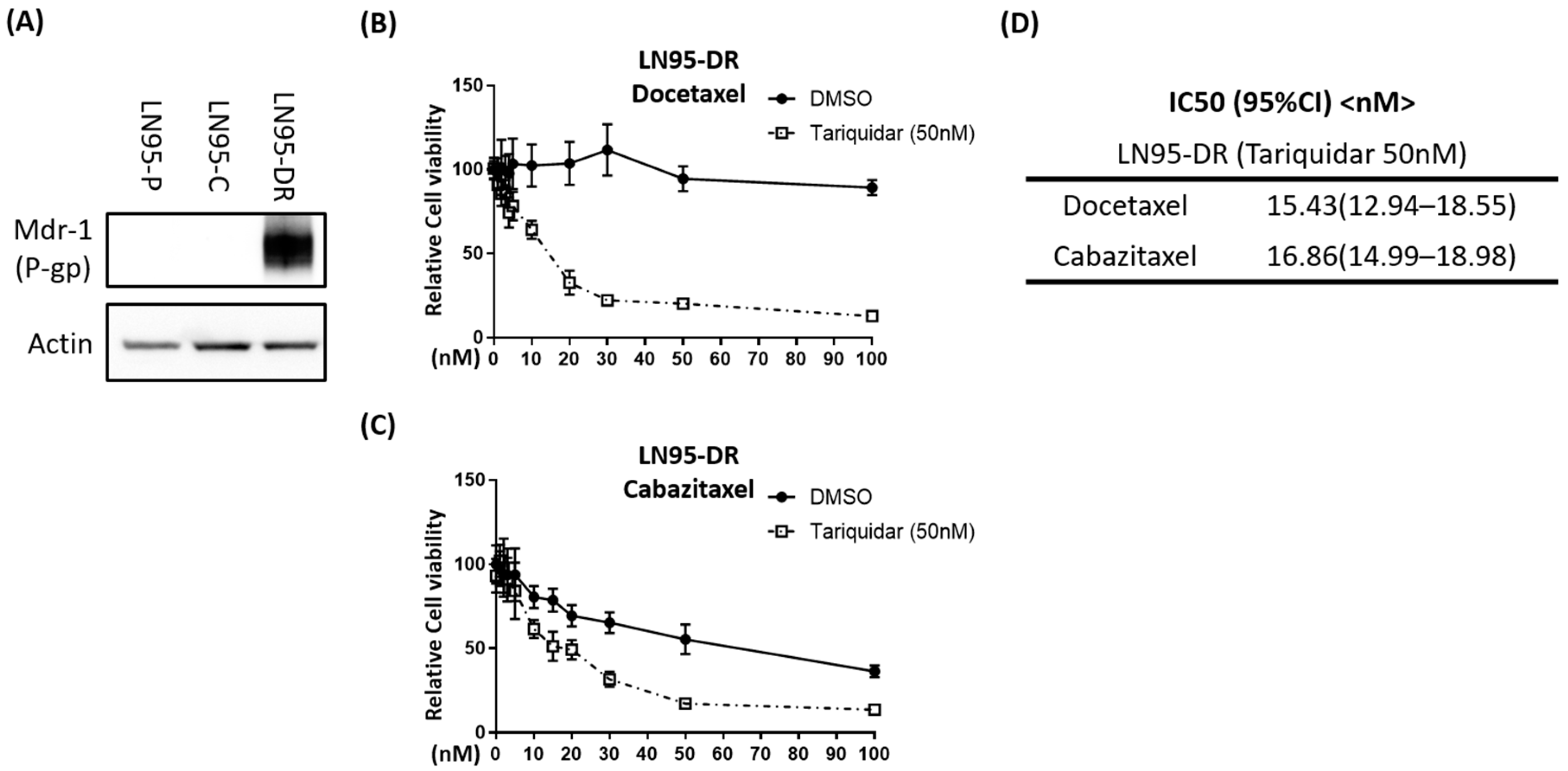
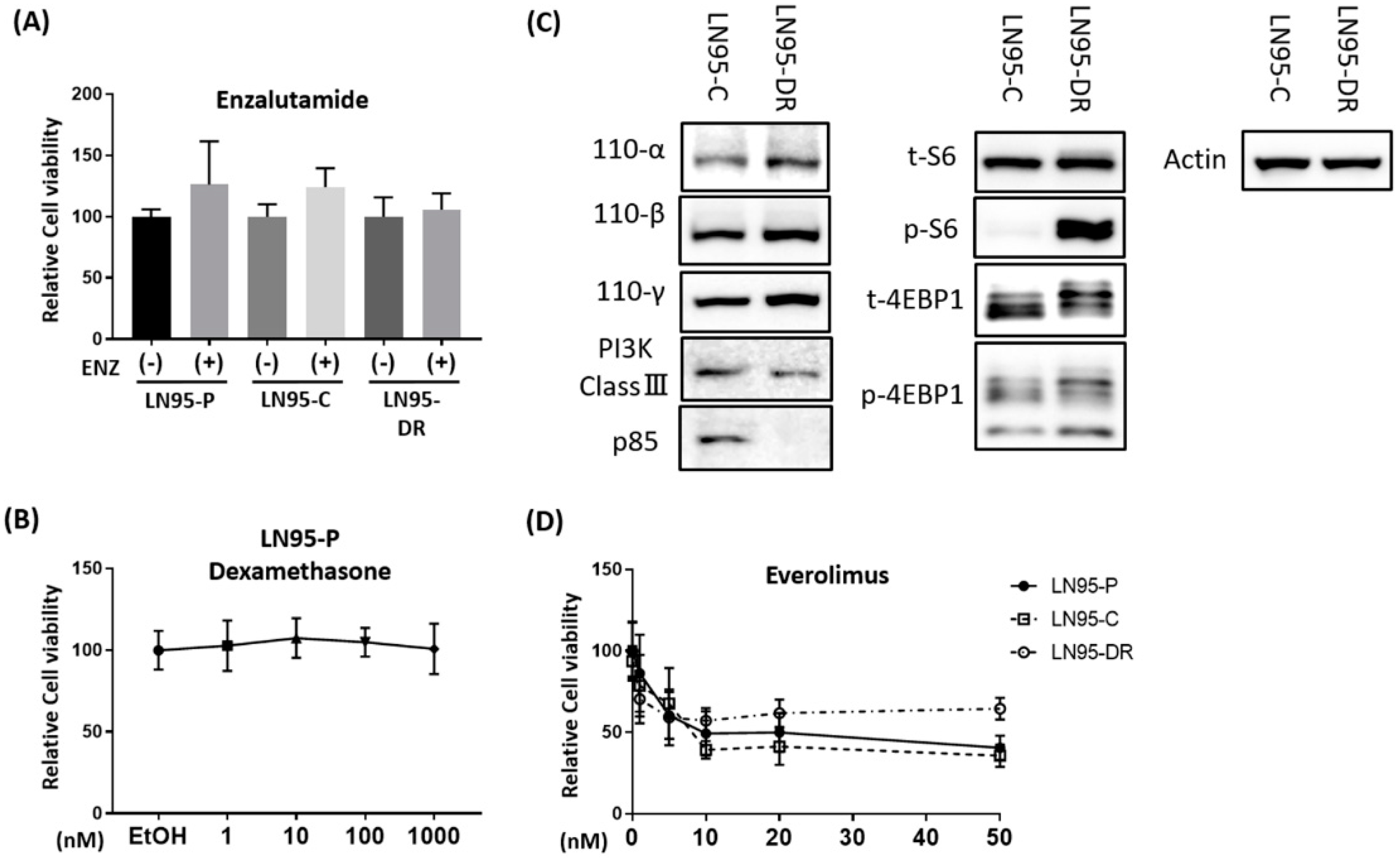
3.2. P-gp Was Overexpressed in LNCaP95-DR Cells and Tariquidar Restored Sensitivity to Docetaxel and Cabazitaxel
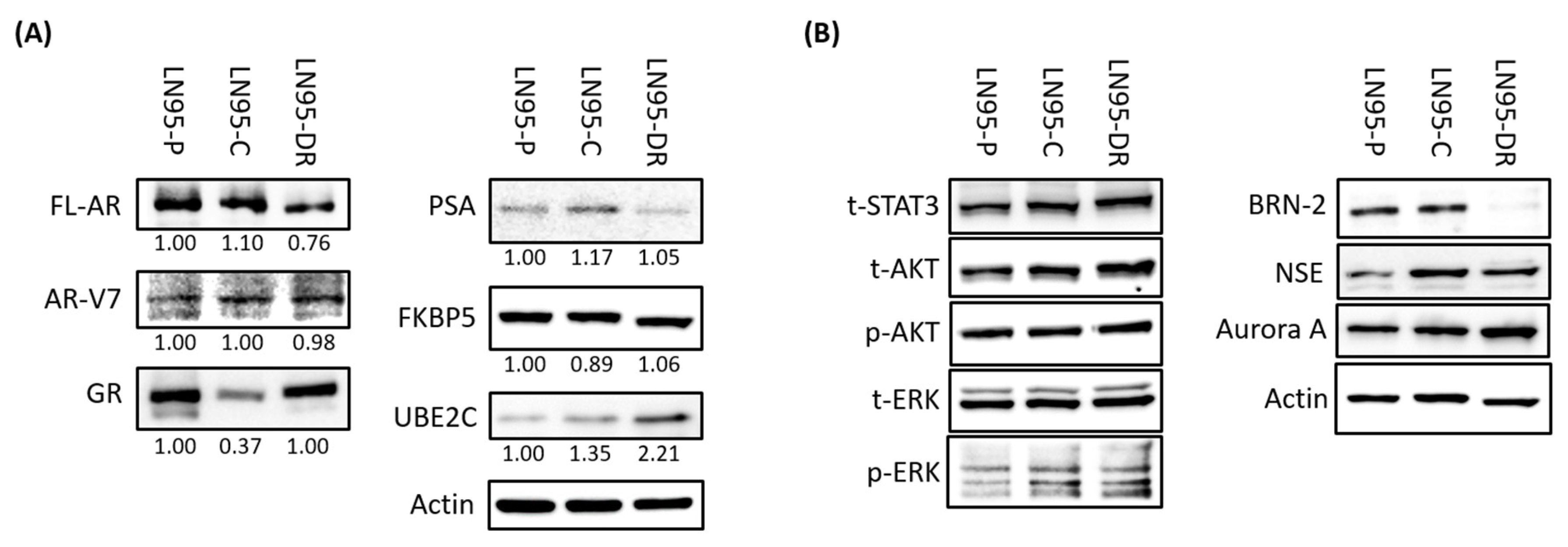
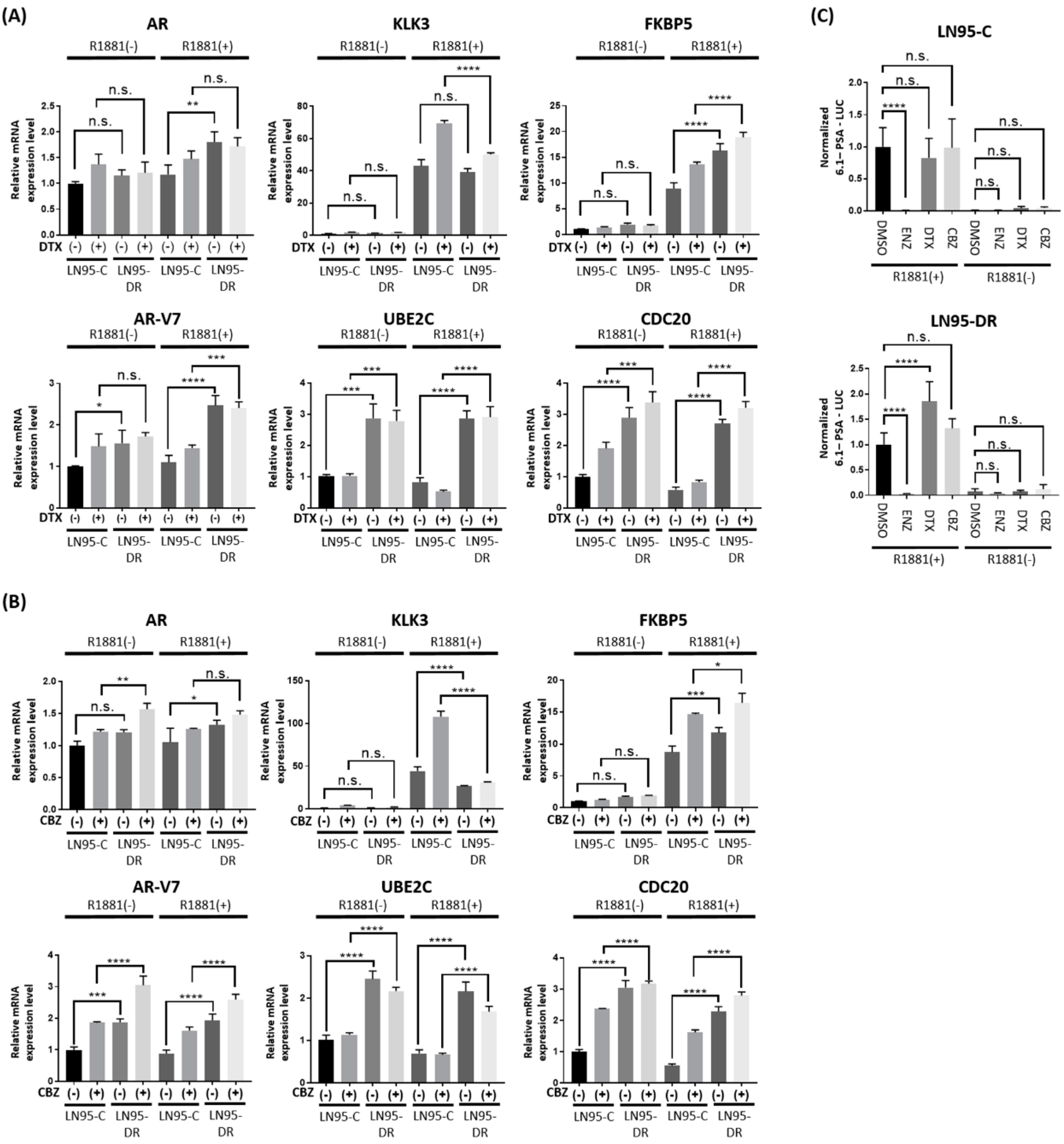
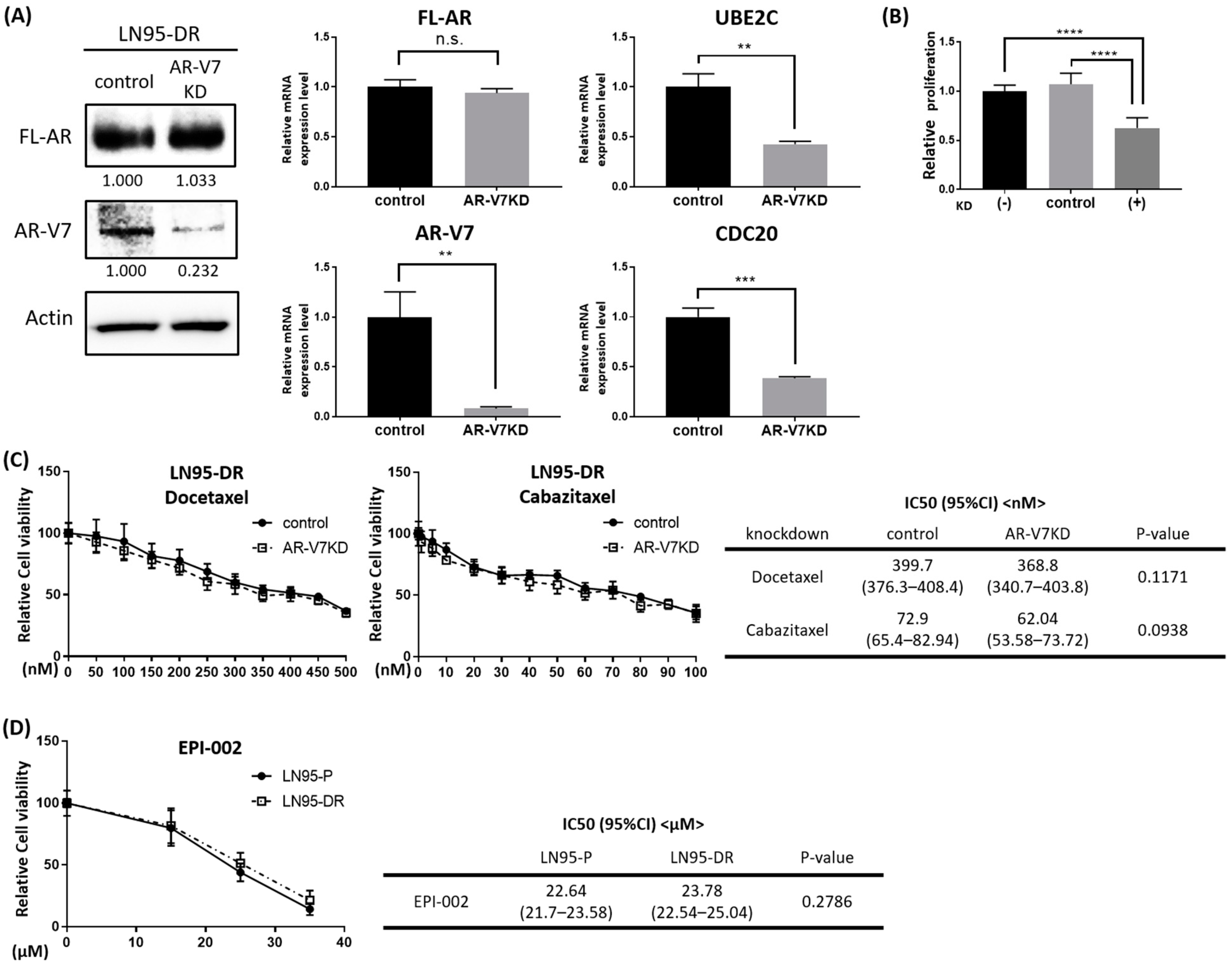
3.3. Expression of AR-V7-Regulated Genes Was Increased in LNCaP95-DR
3.4. Knockdown of AR-V7 Has No Effect on Sensitivity to Docetaxel and Cabazitaxel
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Rini, B.I.; Small, E.J. Hormone-refractory Prostate Cancer. Curr. Treat. Options Oncol. 2002, 3, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.E.; Scher, H.I. Starving the addiction: New opportunities for durable suppression of AR signaling in prostate cancer. Clin. Cancer Res. 2009, 15, 4792–4798. [Google Scholar] [CrossRef] [PubMed]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinanen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, P.; Visakorpi, T.; Kallioniemi, O.P. Androgen receptor gene amplification: A novel molecular mechanism for endocrine therapy resistance in human prostate cancer. Scand. J. Clin. Lab. Investig. Suppl. 1996, 226, 57–63. [Google Scholar] [CrossRef]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Cato, A.C.; Hittmair, A.; Radmayr, C.; Eberle, J.; Bartsch, G.; Klocker, H. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol. Endocrinol. 1993, 7, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Mawji, N.R.; Bruchovsky, N.; Sadar, M.D. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J. Biol. Chem. 2002, 277, 38087–38094. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, R.C.; O’Malley, B.W. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 2009, 9, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Gioeli, D.; Paschal, B.M. Post-translational modification of the androgen receptor. Mol. Cell. Endocrinol. 2012, 352, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Banuelos, C.A.; Sadar, M.D.; Kyprianou, N. N-terminal targeting of androgen receptor variant enhances response of castration resistant prostate cancer to taxane chemotherapy. Mol. Oncol. 2014, 9, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Banuelos, C.A.; Imamura, Y.; Leung, J.K.; Caley, D.P.; Wang, J.; Mawji, N.R.; Sadar, M.D. Cotargeting Androgen Receptor Splice Variants and mTOR Signaling Pathway for the Treatment of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- David-Beabes, G.L.; Overman, M.J.; Petrofski, J.A.; Campbell, P.A.; de Marzo, A.M.; Nelson, W.G. Doxorubicin-resistant variants of human prostate cancer cell lines DU 145, PC-3, PPC-1, and TSU-PR1: Characterization of biochemical determinants of antineoplastic drug sensitivity. Int. J. Oncol. 2000, 17, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Mizokami, A.; Mamiya, K.; Li, Y.Q.; Zhang, J.; Keller, E.T.; Namiki, M. The establishment of two paclitaxel-resistant prostate cancer cell lines and the mechanisms of paclitaxel resistance with two cell lines. Prostate 2007, 67, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Mahon, K.L.; Henshall, S.M.; Sutherland, R.L.; Horvath, L.G. Pathways of chemotherapy resistance in castration-resistant prostate cancer. Endocr.-Relat. Cancer 2011, 18, R103–R123. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, G.; Buonerba, C.; Autorino, R.; De Placido, S.; Sternberg, C.N. Castration-resistant prostate cancer: Current and emerging treatment strategies. Drugs 2010, 70, 983–1000. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Lombard, A.P.; Liu, C.; Armstrong, C.M.; Cucchiara, V.; Gu, X.; Lou, W.; Evans, C.P.; Gao, A.C. ABCB1 Mediates Cabazitaxel-Docetaxel Cross-Resistance in Advanced Prostate Cancer. Mol. Cancer Ther. 2017, 16, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Ploussard, G.; Terry, S.; Maille, P.; Allory, Y.; Sirab, N.; Kheuang, L.; Soyeux, P.; Nicolaiew, N.; Coppolani, E.; Paule, B.; et al. Class III beta-tubulin expression predicts prostate tumor aggressiveness and patient response to docetaxel-based chemotherapy. Cancer Res. 2010, 70, 9253–9264. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; Jayaram, A.; Winquist, E.; McLaughlin, B.; Lu, D.; Fleisher, M.; Orr, S.; Lowes, L.; et al. Assessment of the Validity of Nuclear-Localized Androgen Receptor Splice Variant 7 in Circulating Tumor Cells as a Predictive Biomarker for Castration-Resistant Prostate Cancer. JAMA Oncol. 2018, 4, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Thadani-Mulero, M.; Portella, L.; Sun, S.; Sung, M.; Matov, A.; Vessella, R.L.; Corey, E.; Nanus, D.M.; Plymate, S.R.; Giannakakou, P. Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer Res. 2014, 74, 2270–2282. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Wei, M.; Yamano, S.; Kakehashi, A.; Tamada, S.; Nakatani, T.; Wanibuchi, H. DDX39 acts as a suppressor of invasion for bladder cancer. Cancer Sci. 2012, 103, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Sadar, M.D. Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. J. Biol. Chem. 1999, 274, 7777–7783. [Google Scholar] [CrossRef] [PubMed]
- Cleutjens, K.B.; van der Korput, H.A.; van Eekelen, C.C.; van Rooij, H.C.; Faber, P.W.; Trapman, J. An androgen response element in a far upstream enhancer region is essential for high, androgen-regulated activity of the prostate-specific antigen promoter. Mol. Endocrinol. 1997, 11, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Martin, C.; Berridge, G.; Mistry, P.; Higgins, C.; Charlton, P.; Callaghan, R. The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br. J. Pharmacol. 1999, 128, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Steiner, J.; Mellows, G.; Laguda, B.; Norris, D.; Bevan, P. Phase I trial of XR9576 in healthy volunteers demonstrates modulation of P-glycoprotein in CD56+ lymphocytes after oral and intravenous administration. Clin. Cancer Res. 2000, 6, 4186–4191. [Google Scholar] [PubMed]
- Agrawal, M.; Abraham, J.; Balis, F.M.; Edgerly, M.; Stein, W.D.; Bates, S.; Fojo, T.; Chen, C.C. Increased 99mTc-sestamibi accumulation in normal liver and drug-resistant tumors after the administration of the glycoprotein inhibitor, XR9576. Clin. Cancer Res. 2003, 9, 650–656. [Google Scholar] [PubMed]
- Abraham, J.; Edgerly, M.; Wilson, R.; Chen, C.; Rutt, A.; Bakke, S.; Robey, R.; Dwyer, A.; Goldspiel, B.; Balis, F.; et al. A phase I study of the P-glycoprotein antagonist tariquidar in combination with vinorelbine. Clin. Cancer Res. 2009, 15, 3574–3582. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Attard, G.; Balk, S.P.; Bevan, C.; Burnstein, K.; Cato, L.; Cherkasov, A.; De Bono, J.S.; Dong, Y.; Gao, A.C.; et al. Role of Androgen Receptor Variants in Prostate Cancer: Report from the 2017 Mission Androgen Receptor Variants Meeting. Eur. Urol. 2018, 73, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Bitting, R.L.; Armstrong, A.J. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr.-Relat. Cancer 2013, 20, R83–99. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Reid, A.H.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Hornberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikstrom, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, S.; Sowalsky, A.G.; Voznesensky, O.S.; Mostaghel, E.A.; Nelson, P.S.; Cai, C.; Balk, S.P. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin. Cancer Res. 2014, 20, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Xie, N.; Sun, S.; Plymate, S.; Mostaghel, E.; Dong, X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014, 33, 3140–3150. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C. Development of Novel Small Molecule Inhibitor of Androgen Receptor to Treat Castration-Resistant Prostate Cancer. Ph.D Thesis, The University of British Columbia, Vancouver, BC, Canada, August 2015. [Google Scholar]
- Yang, Y.C.; Meimetis, L.G.; Tien, A.H.; Mawji, N.R.; Carr, G.; Wang, J.; Andersen, R.J.; Sadar, M.D. Spongian diterpenoids inhibit androgen receptor activity. Mol. Cancer Ther. 2013, 12, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, C.; Nadiminty, N.; Lou, W.; Tummala, R.; Evans, C.P.; Gao, A.C. Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Mol. Cancer Ther. 2013, 12, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, C.; Armstrong, C.; Lou, W.; Sandher, A.; Gao, A.C. Antiandrogens Inhibit ABCB1 Efflux and ATPase Activity and Reverse Docetaxel Resistance in Advanced Prostate Cancer. Clin. Cancer Res. 2015, 21, 4133–4142. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.A.; Rodriguez-Barrueco, R.; Bonal, D.M.; Charytonowicz, E.; Gladoun, N.; de la Iglesia-Vicente, J.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D.; Dritselis, A.; Kirkpatrick, P.; Oh, W.K. Cabazitaxel. Nat. Rev. Drug Discov. 2010, 9, 677–678. [Google Scholar] [CrossRef] [PubMed]
- Onstenk, W.; Sieuwerts, A.M.; Kraan, J.; Van, M.; Nieuweboer, A.J.; Mathijssen, R.H.; Hamberg, P.; Meulenbeld, H.J.; De Laere, B.; Dirix, L.Y.; et al. Efficacy of Cabazitaxel in Castration-resistant Prostate Cancer Is Independent of the Presence of AR-V7 in Circulating Tumor Cells. Eur. Urol. 2015, 68, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.L.; Horbinski, C.M.; Garzotto, M.; Qian, D.Z.; Beer, T.M.; Kyprianou, N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010, 70, 7992–8002. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Liu, H.; Kim, S.; Guo, M.; Navarro, V.; Bander, N.H. Docetaxel down-regulates the expression of androgen receptor and prostate-specific antigen but not prostate-specific membrane antigen in prostate cancer cell lines: Implications for PSA surrogacy. Prostate 2009, 69, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Chen, S.; Wang, Y.; Watahiki, A.; Bohrer, L.; Sun, Z.; Wang, Y.; Huang, H. Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res. 2009, 69, 8386–8394. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Sakamoto, J.; Namikawa, T.; Okamoto, K.; Okabayashi, T.; Ichikawa, K.; Araki, K. Pharmacokinetic study of paclitaxel in malignant ascites from advanced gastric cancer patients. World J. Gastroenterol. 2006, 12, 1412–1415. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Kearns, C.M.; Giani, A.; Capri, G.; Vigano, L.; Lacatelli, A.; Bonadonna, G.; Egorin, M.J. Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J. Clin. Oncol. 1995, 13, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Brunsvig, P.F.; Andersen, A.; Aamdal, S.; Kristensen, V.; Olsen, H. Pharmacokinetic analysis of two different docetaxel dose levels in patients with non-small cell lung cancer treated with docetaxel as monotherapy or with concurrent radiotherapy. BMC Cancer 2007, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Ferron, G.M.; Dai, Y.; Semiond, D. Population pharmacokinetics of cabazitaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, J.L.; Taylor, M.N.; Antonarakis, E.S. Novel Insights into Molecular Indicators of Response and Resistance to Modern Androgen-Axis Therapies in Prostate Cancer. Curr. Urol. Rep. 2016, 17, 29. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimizu, Y.; Tamada, S.; Kato, M.; Hirayama, Y.; Takeyama, Y.; Iguchi, T.; Sadar, M.D.; Nakatani, T. Androgen Receptor Splice Variant 7 Drives the Growth of Castration Resistant Prostate Cancer without Being Involved in the Efficacy of Taxane Chemotherapy. J. Clin. Med. 2018, 7, 444. https://doi.org/10.3390/jcm7110444
Shimizu Y, Tamada S, Kato M, Hirayama Y, Takeyama Y, Iguchi T, Sadar MD, Nakatani T. Androgen Receptor Splice Variant 7 Drives the Growth of Castration Resistant Prostate Cancer without Being Involved in the Efficacy of Taxane Chemotherapy. Journal of Clinical Medicine. 2018; 7(11):444. https://doi.org/10.3390/jcm7110444
Chicago/Turabian StyleShimizu, Yasuomi, Satoshi Tamada, Minoru Kato, Yukiyoshi Hirayama, Yuji Takeyama, Taro Iguchi, Marianne D. Sadar, and Tatsuya Nakatani. 2018. "Androgen Receptor Splice Variant 7 Drives the Growth of Castration Resistant Prostate Cancer without Being Involved in the Efficacy of Taxane Chemotherapy" Journal of Clinical Medicine 7, no. 11: 444. https://doi.org/10.3390/jcm7110444
APA StyleShimizu, Y., Tamada, S., Kato, M., Hirayama, Y., Takeyama, Y., Iguchi, T., Sadar, M. D., & Nakatani, T. (2018). Androgen Receptor Splice Variant 7 Drives the Growth of Castration Resistant Prostate Cancer without Being Involved in the Efficacy of Taxane Chemotherapy. Journal of Clinical Medicine, 7(11), 444. https://doi.org/10.3390/jcm7110444