Shared Features of Endothelial Dysfunction between Sepsis and Its Preceding Risk Factors (Aging and Chronic Disease)

 ,
,
Abstract
1. Introduction
2. Search Strategy and Selection Criteria
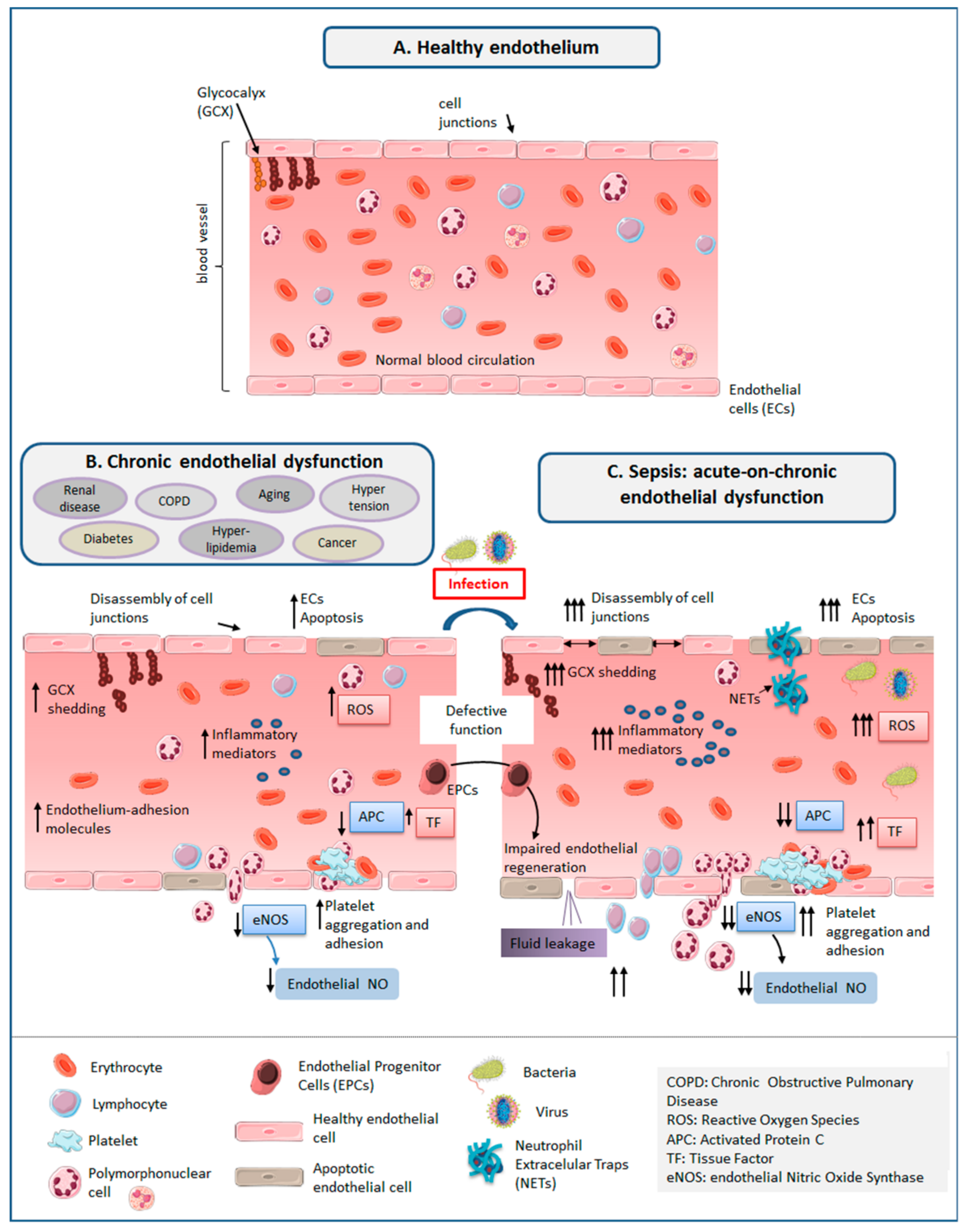
3. The Healthy Endothelium
3.1. Glycocalyx (GCX)
3.2. Endothelial Cells (ECs)
4. ED in Sepsis
4.1. Increased Oxidative Stress and Systemic Inflammation
4.2. GCX Degradation and Shedding
4.3. Disassembly of Intercellular Junctions, Endothelial Cell Death, Blood–Tissue Barrier Disruption
4.4. Enhanced Leukocyte Adhesion and Extravasation
4.5. Induction of A Pro-Coagulant and Anti-Fibrinolytic State
5. ED Associated to Aging and Chronic Disease
5.1. Increased Oxidative Stress and Systemic Inflammation
5.2. GCX Degradation and Shedding
5.3. Disassembly of Intercellular Junctions, Endothelial Cell Death, Blood-Tissue Barrier Disruption
5.4. Enhanced Leukocyte Adhesion and Extravasation
5.5. Induction of A Pro-Coagulant and Anti-Fibrinolytic State
6. Impact of Aging and Chronic Disease on the Mechanisms of Endothelial Repair
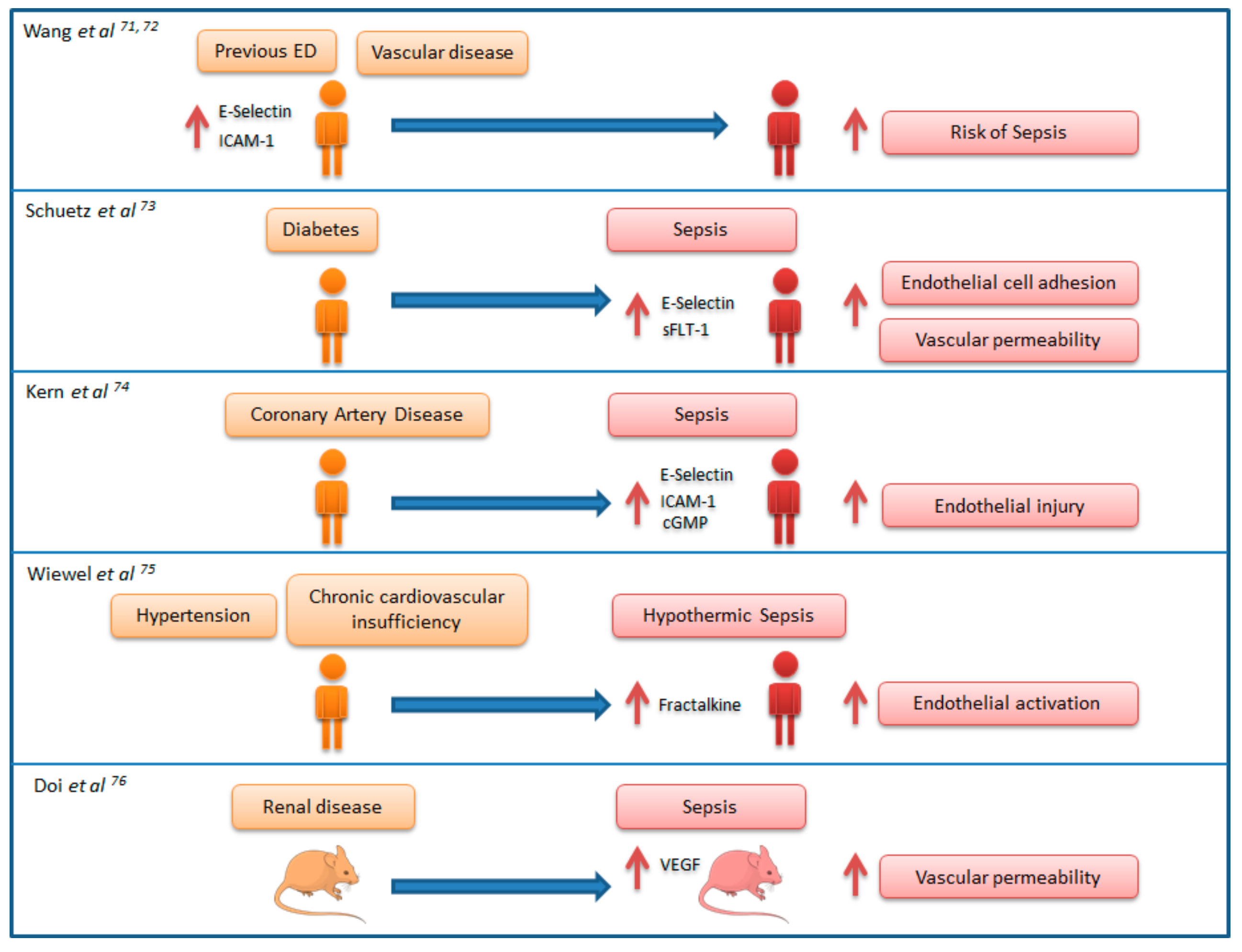
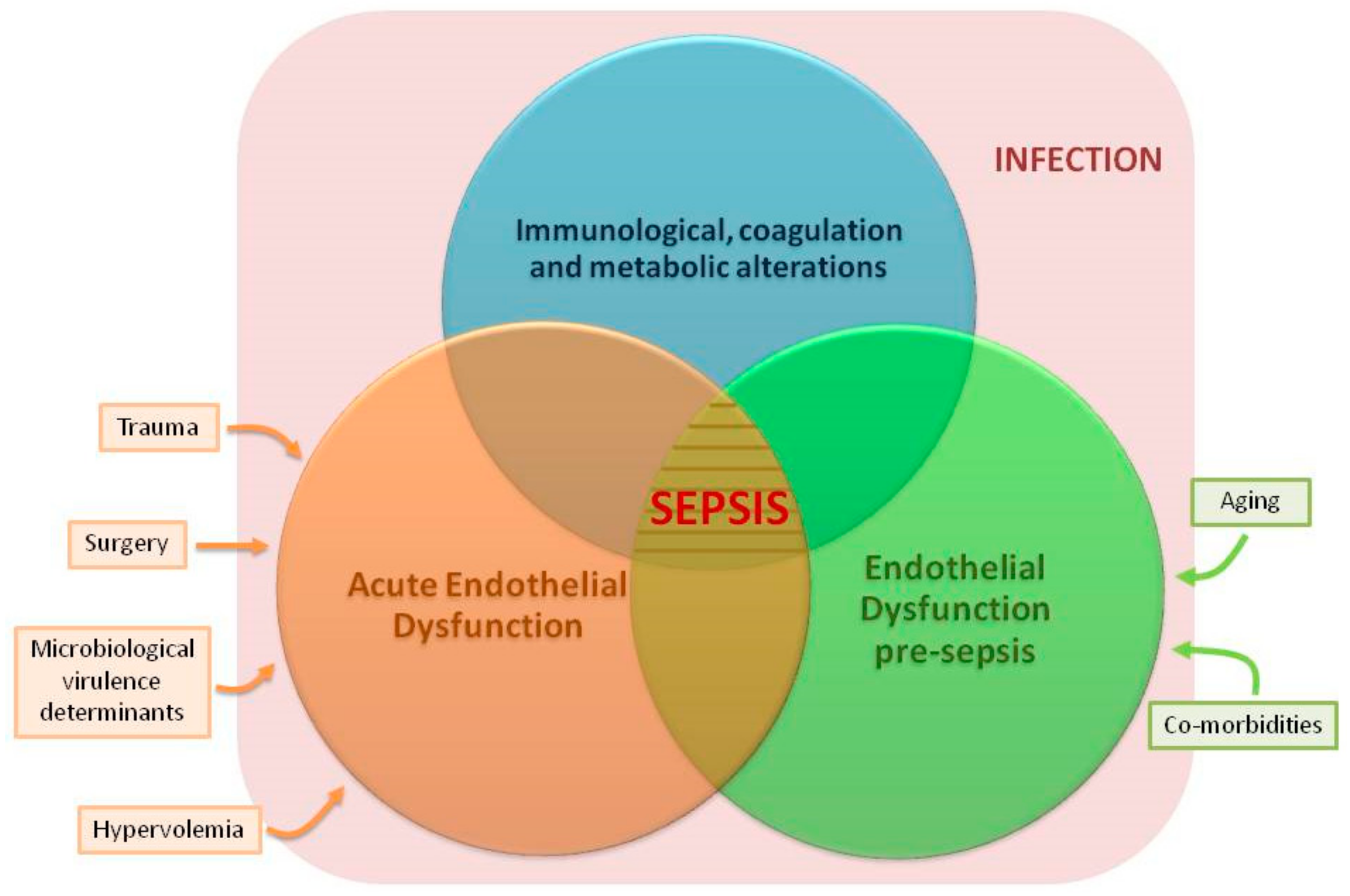
7. Link between Chronic Endothelial Dysfunction and Sepsis: “Proof of Concept” Studies
8. Other Factors Inducing ED before Sepsis
9. Implications for Clinical Practice and Future Research
9.1. Quantifying ED to Predict or Early Detect Sepsis in Infected Patients
9.2. Endothelium-Protective Therapies for the Prevention and Treatment of Sepsis
9.3. Other Potential Research Avenues
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hotchkiss, R.S.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.-L. Sepsis and septic shock. Nat. Rev. Dis. Primers 2016, 2, 16045. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Slutsky, A.S. Sepsis and endothelial permeability. N. Engl. J. Med. 2010, 363, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascón, G.A.; Hernandez, G.; Murray, P.; De Backer, D. ADQI XIV Workgroup. The endothelium in sepsis. Shock 2016, 45, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Novosad, S.A.; Sapiano, M.R.P.; Grigg, C.; Lake, J.; Robyn, M.; Dumyati, G.; Felsen, C.; Blog, D.; Dufort, E.; Zansky, S.; et al. Vital signs: Epidemiology of sepsis: Prevalence of health care factors and opportunities for prevention. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Rhee, C.; Dantes, R.; Epstein, L.; Murphy, D.J.; Seymour, C.W.; Iwashyna, T.J.; Kadri, S.S.; Angus, D.C.; Danner, R.L.; Fiore, A.E.; et al. CDC prevention epicenter program incidence and trends of sepsis in US hospitals using clinical vs. claims data, 2009–2014. JAMA 2017, 318, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, J.P.; Safford, M.M.; Shapiro, N.I.; Baddley, J.W.; Wang, H.E. Application of the Third International Consensus Definitions for Sepsis (Sepsis-3) Classification: A retrospective population-based cohort study. Lancet Infect. Dis. 2017, 17, 661–670. [Google Scholar] [CrossRef]
- Pool, R.; Gomez, H.; Kellum, J.A. Mechanisms of organ dysfunction in sepsis. Crit. Care Clin. 2018, 34, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Lubkin, A.; Torres, V.J. Bacteria and endothelial cells: A toxic relationship. Curr. Opin. Microbiol. 2017, 35, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S. Vascular endothelium in diabetes. Am. J. Physiol. Renal Physiol. 2017, 312, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Münzel, T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef] [PubMed]
- Konukoglu, D.; Uzun, H. Endothelial dysfunction and hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [PubMed]
- Malerba, M.; Nardin, M.; Radaeli, A.; Montuschi, P.; Carpagnano, G.E.; Clini, E. The potential role of endothelial dysfunction and platelet activation in the development of thrombotic risk in COPD patients. Expert Rev. Hematol. 2017, 10, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Vairappan, B. Endothelial dysfunction in cirrhosis: Role of inflammation and oxidative stress. World J. Hepatol. 2015, 7, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Colbert, J.F.; Schmidt, E.P. Endothelial and microcirculatory function and dysfunction in sepsis. Clin. Chest Med. 2016, 37, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Koczera, P.; Zechendorf, E.; Schuerholz, T. The endothelial glycocalyx: New diagnostic and therapeutic approaches in sepsis. Biomed. Res. Int. 2016, 2016, 3758278. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Voumvourakis, A.; Makavos, G.; Triantafyllidi, H.; Pavlidis, G.; Katogiannis, K.; Benas, D.; Vlastos, D.; Trivilou, P.; Varoudi, M.; et al. Association of impaired endothelial glycocalyx with arterial stiffness, coronary microcirculatory dysfunction, and abnormal myocardial deformation in untreated hypertensives. J. Clin. Hypertens. (Greenwich) 2018. [Google Scholar] [CrossRef] [PubMed]
- Groen, B.B.L.; Hamer, H.M.; Snijders, T.; van Kranenburg, J.; Frijns, D.; Vink, H.; Van Loon, L.J.C. Skeletal muscle capillary density and microvascular function are compromised with aging and type 2 diabetes. J. Appl. Physiol. 2014, 116, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, M.C.; Mooij, H.L.; Nieuwdorp, M.; van Lith, B.; Marck, R.; Vink, H.; Kastelein, J.J.P.; Stroes, E.S.G. Partial recovery of the endothelial glycocalyx upon rosuvastatin therapy in patients with heterozygous familial hypercholesterolemia. J. Lipid Res. 2009, 50, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Drake-Holland, A.J.; Noble, M.I. The important new drug target in cardiovascular medicine—The vascular glycocalyx. Cardiovasc. Hematol. Disord. Drug Targets 2009, 9, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; O’Neil, G.L.; Harding, I.C.; Cheng, M.J.; Mensah, S.A.; Ebong, E.E. Glycocalyx in atherosclerosis-relevant endothelium function and as a therapeutic target. Curr. Atheroscler. Rep. 2017, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Vlahu, C.A.; Lemkes, B.A.; Struijk, D.G.; Koopman, M.G.; Krediet, R.T.; Vink, H. Damage of the endothelial glycocalyx in dialysis patients. J. Am. Soc. Nephrol. 2012, 23, 1900–1908. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Marinou, M.; Vlastos, D.; Kourea, K.; Andreadou, I.; Liarakos, N.; Triantafyllidi, H.; Pavlidis, G.; Tsougos, E.; Parissis, J.; et al. Effects of varenicline and nicotine replacement therapy on arterial elasticity, endothelial glycocalyx and oxidative stress during a 3-month smoking cessation program. Atherosclerosis 2017, 262, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Schiefer, J.; Lebherz-Eichinger, D.; Erdoes, G.; Berlakovich, G.; Bacher, A.; Krenn, C.G.; Faybik, P. Alterations of endothelial glycocalyx during orthotopic liver transplantation in patients with end-stage liver disease. Transplantation 2015, 99, 2118–2123. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Savery, M.D.; Gassmann, M.; Baum, O.; Damiano, E.R.; Pries, A.R. Excessive erythrocytosis compromises the blood-endothelium interface in erythropoietin-overexpressing mice. J. Physiol. (Lond.) 2011, 589, 5181–5192. [Google Scholar] [CrossRef] [PubMed]
- Nijst, P.; Cops, J.; Martens, P.; Swennen, Q.; Dupont, M.; Tang, W.H.W.; Mullens, W. Endovascular shedding markers in patients with heart failure with reduced ejection fraction: Results from a single-center exploratory study. Microcirculation 2018, 25. [Google Scholar] [CrossRef] [PubMed]
- Martens, R.J.H.; Vink, H.; van Oostenbrugge, R.J.; Staals, J. Sublingual microvascularglycocalyx dimensions in lacunar stroke patients. Cerebrovasc. Dis. 2013, 35, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Hattori, K.; Suzuki, T.; Matsuda, N. Recent advances in the pathophysiology and molecular basis of sepsis-associated organ dysfunction: Novel therapeutic implications and challenges. Pharmacol. Ther. 2017, 177, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Baker, D.J.; Tachibana, M.; Liu, C.-C.; van Deursen, J.M.; Brott, T.G.; Bu, G.; Kanekiyo, T. Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke 2016, 47, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Dias, H.K.I.; Brown, C.L.R.; Polidori, M.C.; Lip, G.Y.H.; Griffiths, H.R. LDL-lipids from patients with hypercholesterolaemia and Alzheimer’s disease are inflammatory to microvascular endothelial cells: Mitigation by statin intervention. Clin. Sci. 2015, 129, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Li, X.; Zhang, Y.; Gulbins, E.; Zhang, Y. Enhancement of endothelial permeability by free fatty acid through lysosomalcathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget 2016, 7, 73229–73241. [Google Scholar] [PubMed]
- Haidari, M.; Zhang, W.; Willerson, J.T.; Dixon, R.A. Disruption of endothelial adherens junctions by high glucose is mediated by protein kinase C-β-dependent vascular endothelial cadherin tyrosine phosphorylation. Cardiovasc. Diabetol. 2014, 13, 105. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, N.M.; Casselbrant, A.; Joshi, M.; Johansson, B.R.; Sumitran-Holgersson, S. Antibodies to kidney endothelial cells contribute to a “leaky” glomerular barrier in patients with chronic kidney diseases. Am. J. Physiol. Ren. Physiol. 2012, 302, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Gottlieb, E.; Rounds, S. Effects of cigarette smoke on pulmonary endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Polverino, F.; Celli, B.R.; Owen, C.A. COPD as an endothelial disorder: Endothelial injury linking lesions in the lungs and other organs? (2017 Grover Conference Series). Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, C.; Ridley, A.J. Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 2017, 358, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Romanov, Y.A.; Chervontseva, A.M.; Savchenko, V.G.; Smirnov, V.N. Vascular endothelium: Target or victim of cytostatic therapy? Can. J. Physiol. Pharmacol. 2007, 85, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Guipaud, O.; Jaillet, C.; Clément-Colmou, K.; François, A.; Supiot, S.; Milliat, F. The importance of the vascular endothelial barrier in the immune-inflammatory response induced by radiotherapy. Br. J. Radiol. 2018, 91, 20170762. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Endothelial barrier and its abnormalities in cardiovascular disease. Front. Physiol. 2015, 6, 365. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, R.; Mateu, X.; Maseda, E.; Yébenes, J.C.; Aldecoa, C.; De Haro, C.; Ruiz-Rodriguez, J.C.; Garnacho-Montero, J. Non-oncotic properties of albumin. A multidisciplinary vision about the implications for critically ill patients. Expert Rev. Clin. Pharmacol. 2018, 11, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Tarbell, J.M.; Cancel, L.M. The glycocalyx and its significance in human medicine. J. Intern. Med. 2016, 280, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Gane, J.; Stockley, R. Mechanisms of neutrophil transmigration across the vascular endothelium in COPD. Thorax 2012, 67, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.D.; Heffernan, D.S.; Cioffi, W.G.; Reichner, J.S. Neutrophils from critically ill septic patients mediate profound loss of endothelial barrier integrity. Crit. Care 2013, 17, 226. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Nishimura, N.; Rana, K.; Peloquin, J.M.; Califano, J.P.; Montague, C.R.; King, M.R.; Schaffer, C.B.; Reinhart-King, C.A. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Pereira, A.M.; Seiça, R. Endothelial dysfunction—A major mediator of diabetic vascular disease. Biochim. Biophys. Acta 2013, 1832, 2216–2231. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Colgan, S.P.; Shelley, C.S. Hypoxia: The force that drives chronic kidney disease. Clin. Med. Res. 2016, 14, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Favero, G.; Paganelli, C.; Buffoli, B.; Rodella, L.F.; Rezzani, R. Endothelium and its alterations in cardiovascular diseases: Life style intervention. Biomed. Res. Int. 2014, 2014, 801896. [Google Scholar] [CrossRef] [PubMed]
- Wadkin, J.C.R.; Patten, D.A.; Kamarajah, S.K.; Shepherd, E.L.; Novitskaya, V.; Berditchevski, F.; Adams, D.H.; Weston, C.J.; Shetty, S. CD151 supports VCAM-1-mediated lymphocyte adhesion to liver endothelium and is upregulated in chronic liver disease and hepatocellular carcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Gavin, J.B.; Maxwell, L.; Edgar, S.G. Microvascular involvement in cardiac pathology. J. Mol. Cell. Cardiol. 1998, 30, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Frijns, C.J.M.; Kappelle, L.J. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke 2002, 33, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Fortin, C.F.; McDonald, P.P.; Fülöp, T.; Lesur, O. Sepsis, leukocytes, and nitric oxide (NO): An intricate affair. Shock 2010, 33, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, E.E.; Andrews, R.K. Neutrophil extracellular traps (NETs) and infection-related vascular dysfunction. Blood Rev. 2012, 26, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Crouser, E.D.; Matthay, M.A. Endothelial damage during septic shock: Significance and implications for future therapies. Chest 2017, 152, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.C.; Abbas, M.; Khemais-Benkhiat, S.; Burban, M.; Ribeiro, T.P.; Toti, F.; Idris-Khodja, N.; Côrtes, S.F.; Schini-Kerth, V.B. Replicative senescence promotes prothrombotic responses in endothelial cells: Role of NADPH oxidase- and cyclooxygenase-derived oxidative stress. Exp. Gerontol. 2017, 93, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P. Endothelial dysfunction and hypertension. Hypertension 2014, 64, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.; Menke, J.; Sollinger, D.; Schinzel, H.; Thürmel, K. Haemostasis in chronic kidney disease. Nephrol. Dial. Transpl. 2014, 29, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Levi, M. Cancer-related coagulopathies. Thromb. Res. 2014, 133, 70–75. [Google Scholar] [CrossRef]
- Remková, A.; Remko, M. Homocysteine and endothelial markers are increased in patients with chronic liver diseases. Eur. J. Intern. Med. 2009, 20, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Besedina, A. NO-synthase activity in patients with coronary heart disease associated with hypertension of different age groups. J. Med. Biochem. 2016, 35, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, S.; Marlborough, F.; Doubal, F.; Webb, D.J.; Wardlaw, J. Blood markers of coagulation, fibrinolysis, endothelial dysfunction and inflammation in lacunar stroke versus non-lacunar stroke and non-stroke: Systematic review and meta-analysis. Cerebrovasc. Dis. 2014, 37, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-T.; Su, C.-M.; Chen, C.T.; Cheng, H.-H.; Chang, M.-W.; Hung, C.-W.; Hung, S.-C.; Chang, W.-N.; Tsai, N.-W.; Wang, H.-C.; et al. Circulating endothelial progenitor cells may predict outcomes in adult patients with severe sepsis in the emergency department. Clin. Chim. Acta 2016, 455, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Williamson, K.A.; Hamilton, A.; Reynolds, J.A.; Sipos, P.; Crocker, I.; Stringer, S.E.; Alexander, Y.M. Age-related impairment of endothelial progenitor cell migration correlates with structural alterations of heparan sulfate proteoglycans. Aging Cell 2013, 12, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.; Touyz, R.M. Cellular biomarkers of endothelial health: Microparticles, endothelial progenitor cells, and circulating endothelial cells. J. Am. Soc. Hypertens. 2012, 6, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Paschalaki, K.E.; Starke, R.D.; Hu, Y.; Mercado, N.; Margariti, A.; Gorgoulis, V.G.; Randi, A.M.; Barnes, P.J. Dysfunction of endothelial progenitor cells from smokers and chronic obstructive pulmonary disease patients due to increased DNA damage and senescence. Stem Cells 2013, 31, 2813–2826. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, K.; Mund, J.; Case, J.; Gupta, S.; Liu, Z.; Gathirua-Mwangi, W.; McDaniel, A.; Renbarger, J.; Champion, V. Differences in circulating endothelial progenitor cells among childhood cancer survivors treated with and without radiation. J. Hematol. Thromb. 2015, 1. [Google Scholar] [CrossRef]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. (Oxf.) 2018, 222. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta 2008, 1778, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.E.; Shapiro, N.I.; Griffin, R.; Safford, M.M.; Judd, S.; Howard, G. Inflammatory and endothelial activation biomarkers and risk of sepsis: A nested case-control study. J. Crit. Care 2013, 28, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.E.; Shapiro, N.I.; Griffin, R.; Safford, M.M.; Judd, S.; Howard, G. Chronic medical conditions and risk of sepsis. PLoS ONE 2012, 7, e48307. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, P.; Yano, K.; Sorasaki, M.; Ngo, L.; St Hilaire, M.; Lucas, J.M.; Aird, W.; Shapiro, N.I. Influence of diabetes on endothelial cell response during sepsis. Diabetologia 2011, 54, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Kern, H.; Wittich, R.; Rohr, U.; Kox, W.J.; Spies, C.D. Increased endothelial injury in septic patients with coronary artery disease. Chest 2001, 119, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Wiewel, M.A.; Harmon, M.B.; van Vught, L.A.; Scicluna, B.P.; Hoogendijk, A.J.; Horn, J.; Zwinderman, A.H.; Cremer, O.L.; Bonten, M.J.; Schultz, M.J.; et al. Risk factors, host response and outcome of hypothermic sepsis. Crit. Care 2016, 20, 328. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Leelahavanichkul, A.; Hu, X.; Sidransky, K.L.; Zhou, H.; Qin, Y.; Eisner, C.; Schnermann, J.; Yuen, P.S.T.; Star, R.A. Pre-existing renal disease promotes sepsis-induced acute kidney injury and worsens outcome. Kidney Int. 2008, 74, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, A.; Wu, Y. Endothelial glycocalyx layer: A possible therapeutic target for acute lung injury during lung resection. Biomed. Res. Int. 2017, 2017, 5969657. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Bruegger, D.; Potzel, J.; Jacob, M.; Brettner, F.; Vogeser, M.; Conzen, P.; Becker, B.F.; Rehm, M. Hypervolemia increases release of atrial natriuretic peptide and shedding of the endothelial glycocalyx. Crit. Care 2014, 18, 538. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Rodriguez, E.G.; Chang, R.; Holcomb, J.B.; Kao, L.S.; Wade, C.E. PROPPR Study Group elevated syndecan-1 after trauma and risk of sepsis: A secondary analysis of patients from the Pragmatic, Randomized Optimal Platelet and Plasma Ratios (PROPPR) trial. J. Am. Coll. Surg. 2018. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Mastora, Z.; Orfanos, S.E.; Jahaj, E.; Maniatis, N.A.; Koutsoukou, A.; Armaganidis, A.; Kotanidou, A. Elevated biomarkers of endothelial dysfunction/activation at ICU admission are associated with sepsis development. Cytokine 2014, 69, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Kotanidou, A.; Mastora, Z.; Maniatis, N.A.; Albani, P.; Jahaj, E.; Koutsoukou, A.; Armaganidis, A.; Orfanos, S.E. Elevated soluble endothelial protein C receptor levels at ICU admission are associated with sepsis development. Minerva Anestesiol. 2015, 81, 125–134. [Google Scholar] [PubMed]
- Ikegami, K.; Suzuki, Y.; Yukioka, T.; Matsuda, H.; Shimazaki, S. Endothelial cell injury, as quantified by the soluble thrombomodulin level, predicts sepsis/multiple organ dysfunction syndrome after blunt trauma. J. Trauma 1998, 44, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Xing, K.; Murthy, S.; Liles, W.C.; Singh, J.M. Clinical utility of biomarkers of endothelial activation in sepsis—A systematic review. Crit. Care 2012, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Walczak, M.; Suraj, J.; Kus, K.; Kij, A.; Zakrzewska, A.; Chlopicki, S. Towards a comprehensive endothelial biomarkers profiling and endothelium-guided pharmacotherapy. Pharmacol. Rep. 2015, 67, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Paulus, P.; Jennewein, C.; Zacharowski, K. Biomarkers of endothelial dysfunction: Can they help us deciphering systemic inflammation and sepsis? Biomarkers 2011, 16, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; van der Poll, T. Endothelial barrier dysfunction in septic shock. J. Intern. Med. 2015, 277, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.P.; Overdier, K.H.; Sun, X.; Lin, L.; Liu, X.; Yang, Y.; Ammons, L.A.; Hiller, T.D.; Suflita, M.A.; Yu, Y.; et al. Urinary glycosaminoglycans predict outcomes in septic shock and acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2016, 194, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.-Y.; Kung, C.-T.; Tsai, N.-W.; Su, C.-M.; Huang, C.-C.; Lai, Y.-R.; Wang, H.-C.; Cheng, B.-C.; Su, Y.-J.; Lin, W.-C.; et al. Concentration and value of endocan on outcome in adult patients after severe sepsis. Clin. Chim. Acta 2018, 483, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Andaluz-Ojeda, D.; Nguyen, H.B.; Meunier-Beillard, N.; Cicuéndez, R.; Quenot, J.-P.; Calvo, D.; Dargent, A.; Zarca, E.; Andrés, C.; Nogales, L.; et al. Superior accuracy of mid-regional proadrenomedullin for mortality prediction in sepsis with varying levels of illness severity. Ann. Intensive Care 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B., Jr.; Hassan, U.; Seymour, C.; Angus, D.C.; Isbell, T.S; White, K.; Weir, W.; Yeh, L.; Vincent, A.; Bashir, R. Point-of-care sensors for the management of sepsis. Nat. Biomed. Eng. 2018, 2, 640–648. [Google Scholar] [CrossRef]
- Darwish, I.; Liles, W.C. Emerging therapeutic strategies to prevent infection-related microvascular endothelial activation and dysfunction. Virulence 2013, 4, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Marik, P.E.; Khangoora, V.; Rivera, R.; Hooper, M.H.; Catravas, J. Hydrocortisone, vitamin c, and thiamine for the treatment of severe sepsis and septic shock: A retrospective before-after study. Chest 2017, 151, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N.; Khangoora, V.; Marik, P.E.; Catravas, J.D. Hydrocortisone and ascorbic acid synergistically prevent and repair lipopolysaccharide-induced pulmonary endothelial barrier dysfunction. Chest 2017, 152, 954–962. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Rhee et al. (n = 173,690) | Donnelly et al. (n = 1080) | ||
|---|---|---|---|
| Age (mean in years) | 66.5 | Age (mean) | 69.7 |
| Sex (male) | 57.6% | Sex (male) | 59.2% |
| Diabetes | 35.7% | Hypertension | 74.5% |
| Chronic pulmonary disease | 30.9% | Dyslipidaemia | 67.3% |
| Renal disease | 26.8% | Diabetes | 41.8% |
| Congestive heart failure | 25.4% | Chronic kidney disease | 31.5% |
| Cancer | 19.7% | Myocardial infarction | 24.4% |
| Dementia or cerebrovascular disease | 10.3% | Chronic lung disease | 17.4% |
| Liver disease | 10% | Stroke | 12.6% |
| Features of ED | Sepsis | Aging/Chronic Disease |
|---|---|---|
| Increased oxidative stress and systemic inflammation | [3,8,9,10] | [11,12,13,14,15,16] |
| Glycocalyx degradation and shedding | [2,3,17,18] | [19,20,21,22,23,24,25,26,27,28,29] |
| Disassembly of intercellular junctions, endothelial cell death, blood-tissue barrier disruption | [2,3,9,18,30,31] | [11,32,33,34,35,36,37,38,39,40,41,42,43,44] |
| Enhanced leukocyte adhesion and extravasation | [3,18,45,46] | [14,15,23,41,47,48,49,50,51,52,53] |
| Induction of a pro-coagulant and anti-fibrinolytic state | [3,8,17,30,54,55,56] | [12,15,16,23,41,48,57,58,59,60,61,62,63] |
| Impairment in the mechanisms of endothelial repair | [64] | [65,66,67,68] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bermejo-Martin, J.F.; Martín-Fernandez, M.; López-Mestanza, C.; Duque, P.; Almansa, R. Shared Features of Endothelial Dysfunction between Sepsis and Its Preceding Risk Factors (Aging and Chronic Disease). J. Clin. Med. 2018, 7, 400. https://doi.org/10.3390/jcm7110400
Bermejo-Martin JF, Martín-Fernandez M, López-Mestanza C, Duque P, Almansa R. Shared Features of Endothelial Dysfunction between Sepsis and Its Preceding Risk Factors (Aging and Chronic Disease). Journal of Clinical Medicine. 2018; 7(11):400. https://doi.org/10.3390/jcm7110400
Chicago/Turabian StyleBermejo-Martin, Jesus F., Marta Martín-Fernandez, Cristina López-Mestanza, Patricia Duque, and Raquel Almansa. 2018. "Shared Features of Endothelial Dysfunction between Sepsis and Its Preceding Risk Factors (Aging and Chronic Disease)" Journal of Clinical Medicine 7, no. 11: 400. https://doi.org/10.3390/jcm7110400
APA StyleBermejo-Martin, J. F., Martín-Fernandez, M., López-Mestanza, C., Duque, P., & Almansa, R. (2018). Shared Features of Endothelial Dysfunction between Sepsis and Its Preceding Risk Factors (Aging and Chronic Disease). Journal of Clinical Medicine, 7(11), 400. https://doi.org/10.3390/jcm7110400