PLP1 Mutations in Patients with Multiple Sclerosis: Identification of a New Mutation and Potential Pathogenicity of the Mutations


Abstract
:1. Introduction
2. Experimental Methods
2.1. Patients and Controls
2.2. DNA Extraction and PLP1 Sequencing
2.3. PLP1-Containing Vectors
2.4. Transfection of Cos-7 Cells
2.5. Immunocytochemical Analysis of Transfected Cells
2.6. Statistical Analyses
2.7. HLA Binding Predictions
3. Results
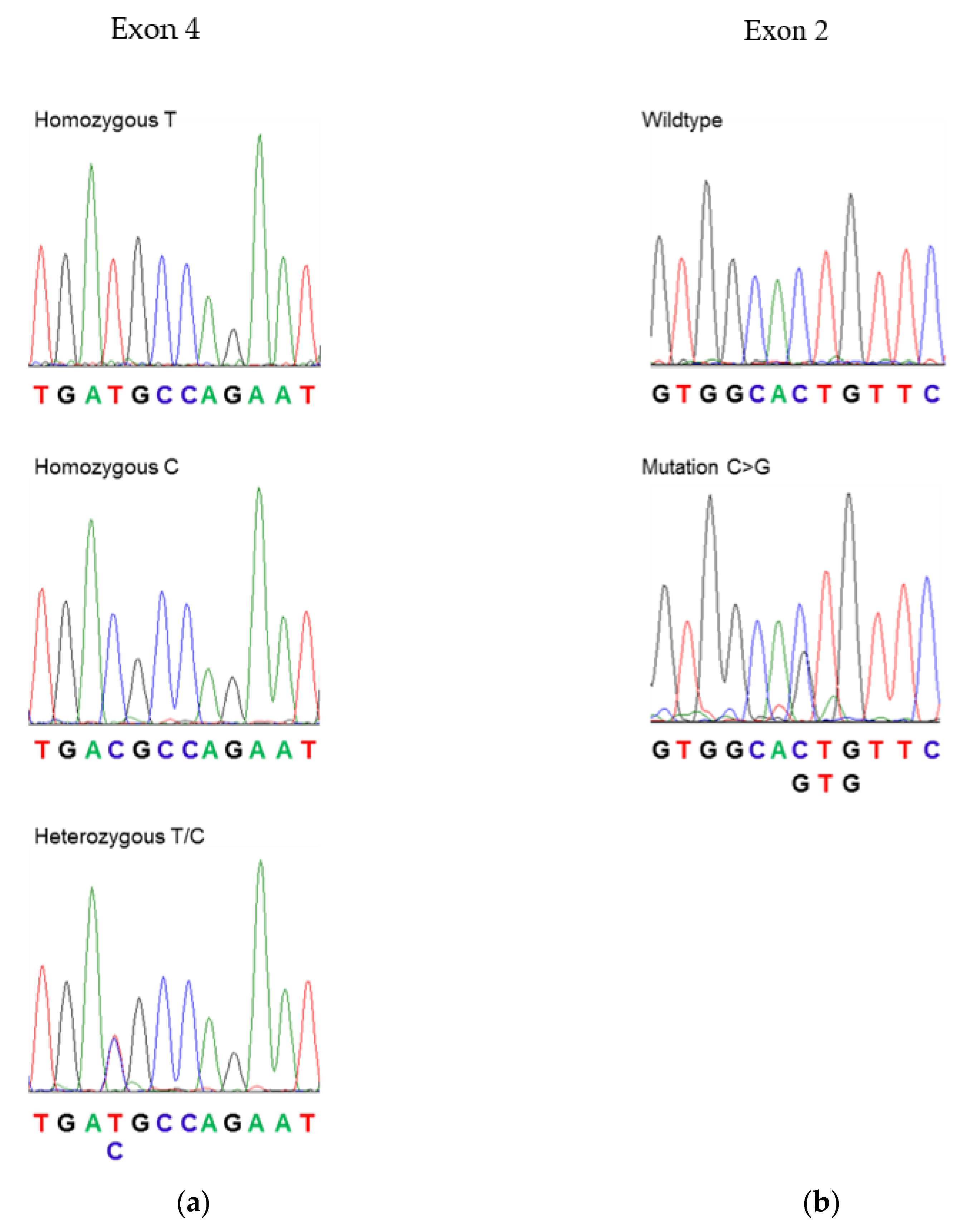
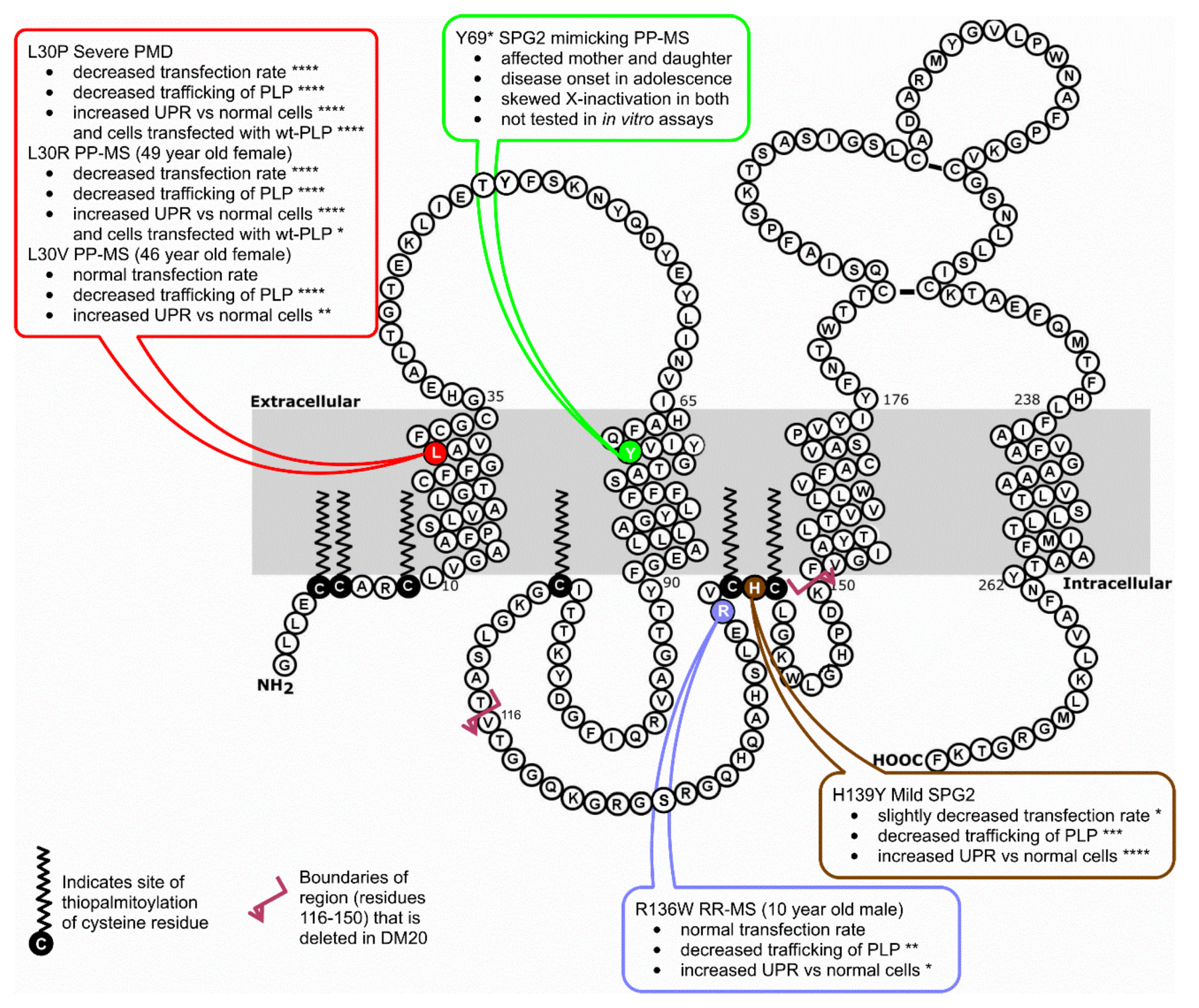
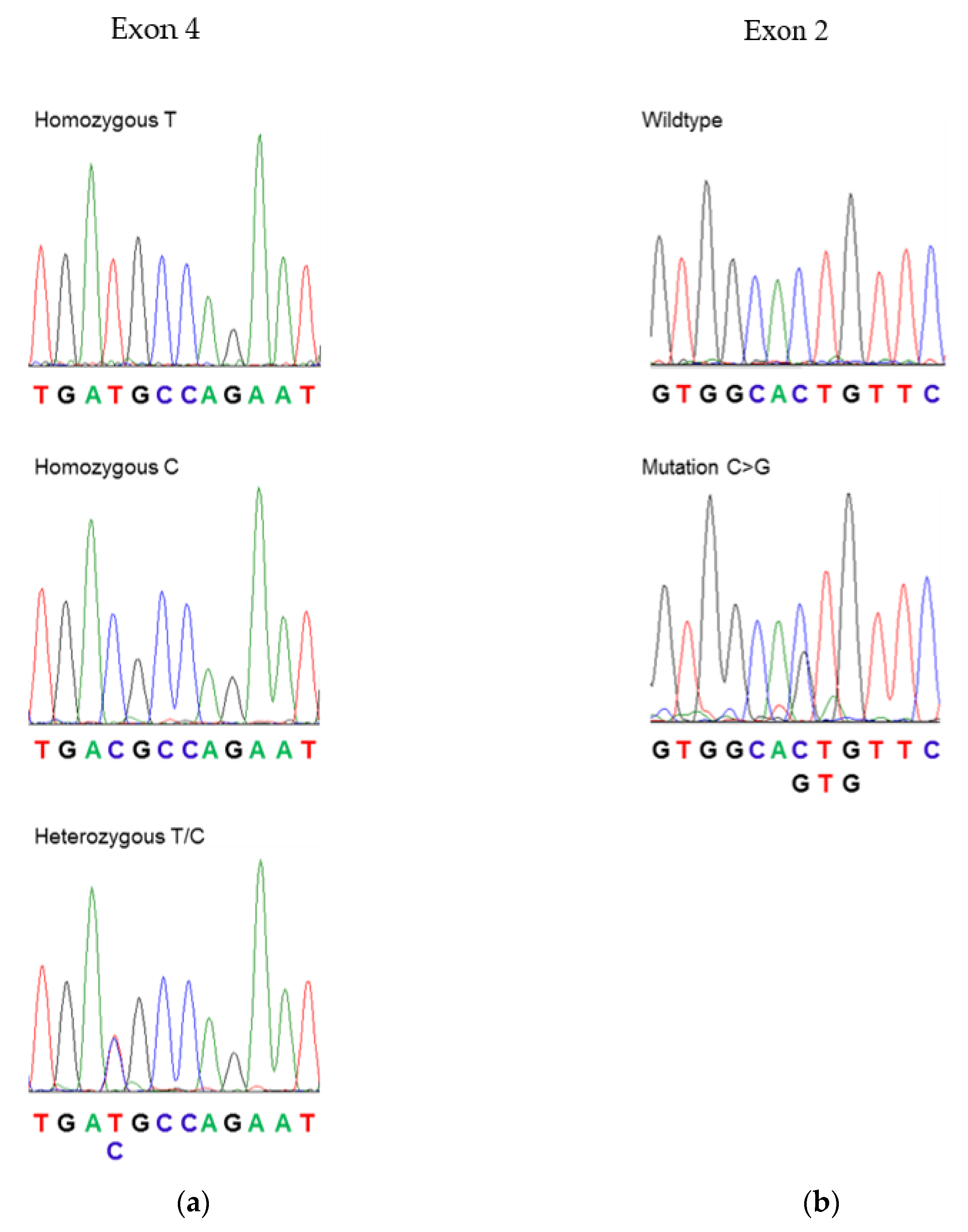
3.1. Sequencing of PLP1 in Human Subjects
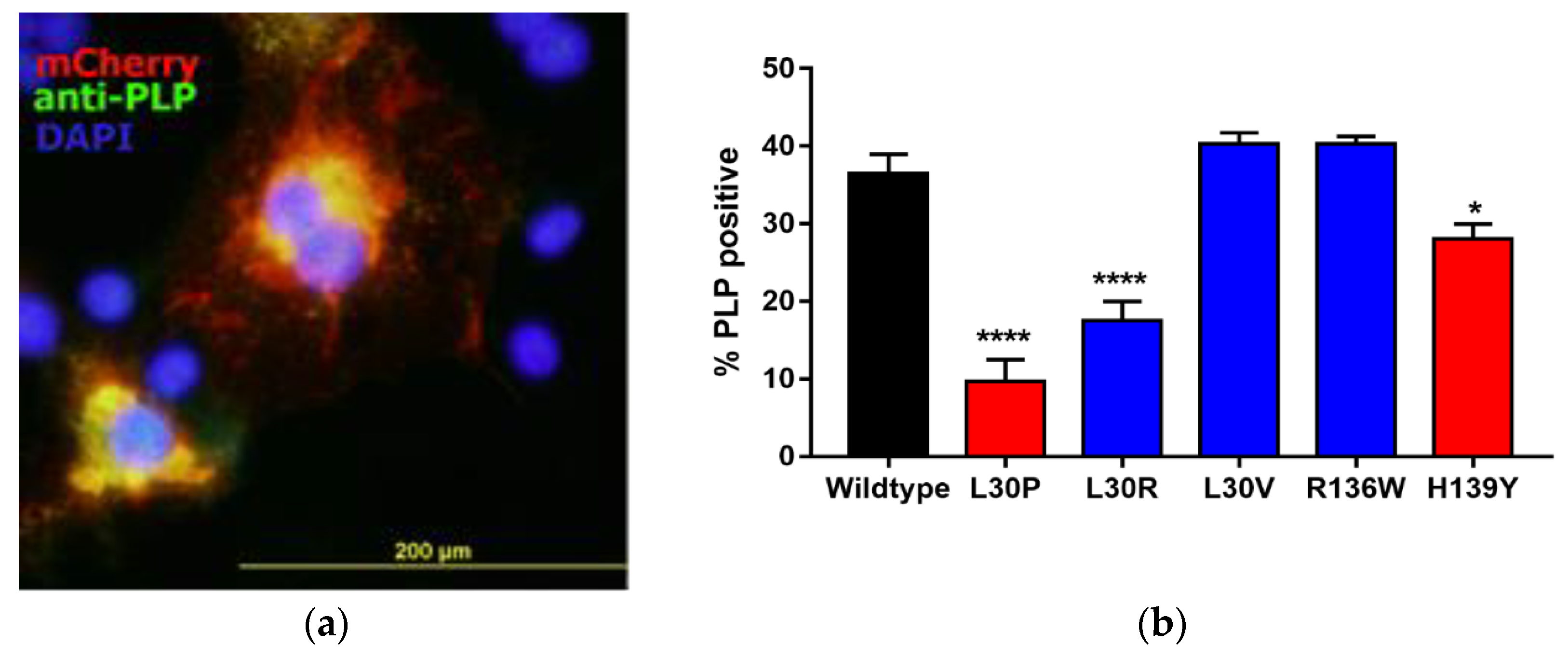
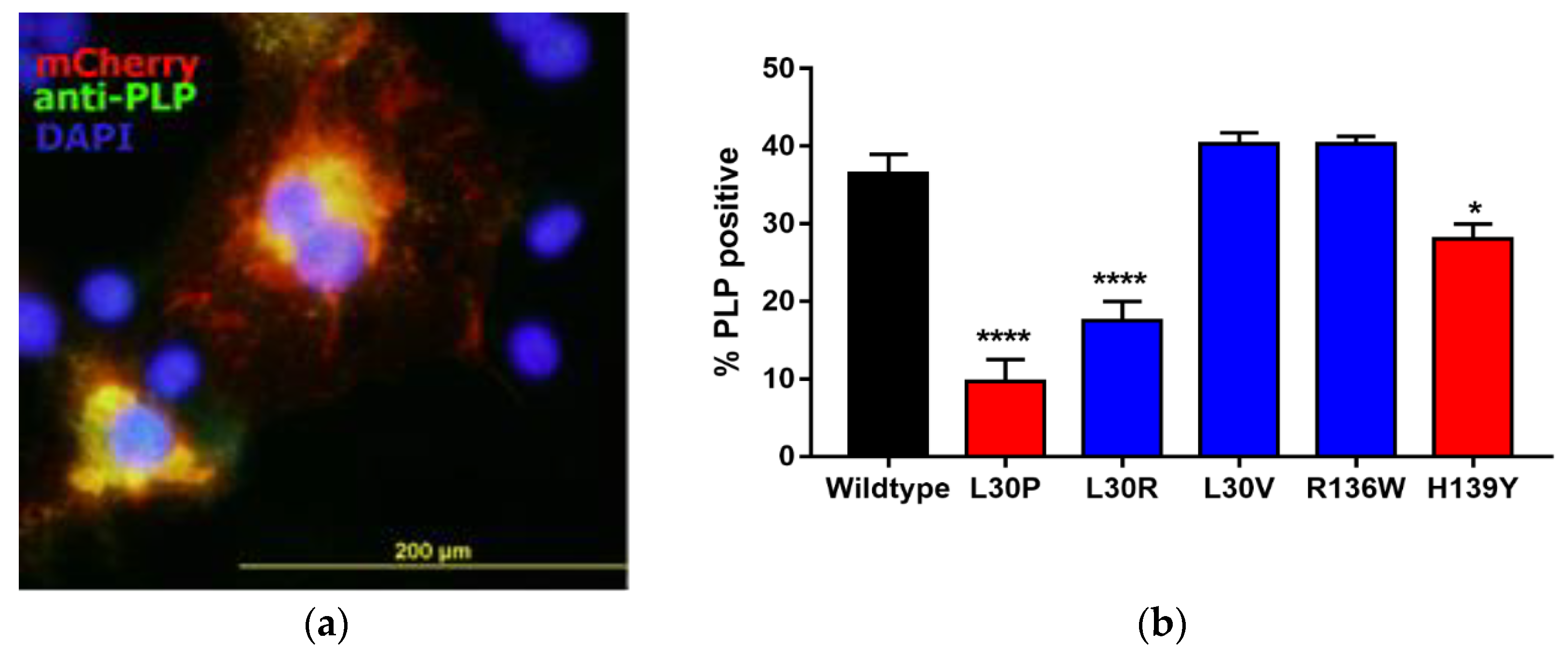
3.2. Effects of Point Mutations in PLP1 on PLP Expression in Cos-7 Cells
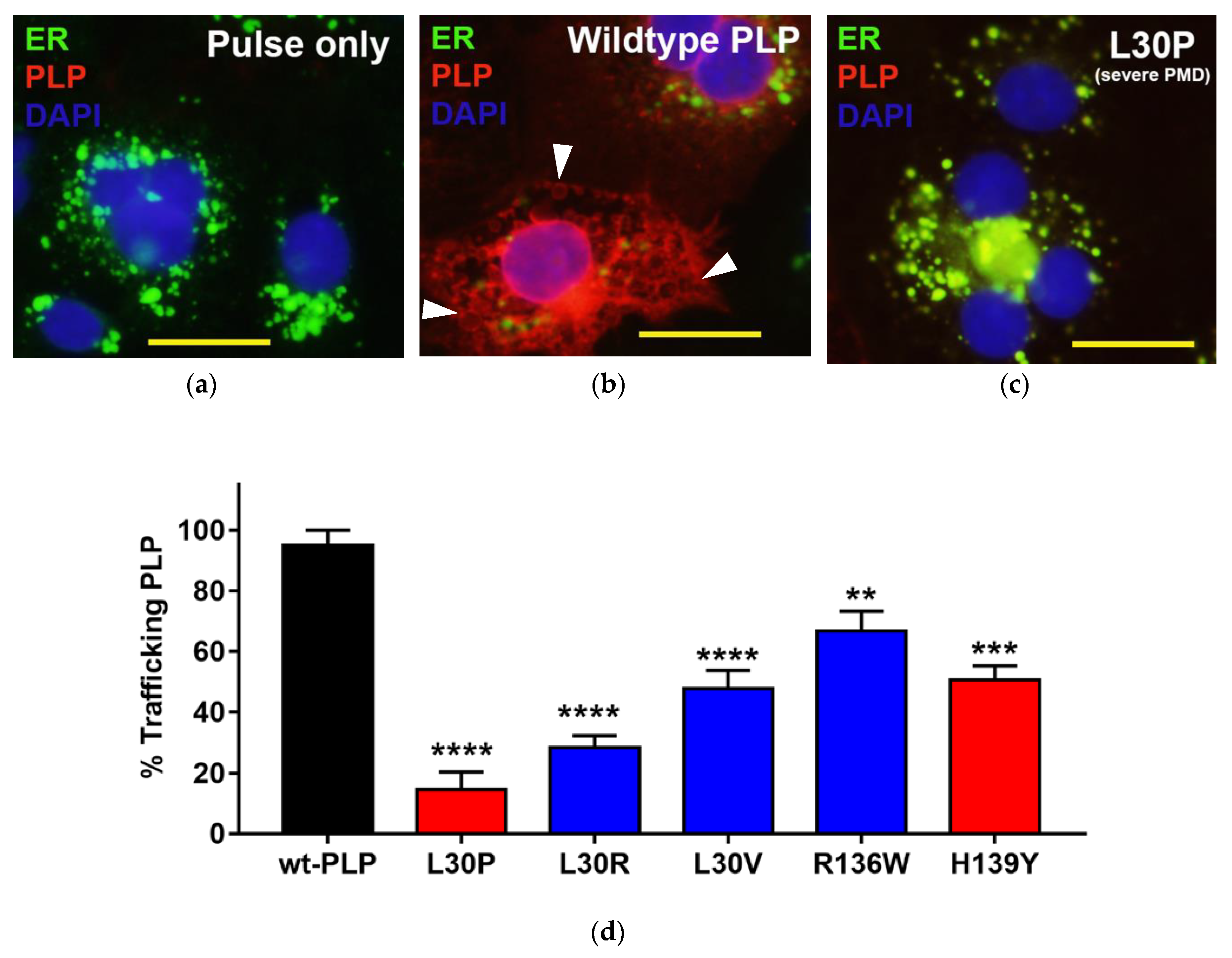
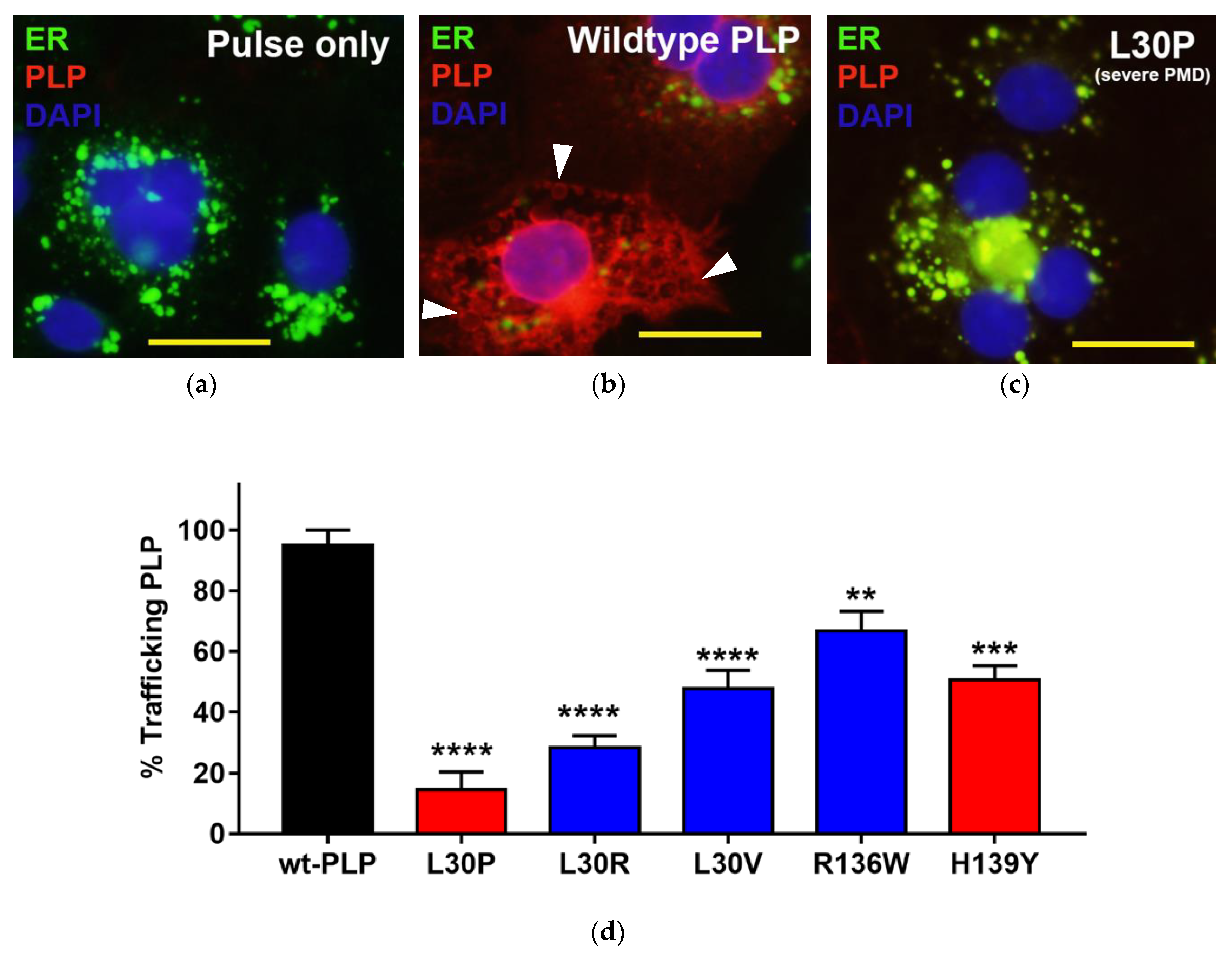
3.3. Effects of Point Mutations in PLP1 on the Trafficking Pattern of PLP in Cos-7 Cells
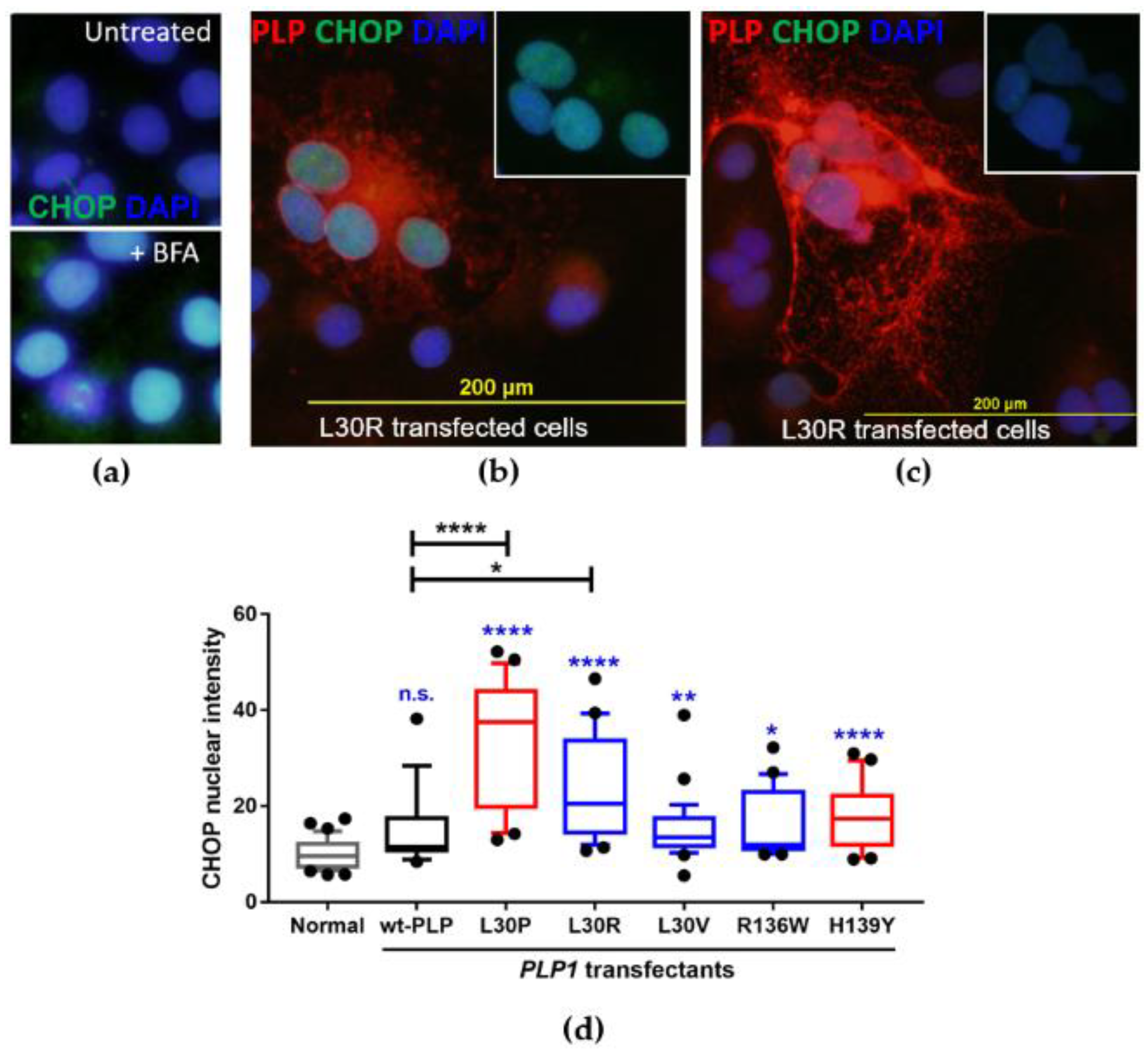
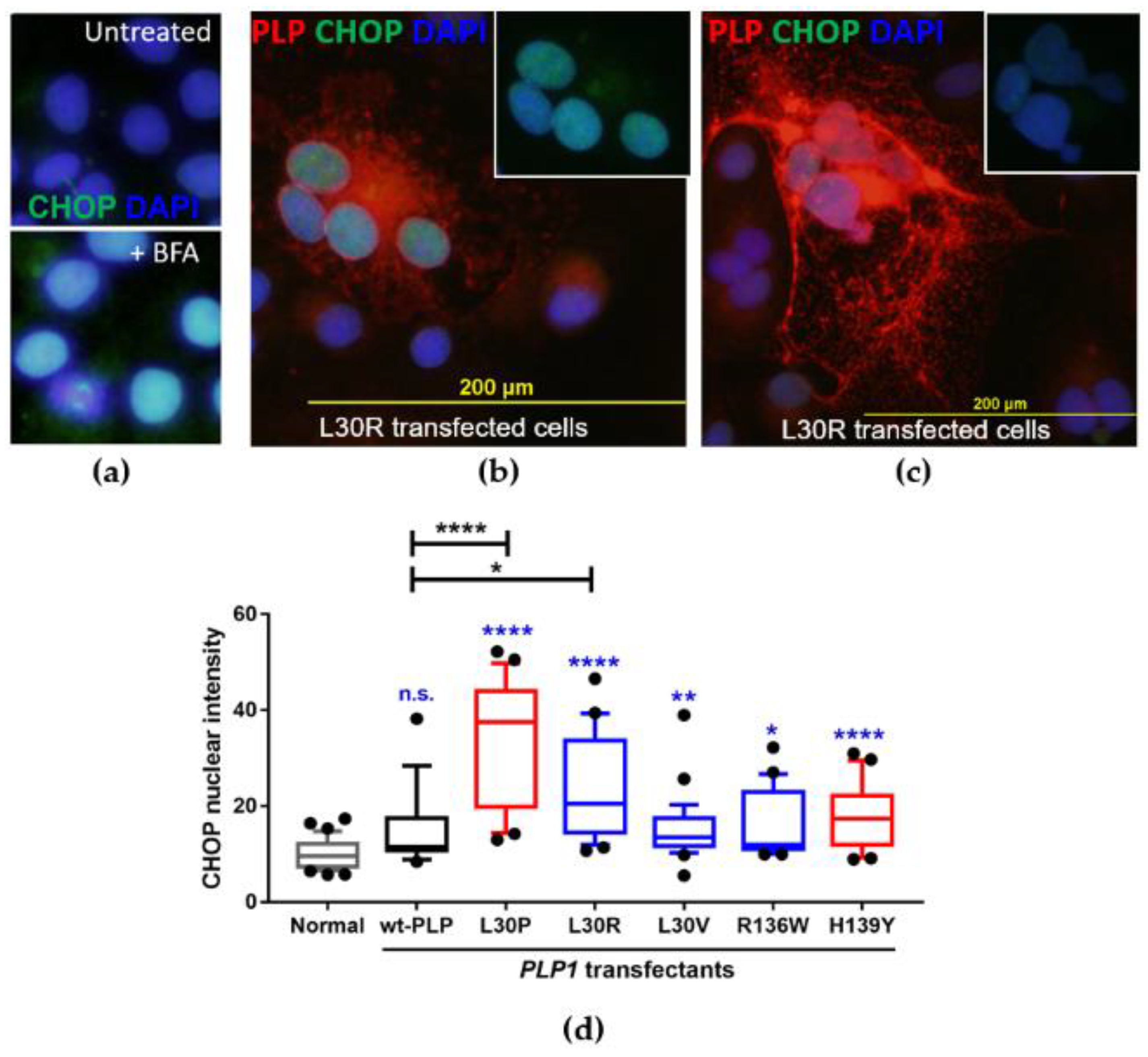
3.4. Investigation of the Induction of the Unfolded Protein Response in PLP1-Transfected Cos-7 Cells
3.5. L30V Mutation Could Potentially Change Immune Responsiveness to PLP
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Garbern, J.Y. Pelizaeus-Merzbacher disease: Genetic and cellular pathogenesis. Cell. Mol. Life Sci. 2007, 64, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Gorman, M.P.; Golomb, M.R.; Walsh, L.E.; Hobson, G.M.; Garbern, J.Y.; Kinkel, R.P.; Darras, B.T.; Urion, D.K.; Eksioglu, Y.Z. Steroid-responsive neurologic relapses in a child with a proteolipid protein-1 mutation. Neurology 2007, 68, 1305–1307. [Google Scholar] [CrossRef] [PubMed]
- Warshawsky, I.; Rudick, R.A.; Staugaitis, S.M.; Natowicz, M.R. Primary progressive multiple sclerosis as a phenotype of a PLP1 gene mutation. Ann. Neurol. 2005, 58, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Groh, J.; Friedman, H.C.; Orel, N.; Ip, C.W.; Fischer, S.; Spahn, I.; Schäffner, E.; Hörner, M.; Stadler, D.; Buttmann, M.; et al. Pathogenic inflammation in the CNS of mice carrying human PLP1 mutations. Hum. Mol. Genet. 2016, 25, 4686–4702. [Google Scholar] [PubMed]
- Grinspan, J.B.; Coulalaglou, M.; Beesley, J.S.; Carpio, D.F.; Scherer, S.S. Maturation-dependent apoptotic cell death of oligodendrocytes in myelin-deficient rats. J. Neurosci. Res. 1998, 54, 623–634. [Google Scholar] [CrossRef]
- Cerghet, M.; Bessert, D.A.; Nave, K.A.; Skoff, R.P. Differential expression of apoptotic markers in jimpy and in Plp overexpressors: Evidence for different apoptotic pathways. J. Neurocytol. 2001, 30, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.; Friedrich, V.L., Jr.; Lazzarini, R.A. Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J. Neurosci. Res. 1994, 37, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.; Friedrich., V.L., Jr.; Lazzarini, R.A. Intracellular transport and sorting of the oligodendrocyte transmembrane proteolipid protein. J. Neurosci. Res. 1994, 37, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.; Southwood, C.M.; Lazzarini, R.A. Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J. Cell Biol. 1998, 140, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Dhaunchak, A.S.; Nave, K.A. A common mechanism of PLP/DM20 misfolding causes cysteine-mediated endoplasmic reticulum retention in oligodendrocytes and Pelizaeus-Merzbacher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 17813–17818. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Harding, H.P.; Ron, D.; Popko, B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J. Cell Biol. 2005, 169, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Parratt, J.D. Oligodendrocytes and the early multiple sclerosis lesion. Ann. Neurol. 2012, 72, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, G.P.; Pedersen, J.; Klingenberg, O.; Lygren, I.; Orstavik, K.H. Increased skewing of X chromosome inactivation with age in both blood and buccal cells. Cytogenet. Genome Res. 2007, 116. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Knudsen, G.P.; Bathum, L.; Naumova, A.K.; Sorensen, T.I.; Brix, T.H.; Svendsen, A.J.; Christensen, K.; Kyvik, K.O.; Ørstavik, K.H. Twin study of genetic and aging effects on X chromosome inactivation. Eur. J. Hum. Genet. 2005, 13, 599–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Confavreux, C.; Vukusic, S. The clinical course of multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 343–369. [Google Scholar] [PubMed]
- Kurtzke, J.F. Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 1983, 33, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Sette, A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 2005, 6, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Sidney, J.; Dow, C.; Mothe, B.; Sette, A.; Peters, B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 2008, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Sidney, J.; Kim, Y.; Sette, A.; Lund, O.; Nielsen, M.; Bjoern, P. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010, 11, 568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Gow, A. The Cos-7 cell in vitro paradigm to study myelin proteolipid protein 1 gene mutations. Methods Mol. Biol. 2003, 217, 263–275. [Google Scholar] [PubMed]
- Cailloux, F.; Gauthier-Barichard, F.; Mimault, C.; Isabelle, V.; Courtois, V.; Dastugue, B.; Boespflug-Tanguy, O. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Eur. J. Human. Genet. 2000, 8, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Saugier-Veber, P.; Munnich, A.; Bonneau, D.; Rozet, J.M.; Le Merrer, M.; Gil, R.; Boespflug-Tanguy, O. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat. Genet. 1994, 6, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Southwood, C.M.; Garbern, J.; Jiang, W.; Gow, A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron 2002, 36, 585–596. [Google Scholar] [CrossRef]
- Gow, A.; Wrabetz, L. CHOP and the endoplasmic reticulum stress response in myelinating glia. Curr. Opin. Neurobiol. 2009, 19, 505–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samali, A.; Fitzgerald, U.; Deegan, S.; Gupta, S. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int. J. Cell Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Stys, P.K.; Zamponi, G.W.; van Minnen, J.; Geurts, J.J.G. Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 2012, 13, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodes, M.E.; Zimmerman, A.W.; Aydanian, A.; Naidu, S.; Miller, N.R.; Garcia Oller, J.L.; Barker, B.; Aleck, K.A.; Hurley, T.D.; Dlouhy, S.R. Different mutations in the same codon of the proteolipid protein gene, PLP, may help in correlating genotype with phenotype in Pelizaeus-Merzbacher disease/X-linked spastic paraplegia (PMD/SPG2). Am. J. Med. Genet. 1999, 82, 132–139. [Google Scholar] [CrossRef]
- Cazzola, M.; May, A.; Bergamaschi, G.; Cerani, P.; Rosti, V.; Bishop, D.F. Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked sideroblastic anemia in carrier females. Blood 2000, 96, 4363–4365. [Google Scholar] [PubMed]
- Knudsen, G.P.; Harbo, H.F.; Smestad, C.; Celius, E.G.; Akesson, E.; Oturai, A.; Ryder, L.P.; Spurkland, A.; Ørstavik, K.H. X chromosome inactivation in females with multiple sclerosis. Eur. J. Neurol. 2007, 14, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Rubegni, A.; Battisti, C.; Tessa, A.; Cerase, A.; Doccini, S.; Santorelli, F.M.; Federico, A. SPG2 mimicking multiple sclerosis in a family identified using next generation sequencing. J. Neurol. Sci. 2017, 375, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Pham-Dinh, D.; Birling, M.C.; Roussel, G.; Dautigny, A.; Nussbaum, J.L. Proteolipid DM-20 predominates over PLP in peripheral nervous system. Neuroreport 1991, 2, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Dimova, N.; Sperle, K.; Huang, Z.; Lock, L.; McCulloch, M.C.; Edgar, J.M.; Hobson, G.M.; Cambi, F. Deletion of a splicing enhancer disrupts PLP1/DM20 ratio and myelin stability. Exp. Neurol. 2008, 214, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Dimova, N.; Cambi, F. PLP/DM20 ratio is regulated by hnRNPH and F and a novel G-rich enhancer in oligodendrocytes. Nucleic Acids Res. 2007, 35, 4164–4178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadon, N.L.; Arnheiter, H.; Hudson, L.D. A combination of PLP and DM20 transgenes promotes partial myelination in the jimpy mouse. J. Neurochem. 1994, 63, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.; Lazzarini, R.A. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat. Genet. 1996, 13, 422–428. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Number | Mean Age (Years ± SE) | Mean MS Duration (Years ± SE) | EDSS 1 |
|---|---|---|---|---|
| Healthy controls | 42 | 37.8 ± 1.8 | Not applicable | Not applicable |
| PP-MS | 22 | 53.0 ± 2.7 | 7.8 ± 1.5 | 6.3 ± 0.3 |
| Group | Polymorphism | |||
|---|---|---|---|---|
| Exon 4–D202D | Other Exons | |||
| T/T | C/T | C/C | ||
| PP-MS (n = 22) | 12 (54.5%) | 7 (31.8%) | 3 (13.6%) | 1 (L30V) het 1 |
| Healthy (n = 42) | 24 (57.0%) | 16 (38.1%) | 2 (4.8%) | None |
| Designation | Mutation | Description | |
|---|---|---|---|
| Codon | Amino Acid Change | ||
| Wildtype | None | None | Wildtype PLP sequence |
| L30P | 89T > C | Leucine to proline at position 30 | Severe PMD mutation [24] |
| L30R | 89T > G | Leucine to arginine at position 30 | MS-like disease mutation [3] |
| L30V | 88C > G | Leucine to valine at position 30 | MS-like disease (this study) |
| R136W | 406C > T | Arginine to tryptophan at position 136 | MS-like disease mutation [2] |
| H139Y | 415C > T | Histidine to tyrosine at position 139 | Mild SPG2 mutation [25] 1 |
| Starting Amino Acid | Peptide | DRB1*07:01 | DRB1*13:02 | A*02 | A*29 | B*44 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| L | V | L | V | L | V | L | V | L | V | ||
| 22 | GLCFFGVA(L/V) | 0.6 | – 1 | 8.7 | 5.8 | 0.4 | 0.2 | 9.2 | 8.5 | – | – |
| 23 | LCFFGVA(L/V)F | 0.5 | 0.5 | 15.7 | 14.4 | 33.2 | 9.2 | 1.4 | 6.3 | 12.4 | 18.4 |
| 24 | CFFGVA(L/V)FC | – | – | 12.9 | 11.8 | 17.6 | 7.2 | 0.5 | 1.8 | 15.7 | 24.7 |
| 25 | FFGVA(L/V)FCG | – | 0.9 | – | – | 14.7 | 14.3 | 6.8 | 7.6 | – | – |
| 26 | FGVA(L/V)FCGC | – | – | – | – | 21.7 | 10.2 | – | – | – | – |
| 27 | GVA(L/V)FCGCG | – | – | – | – | – | 24.7 | 14.5 | 15.4 | – | – |
| 28 | VA(L/V)FCGCGH | 1.8 | 3.7 | – | – | – | 29.5 | 11.1 | 14.9 | – | – |
| 29 | A(L/V)FCGCGHE | – | – | – | – | 16.7 | 26.8 | 15.2 | 11.2 | – | – |
| 30 | (L/V)FCGCGHEA | – | – | 10.5 | 7.8 | 7.7 | 19.9 | 3.2 | 3.4 | – | – |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cloake, N.C.; Yan, J.; Aminian, A.; Pender, M.P.; Greer, J.M. PLP1 Mutations in Patients with Multiple Sclerosis: Identification of a New Mutation and Potential Pathogenicity of the Mutations. J. Clin. Med. 2018, 7, 342. https://doi.org/10.3390/jcm7100342
Cloake NC, Yan J, Aminian A, Pender MP, Greer JM. PLP1 Mutations in Patients with Multiple Sclerosis: Identification of a New Mutation and Potential Pathogenicity of the Mutations. Journal of Clinical Medicine. 2018; 7(10):342. https://doi.org/10.3390/jcm7100342
Chicago/Turabian StyleCloake, Nancy C., Jun Yan, Atefeh Aminian, Michael P. Pender, and Judith M. Greer. 2018. "PLP1 Mutations in Patients with Multiple Sclerosis: Identification of a New Mutation and Potential Pathogenicity of the Mutations" Journal of Clinical Medicine 7, no. 10: 342. https://doi.org/10.3390/jcm7100342
APA StyleCloake, N. C., Yan, J., Aminian, A., Pender, M. P., & Greer, J. M. (2018). PLP1 Mutations in Patients with Multiple Sclerosis: Identification of a New Mutation and Potential Pathogenicity of the Mutations. Journal of Clinical Medicine, 7(10), 342. https://doi.org/10.3390/jcm7100342