
IL-23 and Th17 Disease in Inflammatory Arthritis

 ,
, {kind=link}
{kind=link}
Abstract
:1. IL-23 and Th17 cells
2. IL-23 and Inflammatory Arthritis
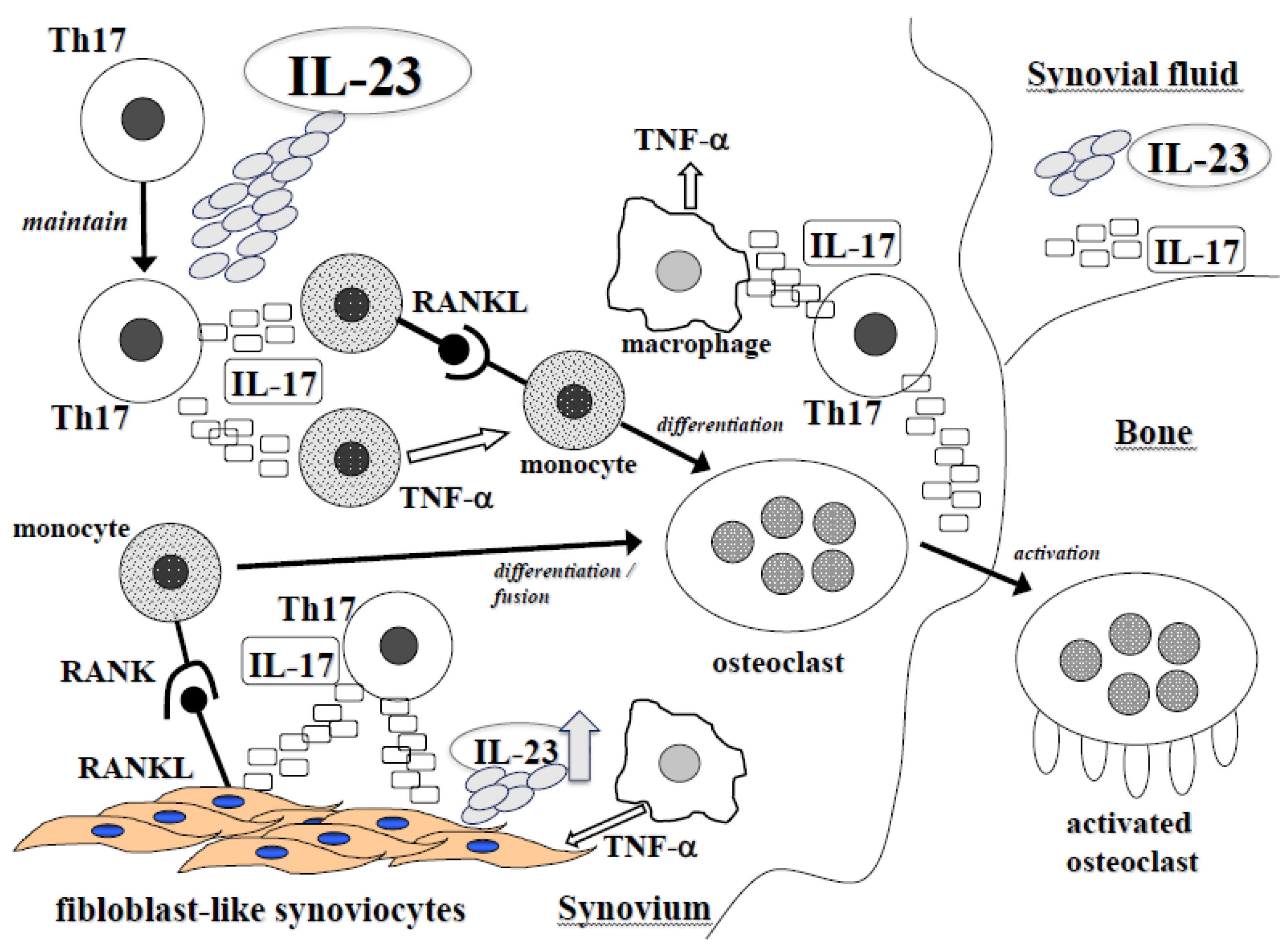
2.1. IL-23 and Rheumatoid Arthritis
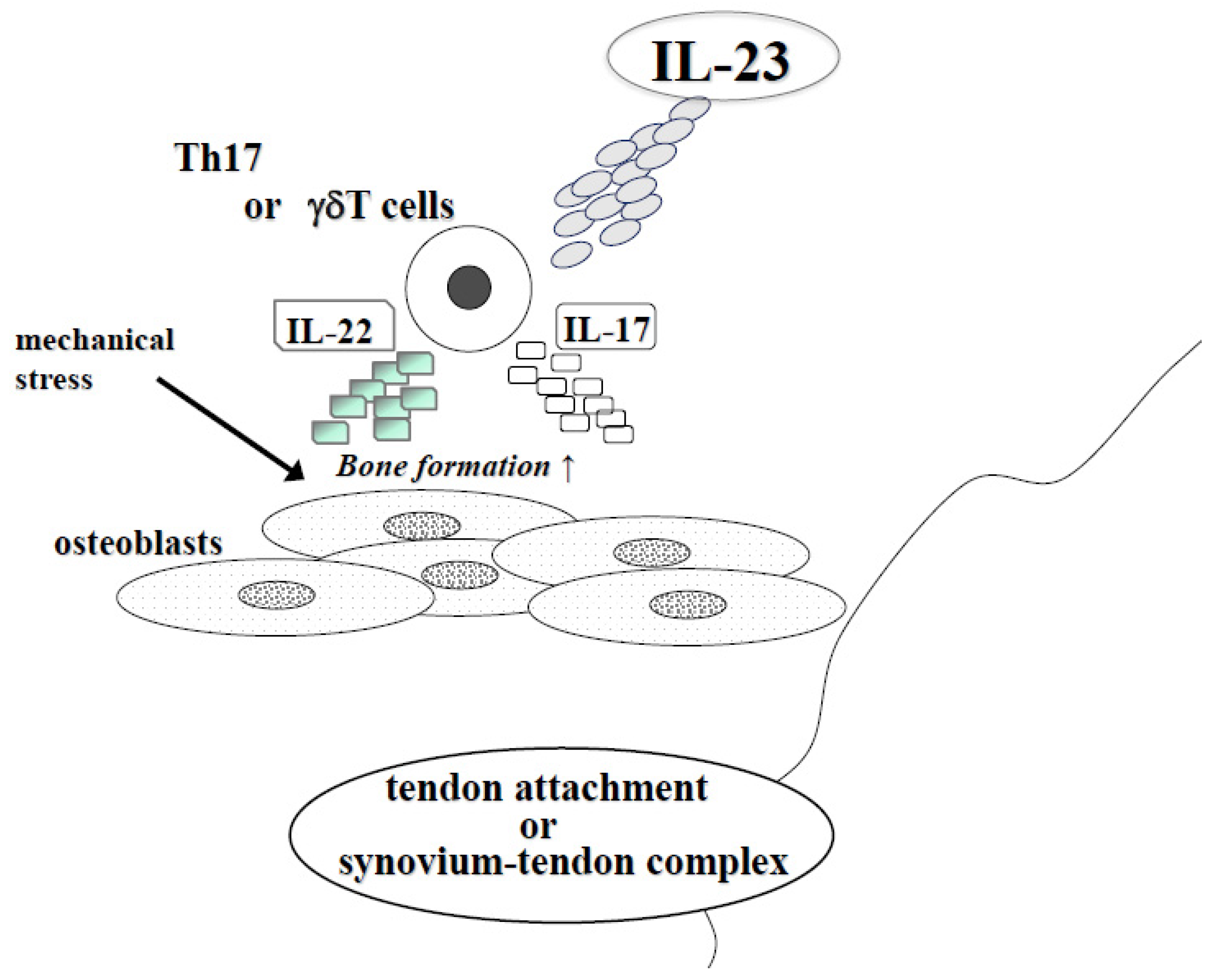
2.2. IL-23 and Spondylarthritis (AS and PsA)
3. Conclusions
Author Contributions
Conflicts of Interest
References
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Ghilardi, N.; Xie, M.H.; de Sauvage, F.J.; Gurney, A.L. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Vanden Eijnden, S.; Goriely, S.; De Wit, D.; Willems, F.; Goldman, M. IL-23 up-regulates IL-10 and induces IL-17 synthesis by polyclonally activated naive T cells in human. Eur. J. Immunol. 2005, 35, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Brombacher, F.; Kastelein, R.A.; Alber, G. Novel IL-12 family members shed light on the orchestration of Th1 responses. Trends Immunol. 2003, 24, 207–212. [Google Scholar] [CrossRef]
- Lankford, C.S.; Frucht, D.M. A unique role for IL-23 in promoting cellular immunity. J. Leukoc. Biol. 2003, 73, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Wiekowski, M.T.; Leach, M.W.; Evans, E.W.; Sullivan, L.; Chen, S.C.; Vassileva, G.; Bazan, J.F.; Gorman, D.M.; Kastelein, R.A.; Narula, S.; et al. Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. J. Immunol. 2001, 166, 7563–7570. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Filì, L.; Ferri, S.; Frosali, F.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tato, C.M.; Muul, L.; Laurence, A.; O’Shea, J.J. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007, 56, 2936–2946. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Chen, Y.; Kanno, Y.; Joyce-Shaikh, B.; Vahedi, G.; Hirahara, K.; Blumenschein, W.M.; Sukumar, S.; Haines, C.J.; Sadekova, S.; et al. Interleukin-23-Induced Transcription Factor Blimp-1 Promotes Pathogenicity of T Helper 17 Cells. Immunity 2016, 44, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Louten, J.; Boniface, K.; de Waal Malefyt, R. Development and function of TH17 cells in health and disease. J. Allergy Clin. Immunol. 2009, 123, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, D.V.; Di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar] [PubMed]
- Rifas, L.; Avioli, L.V. A novel T cell cytokine stimulates interleukin-6 in human osteoblastic cells. J. Bone Miner. Res. 1999, 14, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Yago, T.; Nanke, Y.; Ichikawa, N.; Kobashigawa, T.; Mogi, M.; Kamatani, N.; Kotake, S. IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-alpha antibody: A novel mechanism of osteoclastogenesis by IL-17. J. Cell. Biochem. 2009, 108, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Raza, K.; Falciani, F.; Curnow, S.J.; Ross, E.J.; Lee, C.Y.; Akbar, A.N.; Lord, J.M.; Gordon, C.; Buckley, C.D.; Salmon, M. Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res. Ther. 2005, 7, R784–R795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yago, T.; Nanke, Y.; Kawamoto, M.; Furuya, T.; Kobashigawa, T.; Kamatani, N.; Kotake, S. IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis Res. Ther. 2007, 9, R96. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Nanke, Y.; Mogi, M.; Kawamoto, M.; Furuya, T.; Yago, T.; Kobashigawa, T.; Togari, A.; Kamatani, N. IFN-γ-producing human T cells directly induce osteoclastogenesis from human monocytes via the expression of RANKL. Eur. J. Immunol. 2005, 35, 3353–3363. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, F.; Asmawidjaja, P.S.; Mus, A.M.; Corneth, O.; Kikly, K.; Lubberts, E. IL-23 dependent and independent stages of experimental arthritis: No clinical effect of therapeutic IL-23p19 inhibition in collagen-induced arthritis. PLoS ONE 2013, 8, e57553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.R.; Cho, M.L.; Kim, K.W.; Juhn, J.Y.; Hwang, S.Y.; Yoon, C.H.; Park, S.H.; Lee, S.H.; Kim, H.Y. Up-regulation of IL-23p19 expression in rheumatoid arthritis synovial fibroblasts by IL-17 through PI3-kinase-, NF-kappaB- and p38 MAPK-dependent signalling pathways. Rheumatology 2007, 46, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.L.; Chen, C.H.; Chu, S.J.; Chen, J.H.; Lai, J.H.; Sytwu, H.K.; Chang, D.M. Interleukin (IL)-23 p19 expression induced by IL-1beta in human fibroblast-like synoviocytes with rheumatoid arthritis via active nuclear factor-kappaB and AP-1 dependent pathway. Rheumatology 2007, 46, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.; Nadiv, O.; Luknar-Gabor, N.; Agar, G.; Beer, Y.; Katz, Y. Synergism between tumor necrosis factor alpha and interleukin-17 to induce IL-23 p19 expression in fibroblast-like synoviocytes. Mol. Immunol. 2009, 46, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Wendling, D.; Abbas, W.; Godfrin-Valnet, M.; Kumar, A.; Guillot, X.; Khan, K.A.; Vidon, C.; Coquard, L.; Toussirot, E.; Prati, C.; et al. Dysregulated serum IL-23 and SIRT1 activity in peripheral blood mononuclear cells of patients with rheumatoid arthritis. PLoS ONE 2015, 10, e0119981. [Google Scholar] [CrossRef] [PubMed]
- Zaky, D.S.; El-Nahrery, E.M. Role of interleukin-23 as a biomarker in rheumatoid arthritis patients and its correlation with disease activity. Int. Immunopharmacol. 2016, 31, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, H.S.; Park, M.K.; Cho, M.L.; Lee, S.H.; Kim, H.Y. The clinical role of IL-23p19 in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2007, 36, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Agarwal, S.K.; Ilivanova, E.; Xu, X.L.; Miao, Y.; Zhuang, Y.; Nnane, I.; Radziszewski, W.; Greenspan, A.; Beutler, A.; et al. A randomised phase II study evaluating the efficacy and safety of subcutaneously administered ustekinumab and guselkumab in patients with active rheumatoid arthritis despite treatment with methotrexate. Ann. Rheum. Dis. 2017, 76, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Keijsers, R.R.; Joosten, I.; van Erp, P.E.; Koenen, H.J.; van de Kerkhof, P.C. Cellular sources of IL-17 in psoriasis: A paradigm shift? Exp. Dermatol. 2014, 23, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.P.; Raychaudhuri, S.K. Mechanistic rationales for targeting interleukin-17A in spondyloarthritis. Arthritis Res. Ther. 2017, 19, 51. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.K.; Saxena, A.; Raychaudhuri, S.P. Role of IL-17 in the pathogenesis of psoriatic arthritis and axial spondyloarthritis. Clin. Rheumatol. 2015, 34, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Australo-Anglo-American Spondyloarthritis Consortium (TASC); Reveille, J.D.; Sims, A.M.; Danoy, P.; Evans, D.M.; Leo, P.; Pointon, J.J.; Jin, R.; Zhou, X.; Bradbury, L.A.; et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat. Genet. 2010, 42, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.I.; Liu, Y.; Danoy, P.A.; Wu, X.; Thomas, G.P.; Jiang, L.; Sun, L.; Wang, N.; Han, J.; Han, H.; et al. Association of STAT3 and TNFRSF1A with ankylosing spondylitis in Han Chinese. Ann. Rheum. Dis. 2011, 70, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Pan, F.; Gao, J.; Ge, R.; Duan, Z.; Zeng, Z.; Liao, F.; Xia, G.; Wang, S.; Xu, S.; et al. Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin. Rheumatol. 2011, 30, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Appel, H.; Maier, R.; Wu, P.; Scheer, R.; Hempfing, A.; Kayser, R.; Thiel, A.; Radbruch, A.; Loddenkemper, C.; Sieper, J. Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res. Ther. 2011, 13, R95. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4-CD8- entheseal resident T cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Sukhov, A.; Adamopoulos, I.E.; Maverakis, E. Interactions of the Immune System with Skin and Bone Tissue in Psoriatic Arthritis: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 51, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Mellins, E.D.; Gershwin, M.E.; Nestle, F.O.; Adamopoulos, I.E. The IL-23/IL-17 axis in psoriatic arthritis. Autoimmun. Rev. 2014, 13, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.P.; Raychaudhuri, S.K.; Genovese, M.C. IL-17 receptor and its functional significance in psoriatic arthritis. Mol. Cell. Biochem. 2012, 359, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Celis, R.; Planell, N.; Fernández-Sueiro, J.L.; Sanmartí, R.; Ramírez, J.; González-Álvaro, I.; Pablos, J.L.; Cañete, J.D. Synovial cytokine expression in psoriatic arthritis and associations with lymphoid neogenesis and clinical features. Arthritis Res. Ther. 2012, 14, R93. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Raychaudhuri, S.K.; Raychaudhuri, S.P. Functional role of IL-22 in psoriatic arthritis. Arthritis Res. Ther. 2012, 14, R65. [Google Scholar] [CrossRef] [PubMed]
- Jadon, D.R.; Nightingale, A.L.; McHugh, N.J.; Lindsay, M.A.; Korendowych, E.; Sengupta, R. Serum soluble bone turnover biomarkers in psoriatic arthritis and psoriatic spondyloarthropathy. J. Rheumatol. 2015, 42, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Prajzlerova, K.; Grobelna, K.; Pavelka, K.; Senolt, L.; Filkova, M. An update on biomarkers in axial spondyloarthritis. Autoimmun. Rev. 2016, 15, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Brandt, J.; Listing, J.; Zink, A.; Alten, R.; Golder, W.; Gromnica-Ihle, E.; Kellner, H.; Krause, A.; Schneider, M.; et al. Treatment of active ankylosing spondylitis with infliximab: A randomised controlled multicentre trial. Lancet 2002, 359, 1187–1193. [Google Scholar] [CrossRef]
- Baeten, D.; Sieper, J.; Braun, J.; Baraliakos, X.; Dougados, M.; Emery, P.; Deodhar, A.; Porter, B.; Martin, R.; Andersson, M.; et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N. Engl. J. Med. 2015, 373, 2534–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poddubnyy, D.; Hermann, K.G.; Callhoff, J.; Listing, J.; Sieper, J. Ustekinumab for the treatment of patients with active ankylosing spondylitis: Results of a 28-week, prospective, open-label, proof-of-concept study (TOPAS). Ann. Rheum. Dis. 2014, 73, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; McInnes, I.B. Secukinumab: A New Treatment Option for Psoriatic Arthritis. Rheumatol. Ther. 2016, 3, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; van der Heijde, D.; Ritchlin, C.T.; Okada, M.; Cuchacovich, R.S.; Shuler, C.L.; Lin, C.Y.; Braun, D.K.; Lee, C.H.; Gladman, D.D.; et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: Results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann. Rheum. Dis. 2017, 76, 79–87. [Google Scholar] [PubMed]
- Kavanaugh, A.; Puig, L.; Gottlieb, A.B.; Ritchlin, C.; You, Y.; Li, S.; Song, M.; Randazzo, B.; Rahman, P.; McInnes, I.B. Efficacy and safety of ustekinumab in psoriatic arthritis patients with peripheral arthritis and physician-reported spondylitis: Post-hoc analyses from two phase III, multicentre, double-blind, placebo-controlled studies (PSUMMIT-1/PSUMMIT-2). Ann. Rheum. Dis. 2016, 75, 1984–1988. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Armstrong, A.W.; Foley, P.; Song, M.; Wasfi, Y.; Randazzo, B.; Li, S.; Shen, Y.K.; Gordon, K.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J. Am. Acad. Dermatol. 2017, 76, 418–431. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yago, T.; Nanke, Y.; Kawamoto, M.; Kobashigawa, T.; Yamanaka, H.; Kotake, S. IL-23 and Th17 Disease in Inflammatory Arthritis. J. Clin. Med. 2017, 6, 81. https://doi.org/10.3390/jcm6090081
Yago T, Nanke Y, Kawamoto M, Kobashigawa T, Yamanaka H, Kotake S. IL-23 and Th17 Disease in Inflammatory Arthritis. Journal of Clinical Medicine. 2017; 6(9):81. https://doi.org/10.3390/jcm6090081
Chicago/Turabian StyleYago, Toru, Yuki Nanke, Manabu Kawamoto, Tsuyoshi Kobashigawa, Hisashi Yamanaka, and Shigeru Kotake. 2017. "IL-23 and Th17 Disease in Inflammatory Arthritis" Journal of Clinical Medicine 6, no. 9: 81. https://doi.org/10.3390/jcm6090081
APA StyleYago, T., Nanke, Y., Kawamoto, M., Kobashigawa, T., Yamanaka, H., & Kotake, S. (2017). IL-23 and Th17 Disease in Inflammatory Arthritis. Journal of Clinical Medicine, 6(9), 81. https://doi.org/10.3390/jcm6090081