
Minimal Residual Disease in Acute Myeloid Leukemia: Still a Work in Progress?

Abstract
:
1. Introduction
2. Has MRD Evaluation Superseded Baseline Risk Assessment and Disease Staging?
3. Molecular Biology Techniques
3.1. RUNX1/RUNX1T1 and CBFB/MYH11
3.2. NPM1
3.3. WT1
3.4. Other Molecular Markers
4. Multiparameter Flow Cytometry
5. Combined Approaches
6. Issues in the Implementation of MRD Assessment as the New Standard to Evaluate Clinical Response
6.1. Inter-Laboratory Standardization
6.2. Choice of Methodology
6.3. Best Source for MRD and Sample Quality
6.4. Significance of MRD Evaluation as a Decision-Making Tool
6.5. Safety and Efficacy of Treatment Alternatives
6.6. Effectiveness of allo-HSCT in Eradicating MRD
6.7. Best Time-Points for MRD Evaluation
7. “Real-Life” Use of MRD as a Decision-Making Tool in 2017
8. Some Final Considerations about Future Developments
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rowe, J.M. AML in 2016: Where are we now? Best Pract. Res. Clin. Haematol. 2016, 29, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 217 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.R.; Tallman, M.S.; Abboud, C.N.; Altman, J.K.; Appelbaum, F.R.; Arber, D.A.; Attar, E.; Borate, U.; Coutre, S.E.; Damon, L.E.; et al. Acute myeloid leukemia. Version 2.2013. J. Natl. Compr. Cancer Netw. 2013, 11, 1047–1055. [Google Scholar] [CrossRef]
- Grimwade, D.; Hills, R.K. Independent prognostic factors for AML outcome. Hematology 2009, 2009, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Freeman, S.D. Defining minimal residual disease in acute myeloid leukemia: Which platforms are ready for “prime-time”? Hematology 2014, 2014, 222–233. [Google Scholar]
- Ossenkoppele, G.J.; Schuurhuis, G.J. MRD in AML: Does it already guide therapy decision-making? Hematology 2016, 2016, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Hourigan, C.S.; Gale, R.P.; Gormley, N.J.; Ossenkoppele, G.J.; Walter, R.B. Measurable residual disease testing in acute myeloid leukemia. Leukemia 2017. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.J.; Gratwohl, A.; Schlenk, R.F.; Sierra, J.; Bornhauser, M.; Juliusson, G.; Racil, Z.; Rowe, J.M.; Russel, N.; Mohty, M.; et al. The European LeukemiaNet AMl Working Party consensus statement on allogeneic HSCT for patients with AML in remission: An integrated-risk adapted approach. Nat. Rev. Clin. Oncol. 2012, 9, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Koreth, J.; Schlenk, R.; Kopecky, K.J.; Honda, S.; Sierra, J.; Djulbegovic, B.J.; Wadleigh, M.; DeAngelo, D.J.; Stone, R.M.; Sakamaki, H.; et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: Systematic review and meta-analysis of prospective clinical trials. JAMA 2009, 301, 2349–2361. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.J.; Blaise, D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood 2016, 127, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.J.; Van Putten, W.L.; Verdonck, L.F.; Theobald, M.; Jacky, E.; Daenen, S.M.; van Marwijk, K.M.; Wijermans, P.; Schouten, H.; Huijgens, P.C.; et al. Results of a HOVON/SAKK donor versus no-donor analysis of myeloablative HLA-identical sibling stem cell transplantation in first remission acute myeloid leukemia in young and middle-aged adults: Benefits for whom? Blood 2007, 109, 3658–3666. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Hills, R.K. Who should be transplanted in first remission of acute myeloid leukemia? Curr. Treat. Options Oncol. 2011, 12, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.; Majeti, R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kahler, A.K.; Handsaker, R.F.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukemic hematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Mosna, F.; Gottardi, M. Stem cell modeling of core binding factor acute myeloid leukemia. Stem Cells Int. 2016, 2016, 7625827. [Google Scholar] [CrossRef] [PubMed]
- Del Principe, M.I.; Buccisano, F.; Maurillo, L.; Sconocchia, G.; Cefalo, M.; Consalvo, M.I.; Sarlo, C.; Conti, C.; De Santis, G.; De Bellis, E.; et al. Minimal residual disease in acute myeloid leukemia of adults: Determination, prognostic impact and clinical applications. Med. J. Hematol. Infect. Dis. 2016, 8, e2016052. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Saglio, G. WT1 as a universal marker for minimal residual disease detection and quantification in myeloid leukemias and in myelodysplastic syndrome. Acta Haematol. 2004, 112, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Stentoft, J.; Hokland, P.; Ostergaard, M.; Hasle, H.; Nyvold, C.G. Minimal residual core binding factor AMLs by real time quantitative PCR: Initial response to chemotherapy predicts event free survival and close monitoring of peripheral blood unravels the kinetics of relapse. Leuk. Res. 2006, 30, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Krauter, J.; Gorlich, K.; Ottmann, O.; Lubbert, M.; Dohner, H.; Heit, W.; Kanz, L.; Ganser, A.; Heil, G. Prognostic value of minimal residual disease quantification by real-time reverse transcriptase polymerase chain reaction in patients with core binding factor leukemias. J. Clin. Oncol. 2003, 21, 4413–4422. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Caligiuri, M.A.; Dohner, H.; Archer, K.J.; Schlenk, R.F.; Dohner, K.; Maghraby, E.A.; Bloomfield, C.D. Quantification of CBFbeta/MYH11 fusion transcript by real-time RT-PCR in patients with inv(16) acute myeloid leukemia. Leukemia 2001, 15, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Mosna, F.; Papayannidis, C.; Martinelli, G.; Di Bona, E.; Bonalumi, A.; Tecchio, C.; Candoni, A.; Capelli, D.; Piccin, A.; Forghieri, F.; et al. Complex karyotype, older age, and reduced first-line dose intensity determine poor survival in core binding factor acute myeloid leukemia patients with long-term follow-up. Am. J. Hematol. 2015, 90, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Schoch, C. Quantitative PCR based minimal residual disease detection in core binding factor leukemias: Prognostication and guiding of therapy. Leuk. Res. 2006, 30, 657–658. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S; Ottaviani, E.; Testoni, N.; Montefusco, V.; Visani, G.; Bonifazi, F.; Amabile, M.; Terragna, C.; Ruggeri, D.; Piccaluga, P.P; et al. Real-time quantitation of minimal residual disease in inv(16)-positive acute myeloid leukemia may indicate risk for clinical relapse and may identify patients in a curable state. Blood 2002, 99, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Corbacioglu, A.; Scholl, C.; Schlenk, R.F.; Eiwen, K.; Du, J.; Bullinger, L.; Frohling, S.; Reimer, P.; Rummel, M; Derigs, H.G.; et al. Prognostic impact of minimal residual disease in CBFB-MYH11-positive acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 3724–3729. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.A.; O’Brien, M.A.; Hills, R.K.; Daly, S.B.; Wheatley, K.; Burnett, A.K. Minimal residual disease monitoring by quantitative RT-PCR in core binding factor AML allows risk stratification and predicts relapse: Results of the United Kingdom MRC AML-15 trial. Blood 2012, 120, 2826–2836. [Google Scholar] [CrossRef] [PubMed]
- Leroy, H.; De Batton, S.; Grardel-Duflos, N.; Darre, S.; Leleu, X.; Roumier, C.; Morschhauser, F.; Lai, J.L.; Bauters, F.; Fenaux, P.; et al. Prognostic value of real-time quantative PCR (RQ-PCR) in AML with t(8;21). Leukemia 2005, 19, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, E.; Boissel, N.; Chevret, S.; Delabesse, E.; Renneville, A.; Cornillet, P.; Blanchet, O.; Cayuela, J.M.; Recher, C.; Raffoux, E.; et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood 2013, 121, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.L.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukaemia who benefit from the addition of Gemtuzumab Ozogamicin: Results of the MRC AML 15 Trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Kell, J.; Freeman, S.; Kjeldsen, L.; Hunter, A.E.; Yin, J.; Craddock, C.F.; Dufva, I.H.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 3924–3931. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.N.; Lewgrand, O.; Thojmas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Borthakur, G.; Cortes, J.E.; Estey, E.E.; Jabbour, E.; Faderl, S.; O’Brien, S.; Garcia-Manero, G.; Kadia, T.M.; Wang, X.; Patel, K.; et al. Gemtuzumab ozogamicin with fludarabine, cytarabine, and granulocyte colony stimulating factor (FLAG-GO) as front-line regimen in patients with core binding factor acute myelogeneous leukemia. Am. J. Hematol. 2014, 89, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Mercer, D.; Hu, X.; Liu, H.; Li, M.M. Common leukemia- and lymphoma-associated genetic aberrations in healthy individuals. J. Mol. Diagn. 2011, 13, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogeneous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Martelli, M.F.; Mecucci, C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc + AML): Biologic and clinical features. Blood 2007, 109, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Bullinger, L.; Teleanu, V.; Tschürtz, F.; Gaidzik, V.I.; Kühn, M.W.; Rücker, F.G.; Holzmann, K.; Paschka, P.; Kapp-Schwörer, S.; et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood 2013, 122, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Dohner, K.; Krauter, J.; Frohling, S.; Corbacioglu, A.; Bullinger, L.; Habdank, M.; Spath, D.; Morgan, M.; Benner, A.; et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N. Engl. J. Med. 2008, 348, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of minimal residual disease in standard-risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Balsat, M.; Renneville, A.; Thomas, X.; De Botton, S.; Caillot, D.; Marceau, A.; Lemasle, E.; Marolleau, J.P.; Nibourel, O.; Berthon, C.; et al. Postinduction minimal residual disease predicts outcome and benefit from allogeneic stem cell transplantation in acute myeloid leukemia with NPM1 mutation: A study by the Acute Leukemia French Association Group. J. Clin. Oncol. 2017, 35, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Kern, W.; Tschulik, C.; Weiss, T.; Dicker, F.; Falini, B.; Haferlach, C.; Haferlach, T. Minimal residual disease levels assessed by NPM1 mutation specific RT-PCR provide important prognostic information in AML. Blood 2009, 114, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Hubmann, M.; Köhnke, T.; Hoster, E.; Schneider, S.; Dufour, A.; Zellmeier, E.; Fiegl, M.; Braess, J.; Bohlander, S.K.; Subklewe, M.; et al. Molecular response assessment by quantitative real-time polymerase chain reaction after induction therapy in NPM1-mutated patients identifies those at high risk of relapse. Haematologica 2014, 99, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Schlenk, R.F.; Jensen, K.O.; Tschürtz, F.; Corbacioglu, A.; Gaidzik, V.I.; Paschka, P.; Onken, S.; Eiwen, K.; Habdank, M.; et al. Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: A study from the German-Austrian acute myeloid leukemia study group. J. Clin. Oncol. 2011, 29, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.; Lambert, J.; Nibourel, O.; Pautas, C.; Hayette, S.; Cayuela, J.M.; Terre, C.; Rousselot, P.; Dombret, H.; Chevret, S.; et al. MRD assessed by WT1 and NPM1 transcript levels identifies distinct outcomes in AML patients and is influenced by gemtuzumab ozogamicin. Oncotarget 2014, 5, 6280–6288. [Google Scholar] [CrossRef] [PubMed]
- Ommen, H.B.; Nyvold, C.G.; Braendstrup, K.; Andersen, B.L.; Ommen, I.B.; Hasle, H.; Hokland, P.; Ostergaard, M. Relapse prediction in acute myeloid leukaemia patients in complete remission using WT1 as a molecular marker: Development of a mathematical model to predict time from molecular to clinical relapse and define optimal sampling intervals. Br. J. Haematol. 2008, 141, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Messa, F.; Arruga, F.; Defilippi, I.; Gottardi, E.; Fava, M.; Carturan, S.; Catalano, R.; Bracco, E.; Messa, E.; et al. Early prediction of treatment outcome in acute myeloid leukemia by measurement of WT1 transcript levels in peripheral blood samples collected after chemotherapy. Haematologica 2008, 93, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Renneville, A.; Hermitte, F.; Hills, R.K.; Daly, S.; Jovanovic, J.V.; Gottardi, E.; Fava, M.; Schnittger, S.; Weiss, T.; et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: A European LeukemiaNet study. J. Clin. Oncol. 2009, 27, 5195–5201. [Google Scholar] [CrossRef] [PubMed]
- Candoni, A.; Tiribelli, M.; Toffoletti, E.; Cilloni, D.; Chiarvesio, A.; Michelutti, A.; Simeone, E.; Pipan, C.; Saglio, G.; Fanin, R. Quantitative assessment of WT1 gene expression after allogeneic stem cell transplantation is a useful tool for monitoring minimal residual disease in acute myeloid leukemia. Eur. J. Haematol. 2009, 82, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, S.; Geroldi, S.; Tedone, E.; Luchetti, S.; Grasso, R.; Colombo, N.; Di Grazia, C.; Lamparelli, T.; Gualandi, F.; Ibatici, A.; et al. Leukaemia relapse after allogeneic transplants for acute myeloid leukaemia: Predictive role of WT1 expression. Br. J. Haematol. 2013, 160, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Hokland, P.; Ommen, H. Towards individualized follow-up in adult acute myeloid leukemia in remission. Blood 2011, 117, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; McGowan, K.S.; Lu, K.; Jain, N.; Candia, J.; Hensel, N.F.; Tang, J.; Calvo, K.R.; Battiwalla, M.; Barrett, A.J.; et al. A multigene array for measurable residual disease detection in AML patients undergoing SCT. Bone Marrow Transplant. 2015, 50, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Linch, D.C.; Hills, R.K.; Burnett, A.K.; Khwaja, A.; Gale, R.E. Impact of FLT3ITD mutant allele level on relapse risk in intermediate-risk acute myeloid leukemia. Blood 2014, 124, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Ploen, G.G.; Nederby, L.; Guldberg, P.; Hansen, M.; Ebbesen, L.H.; Jensen, U.B.; Hokland, P.; Aggerholm, A. Persistence of DNMT3A mutations at long-term remission in adult patients with AML. Br. J. Haematol. 2014, 167, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Debarri, H.; Lebon, D.; Roumier, C.; Cheok, M.; Marceau-Renaut, A.; Niboiurel, O.; Geffroy, S.; Helevaut, N.; Rousselot, P.; Gruson, B.; et al. IDH1/2 but not DNMT3A mutations are suitable targets for minimal residual disease monitoring in acute myeloid leukemia patients: A study by the Acute Leukemia French Association. Oncotarget 2015, 6, 42345–42353. [Google Scholar] [PubMed]
- Chaturvedi, A.; Araujo-Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Gorlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Target inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.I.; Teleanu, V.; Papaemmanuil, E.; Weber, D.; Paschka, P.; Hahn, J.; Wallrabenstein, T.; Kolbinger, B.; Kohne, C.H.; Horst, H.A.; et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinic-pathologic and genetic features. Leukemia 2016, 30, 2160–2168. [Google Scholar] [CrossRef] [PubMed]
- Kohlmann, A.; Nadarajah, N.; Alpermann, T.; Grossmann, V.; Schindela, S.; Dicker, F.; Roller, A.; Kern, W.; Haferlach, C.; Schnittger, S.; et al. Monitoring of residual disease by next-generation deep-sequencing of RUNX1 mutations can identify acute myeloid leukemia patients with resistant disease. Leukemia 2014, 28, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Scholl, C.; Schlenk, R.F.; Eiwen, K.; Dohner, H.; Frohling, S.; Dohner, K.; AML Study Group. The prognostic value of MLL-AF9 detection in patients with t(9;11)(p22;q23)-positive acute myeloid leukemia. Haematologica 2005, 90, 1626–1634. [Google Scholar] [PubMed]
- Reading, C.L.; Estey, E.; Huh, Y.; Claxton, D.F.; Sanchez, G.; Terstappen, L.W.; O’Brien, M.C.; Baron, S.; Deisseroth, A.B. Expression of unusual immunophenotype combinations in acute myelogenous leukemia. Blood 1993, 81, 3083–3090. [Google Scholar] [PubMed]
- Sievers, E.L.; Lange, B.J.; Alonzo, T.A.; Gerbing, R.B.; Bernstein, I.D.; Smith, F.O.; Arceci, R.J.; Woods, W.G.; Loken, M.R. Immunophenotypic evidence of leukemia after induction therapy predicts relapse: Results from a prospective Children’s Cancer Group study of 252 patients with acute myeloid leukemia. Blood 2003, 101, 3398–3406. [Google Scholar] [CrossRef] [PubMed]
- Feller, N.; van der Pol, M.A.; van Stijn, A.; Weijers, G.W.; Westra, A.H.; Evertse, B.W.; Ossenkoppele, G.J.; Schuurhuis, G.J. MRD parameters using immunophenotypic detection methods are highly reliable in predicting survival in acute myeloid leukaemia. Leukemia 2004, 18, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Voskova, D.; Schnittger, S.; Schoch, C.; Haferlach, T.; Kern, W. Use of five-color staining improves the sensitivity of multiparameter flow cytometric assessment of minimal residual disease in patients with acute myeloid leukemia. Leuk. Lymphoma 2007, 48, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawali, A.; Gillis, D.; Lewis, I. The role of multiparameter flow cytometry for detection of minimal residual disease in acute myeloid leukemia. Am. J. Clin. Pathol. 2009, 131, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.R.; Stewart, C.C.; Dodge, R.K.; Leget, G.; Sulé, N.; Mrozek, K.; Schiffer, C.A.; Powell, B.L.; Kolitz, J.E.; Moore, J.O.; et al. High frequency of immunophenotype changes in acute myeloid leukemia at relapse: Implications for residual disease detection (Cancer and Leukemia Group B Study 8361). Blood 2001, 97, 3574–3580. [Google Scholar] [CrossRef] [PubMed]
- Terwijn, M.; van Putten, W.L.; Kelder, A.; van der Velden, V.H.; Brooimans, R.A.; Pabst, T.; Maertens, J.; Boeckx, N.; de Greef, G.E.; Valk, P.J.; et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: Data from the HOVON/SAKK AML 42A study. J. Clin. Oncol. 2013, 31, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.D.; Virgo, P.; Couzens, S.; Grimwade, D.; Russel, N.; Hills, R.K.; Burnett, A.K. Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J. Clin. Oncol. 2013, 31, 4123–4131. [Google Scholar] [CrossRef] [PubMed]
- Maurillo, L.; Buccisano, F.; Spagnoli, A.; Del Poeta, G.; Panetta, P.; Neri, B.; Del Principe, M.I.; Mazzone, C.; Consalvo, M.I.; Tamburini, A.; et al. Monitoring of minimal residual disease in adult acute myeloid leukemia using peripheral blood as an alternative source to bone marrow. Haematologica 2007, 92, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Venditti, A.; Buccisano, F.; Del Poeta, G.; Maurillo, L.; Tamburini, A.; Cox, C.; Battaglia, A.; Catalano, G.; Del Moro, B.; Cudillo, L.; et al. Level of minimal residual disease after consolidation therapy predicts outcome in acute myeloid leukemia. Blood 2000, 96, 3948–3952. [Google Scholar] [PubMed]
- Venditti, A.; Maurillo, L.; Buccisano, F.; Del Poeta, G.; Mazzone, C.; Tamburini, A.; Del Principe, M.I.; Consalvo, M.I.; De Fabritiis, P.; Cudillo, L.; et al. Pretransplant minimal residual disease level predicts clinical outcome in patients with acute myeloid leukemia receiving high-dose chemotherapy and autologous stem cell transplantation. Leukemia 2003, 17, 2178–2182. [Google Scholar] [CrossRef] [PubMed]
- Buccisano, F.; Maurillo, L.; Gattei, V.; Del Poeta, G.; Del Principe, M.I.; Cox, M.C.; Panetta, P.; Consalvo, M.I.; Mazzone, C.; Neri, B.; et al. The kinetics of reduction of minimal residual disease impacts on duration of response and survival of patients with acute myeloid leukemia. Leukemia 2006, 20, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Buccisano, F.; Maurillo, L.; Spagnoli, A.; Del Principe, M.I.; Fraboni, D.; Panetta, P.; Ottone, T.; Consalvo, M.I.; Lavorgna, S.; Bulian, P.; et al. Cytogenetic and molecular diagnostic characterization combined to post-consolidation minimal residual disease assessment by flow-cytometry improves risk stratification in adult acute myeloid leukemia. Blood 2010, 116, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Buccisano, F.; Maurillo, L.; Del Principe, M.I.; Del Poeta, G.; Sconocchia, G.; Lo-Coco, F.; Arcese, W.; Amadori, S.; Venditti, A. Prognostic and therapeutic implications of minimal residual disease detection in acute myeloid leukemia. Blood 2012, 119, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Van Tendeloo, V.F.; Berneman, Z.N. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia 2012, 26, 2186–2196. [Google Scholar] [CrossRef] [PubMed]
- Terwijn, M.; Zeijlemaker, W.; Kelder, A.; Rutten, A.P.; Snel, A.N.; Scholten, W.J.; Pabst, T.; Verhoef, G.; Löwenberg, B.; Zweegman, S.; et al. Leukemic Stem Cell Frequency: A Strong Biomarker for Clinical Outcome in Acute Myeloid Leukemia. PLoS ONE 2014, 9, e107587. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; Feller, N.; Kelder, A.; Westra, A.H.; Rombouts, E.; Zweegman, S.; van der Pol, M.A.; Waisfisz, Q.; Ossenkoppele, G.J.; Schuurhuis, G.J. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin. Cancer Res. 2005, 11, 6520–6527. [Google Scholar] [CrossRef] [PubMed]
- Paietta, E. Should minimal residual disease guide therapy in AML? Best Pract. Res. Clin. Haematol. 2015, 28, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; van Dongen, G.A.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Stigter-van Walsum, M; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef] [PubMed]
- Gerber, J.M.; Smith, B.D.; Ngwang, B.; Zhang, H.; Vala, M.S.; Morsberger, L.; Galkin, S.; Collector, M.I.; Perkins, B.; Lewis, M.J.; et al. A clinically relevant population of leukemic CD34(+) CD38(−) cells in acute myeloid leukemia. Blood 2012, 119, 3571–3577. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; Moshaver, B.; Kelder, A.; Feller, N.; Nieuwint, A.W.; Zweegman, S.; Ossenkoppele, G.J.; Schuurhuis, G.J. Aberrant marker expression patterns on the CD34+CD38− stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia 2007, 21, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Zeijlemaker, W.; Kelder, A.; Oussoren-Brockhoff, Y.J.; Scholten, W.J.; Snel, A.N.; Veldhuizen, D.; Cloos, J.; Ossenkoppele, G.J.; Schuurhuis, G.J. A simple one-tube assay for immunophenotypical quantification of leukemic stem cells in acute myeloid leukemia. Leukemia 2016, 30, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Kern, W.; Haferlach, T.; Schoch, C.; Loffler, H.; Gassmann, W.; Heinecke, A.; Sauerland, M.C.; Berdel, W.; Buchner, T.; Hiddemann, W. Early blast clearance by remission induction therapy is a major independent prognostic factor for both achievement of complete remission and long-term outcome in acute myeloid leukemia: Data from the German AML Cooperative Group (AMLCG) 1992 Trial. Blood 2003, 101, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Kern, W.; Voskova, D.; Schoch, C.; Hiddemann, W.; Schnittger, S.; Haferlach, T. Determination of relapse risk based on assessment of minimal residual disease during complete remission by multiparameter flow cytometry in unselected patients with acute myeloid leukemia. Blood 2004, 104, 3078–3085. [Google Scholar] [CrossRef] [PubMed]
- Gianfaldoni, G.; Mannelli, F.; Bencini, S.; Leoni, F.; Baldini, S.; Bosi, A. Peripheral blood blast clearance during induction therapy in acute myeloid leukemia. Blood 2008, 111, 1746–1747. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.A.; Litzow, M.R.; Letendre, L.L.; Wolf, R.C.; Hanson, C.A.; Tefferi, A.; Tallman, M.S. Early peripheral blood blast clearance during induction chemotherapy for acute myeloid leukemia predicts superior relapse-free survival. Blood 2007, 110, 4172–4174. [Google Scholar] [CrossRef] [PubMed]
- Kohnke, T.; Sauter, D.; Ringel, K.; Hoster, E.; Laubender, R.P.; Hubmann, M.; Bohlander, S.K.; Kakadia, P.M.; Schneider, S.; Dufour, A.; et al. Early assessment of minimal residual disease in AML by flow cytometry during aplasia identifies patients at increased risk of relapse. Leukemia 2014, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lowenberg, B. Strategies in the treatment of acute myeloid leukemia. Haematologica 2004, 89, 1029–1032. [Google Scholar] [PubMed]
- Marani, C.; Clavio, M.; Grasso, R.; Colombo, N.; Guolo, F.; Kunkl, A.; Ballerini, F.; Giannoni, L.; Ghiggi, C.; Fugazza, G.; et al. Integrating post induction WT1 quantification and flow-cytometry results improves minimal residual disease stratification in acute myeloid leukemia. Leuk. Res. 2013, 37, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Minervini, M.M.; Melillo, L.; di Nardo, F.; de Waure, C.; Scalzulli, P.R.; Perla, G.; Valente, D.; Sinisi, N.; Cascavilla, N.; et al. Predictive role of minimal residual disease and log clearance in acute myeloid leukemia: A comparison between multiparameter flow cytometry and Wilms tumor 1 levels. Ann. Hematol. 2014, 93, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Carella, A.M.; Minervini, M.M.; di Nardo, F.; Waure, C.D.; Greco, M.M.; Merla, E.; Cillis, G.P.; Di Renzo, N.; Melpignano, A.; et al. Optimal time-points for minimal residual disease monitoring change on the basis of the method used in patients with acute myeloid leukemia who underwent allogeneic stem cell transplantation: A comparison between multiparameter flow cytometry and Wilms’ tumor 1 expression. Leuk. Res. 2015, 39, 138–143. [Google Scholar] [PubMed]
- Capelli, D.; Guolo, F.; Mancini, G.; Coluzzi, S.; Mancini, S.; Gobbi, M.; Viola, N.; Minetto, P.; Maravalle, D.; Mosna, F.; et al. WT1 and Multiparameter Flow Cytometry MRD status after consolidation significantly predict early relapse in cohort of 175 Acute Myeloid Leukemia patients: O104. Bone Marrow Transplant. 2016, 51, S60–S61. [Google Scholar]
- Ouyang, J.; Goswami, M.; Peng, J.; Zuo, Z.; Daver, N.; Borthakur, G.; Tang, G.; Medeiros, L.J.; Jorgensen, J.L.; Ravandi, F.; et al. Comparison of multiparameter flow cytometry immunophenotypic analysis and quantitative RT-PCR for the detection of minimal residual disease of core binding factor acute myeloid leukemia. Am. J. Clin. Pathol. 2016, 145, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Bachas, C.; Schuurhuis, G.J.; Assaraf, Y.G.; Kwidama, Z.J.; Kelder, A.; Wouters, F.; Snel, A.N.; Kaspers, G.J.L.; Cloos, J. The role of minor subpopulations within the leukemic blast compartment of AML patients at initial diagnosis in the development of relapse. Leukemia 2012, 26, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Zeijlemaker, W.; Kelder, A.; Oussoren-Brockhoff, Y.J.; Scholten, W.J.; Snel, A.N.; Veldhuizen, D.; Cloos, J.; Ossenkoppele, G.J.; Schuurhuis, G.J. Peripheral blood minimal residual disease may replace bone marrow minimal residual disease as an immunophenotypical biomarker for impending relapse in acute myeloid leukemia. Leukemia 2016, 30, 708–715. [Google Scholar]
- Vanderhoek, M.; Juckett, M.B.; Perlman, S.B.; Nickles, R.J.; Jeraj, R. Early assessment of treatment response in patients with AML using [(18)F]FLT-PET imaging. Leuk. Res. 2011, 35, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Martens, A.C.; Hagenbeek, A. Detection of minimal disease in acute leukemia using flow cytometry: Studies in a rat model for human acute leukemia. Cytometry 1985, 6, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Buccisano, F.; Maurillo, L.; Piciocchi, A.; Del Principe, M.I.; Sarlo, C.; Cefalo, M.; Ditto, C.; Di Veroli, A.; De Santis, G.; Irno Consalvo, M.; et al. Minimal residual disease negativity in elderly patients with acute myeloid leukemia may indicate different postremission strategies than in younger patients. Ann. Hematol. 2015, 94, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Blaise, D.; Fürst, S.; Crocchiolo, R.; El-Cheikh, J.; Granata, A.; Harbi, S.; Bouabdallah, R.; Devillier, R.; Bramanti, S.; Lemarie, C.; et al. Haploidentical T Cell-replete transplantation with post-transplantation cyclophosphamide for patients in or above the sixth decade of age compared with allogeneic hematopoietic stem cell transplantation from a human leukocyte antigen-matched related or unrelated donor. Biol. Blood Marrow Transplant. 2016, 22, 119–124. [Google Scholar] [PubMed]
- Sorror, M.L.; Maris, M.B.; Storb, R.; Baron, F.; Sandmaier, B.M.; Maloney, D.G.; Storer, B. Hematopoietic Cell Transplantation (HCT)-specific comorbidity index: A new toll for risk assessment before allogeneic HCT. Blood 2005, 106, 2912–2919. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.H.; Zhang, X.H.; Qin, Y.Z.; Liu, D.H.; Jiang, H.; Chen, H.; Jiang, Q.; Xu, L.P.; Lu, J.; Han, W.; et al. MRD-directed risk stratification treatment may improve outcomes of t(8;21) AML in the first complete remission: Results from the AML05 multicenter trial. Blood 2013, 121, 4056–4062. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Buckley, S.A.; Pagel, J.M.; Wood, B.L.; Storer, B.E.; Sandmaier, B.M.; Fang, M.; Gyurkocza, B.; Delaney, C.; Radich, J.P.; et al. Significance of minimal residual disease before myeloablative allogeneic hematopoietic cell transplantation for AML in first and second complete remission. Blood 2013, 122, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Gyurkocza, B.; Storer, B.E.; Godwin, C.D.; Pagel, J.M.; Buckley, S.A.; Sorror, M.L.; Wood, B.L.; Storb, R.; Appelbaum, F.R.; et al. Comparison of minimal residual disease as outcome predictor for AML patients in first complete remission undergoing myeloablative or nonmyeloablative allogeneic hematopoietic cell transplantation. Leukemia 2015, 29, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Araki, D.; Wood, B.L.; Othus, M.; Radich, J.P.; Halpern, A.B.; Zhou, Y.; Mielcarek, M.; Estey, E.H.; Appelbaum, F.R.; Walter, R.B. Allogeneic hematopoietic cell transplantation for acute myeloid leukemia: Time to move toward a minimal residual disease-based definition of complete remission? J. Clin. Oncol. 2016, 34, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Othus, M.; Araki, D.; Wood, B.L.; Radich, J.P.; Halpern, A.B.; Mielcarek, M.; Estey, E.H.; Appelbaum, F.R.; Walter, R.B. Pre- and post-transplant quantification of measurable (“minimal”) residual disease via multiparameter flow cytometry in adult acute myeloid leukemia. Leukemia 2016, 30, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Ommen, H.B.; Schnittger, S.; Jovanovic, J.V.; Ommen, I.B.; Hasle, H.; Ostergaard, M.; Grimwade, D.; Hockland, P. Strikingly different molecular relapse kinetics in NPM1c, PML-RARA, RUNX1-RUNX1T1, and CBFB-MYH11 acute myeloid leukemias. Blood 2010, 115, 198–205. [Google Scholar] [CrossRef] [PubMed]
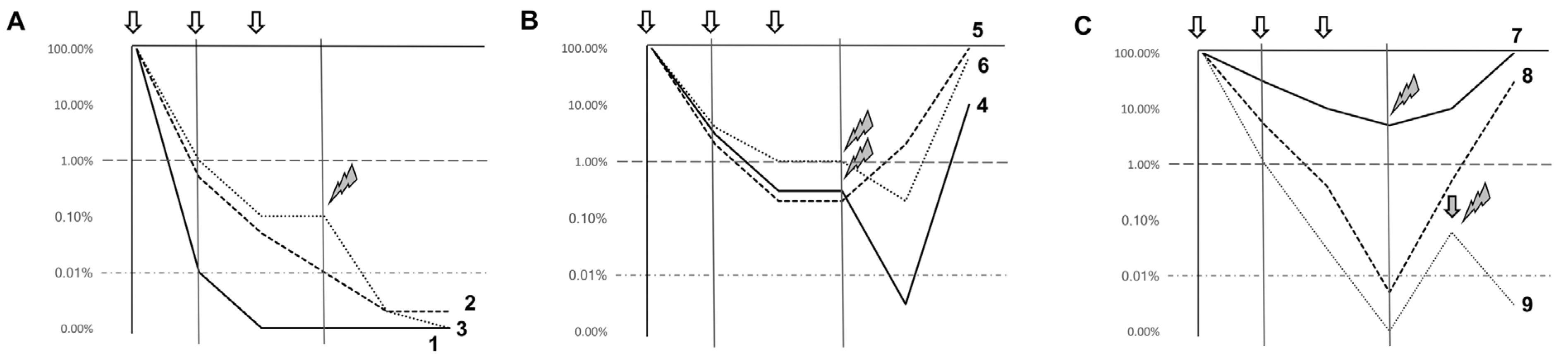

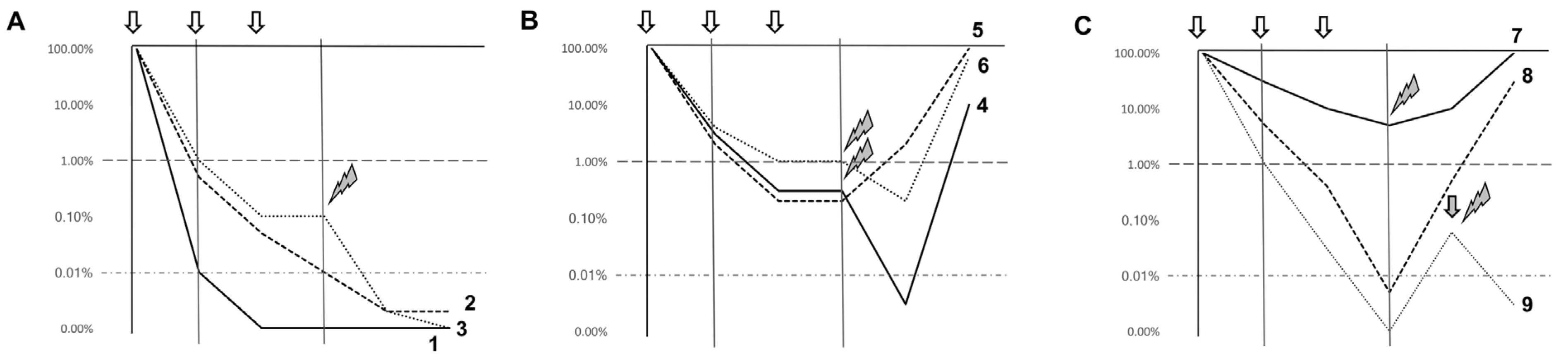
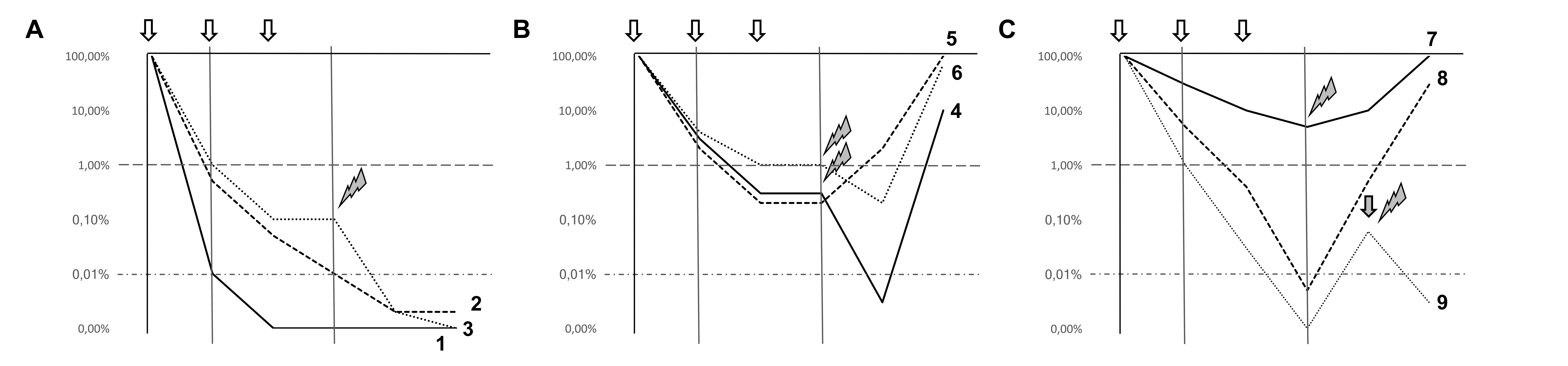{kind=link}
{kind=link}
| Molecular Markers | Frequency (% of All) | Occurrence in Leukemogenesis | Predictive Power for Clinical Relapse | Technique |
|---|---|---|---|---|
| Fusion products | ||||
| RUNX1/RUNX1T1 | 7–10% | Early | Very good | RT-qPCR |
| CBFB/MYH11 | 5–8% | Early | Very good | RT-qPCR |
| MLL/MLLT3 | 2% | Probably late | Good | RT-qPCR |
| Mutations | ||||
| FLT3-ITD | 25–30% | Late | Poor | RT-qPCR/NGS |
| NPM1 | 30% (50% in normal-karyotype) | Late | Very good | RT-qPCR/NGS |
| DNMT3A | 10–15% | Early | Poor | NGS |
| RUNX1 | 10% | Early | Possibly good | NGS |
| IDH1/IDH2 | 8–9% each | Early | Possibly good | NGS |
| Overexpression | ||||
| WT1 | 85–90% | Unknown | Good | RT-qPCR |
| Trial | Nation | ID | MRD-Related Endpoints | Type | Age Limits |
|---|---|---|---|---|---|
| MRC AML 17 | UK | ISRCTN55675535 | Assess the prognostic value of minimal residual disease monitoring (randomization: monitoring vs. not monitoring) | Phase 3 | <60 years |
| MRC AML 19 | UK | ISRCTN31682779 | Assess the prognostic value of minimal residual disease monitoring (randomization: monitoring vs. not monitoring) | Phase 3 | 18–60 years |
| MRC AML 18 | UK | ISRCTN78449203 | Treatment intensification in MRD+ patients after the first cycle, chemotherapy randomization | Phase 3 | >60 years |
| GIMEMA AML1310 | Italy | NCT01452646 | MRD stratification of intermediate-risk karyotype; risk-adapted, MRD-directed therapy (autoSCT vs. SCT) after first consolidation | Phase 2 | 18–60 years |
| CETLAM AML-03 | Spain | NCT01723657 | MRD stratification of intermediate-risk karyotype; risk-adapted, MRD-directed therapy (autoSCT vs. SCT) after first consolidation | Phase 2 | 18–70 years |
| PETHEMA LMA10 | Spain | NCT01296178 | Risk-adapted, MRD-directed therapy(study arms not provided) | Phase 3 | <65 years |
| PETHEMA | Spain | NCT00390715 | Prospective study on the prognostic value of baseline cytogenetics and MRD monitoring | Observational (prospective) | <65 years |
| Nanfang Hospital of Southern Medical University, Guangzhou | China | NCT02870777 | MRD-directed therapy for low- and intermediate-risk AML. Front-line allo-HSCT intensification is programmed for MRD+ patients | Phase 3 | 18–60 years |
| Rochester University | USA | NCT01311258 | Identification by MPFC, among all MRD cells, of the clones eventually responsible for clinical relapse (LIC) | Observational (prospective) | >18 years |
| Az. Ospedaliera Città della Salute e della Scienza Torino | Italy | NCT02714790 | Assess the prognostic role of MRD defined as BM expression of WT1 | Observational (retrospective) | >18 years |
| Medical College of Wisconsin | USA | NCT02349178 | Estimating the efficacy of Clofarabine, Cyclophosphamide and Etoposide in eliminating MRD in AML patients, otherwise in clinical remission, before allo-HSCT | Phase 2 | <40 years |
| Technische Universitat of Dresden RELAZA2 | Germany | EudraCT 2010-022388-37 | 5-Azacitidinetreatment of patients with MDS or AML with significant residual disease or an increase of MRD | Phase 2 | >18 years |
| Ulm University | Germany | NCT01770158 | Maintenance Therapy with Histamine Dihydrochloride and Interleukin-2 in AML MRD+ patients post consolidation therapy | Observational (prospective) | >18 years |
| Washington University | USA | NCT00863434 | Clofarabine and Cytarabine in treating MRD+ (by MPFC) AML patients | Phase 2 | 18–75 years |
| Singapore General Hospital | Singapore | NCT00394381 | Autologous Cytokine-induced Killer cell adoptive immunotherapy for MRD+ patients post autologous HSCT | Phase 1/2 | 12–75 years |
| Institute of Hematology & BloodDisease Hospital, Tianjin | China | NCT03021395 | Efficacy of maintenance Decitabine (after consolidation chemotherapy) in clearing MRD in patients in clinical remission | Phase 1/2 | 14–55 years |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosna, F.; Capelli, D.; Gottardi, M. Minimal Residual Disease in Acute Myeloid Leukemia: Still a Work in Progress? J. Clin. Med. 2017, 6, 57. https://doi.org/10.3390/jcm6060057
Mosna F, Capelli D, Gottardi M. Minimal Residual Disease in Acute Myeloid Leukemia: Still a Work in Progress? Journal of Clinical Medicine. 2017; 6(6):57. https://doi.org/10.3390/jcm6060057
Chicago/Turabian StyleMosna, Federico, Debora Capelli, and Michele Gottardi. 2017. "Minimal Residual Disease in Acute Myeloid Leukemia: Still a Work in Progress?" Journal of Clinical Medicine 6, no. 6: 57. https://doi.org/10.3390/jcm6060057
APA StyleMosna, F., Capelli, D., & Gottardi, M. (2017). Minimal Residual Disease in Acute Myeloid Leukemia: Still a Work in Progress? Journal of Clinical Medicine, 6(6), 57. https://doi.org/10.3390/jcm6060057