
Treatment and Prevention of Bleeds in Haemophilia Patients with Inhibitors to Factor VIII/IX

Abstract
:
1. Introduction
2. Treatment of Acute Bleeding
3. Monitoring Therapeutic Efficacy of Haemostatic Treatment
4. Bleeding Prevention
5. Unresolved Issues and Unmet Needs
6. Concluding Remarks
Author Contributions
Conflicts of Interest
References
- Gouw, S.C.; van den Berg, H.M.; Fischer, K.; Auerswald, G.; Carcao, M.; Chalmers, E.; Chambost, H.; Kurnik, K.; Liesner, R.; Petrini, P.; et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: The RODIN study. Blood 2013, 121, 4046–4055. [Google Scholar] [CrossRef] [PubMed]
- Hay, C.R.; Palmer, B.; Chalmers, E.; Liesner, R.; Maclean, R.; Rangarajan, S.; Williams, M.; Collins, P.W.; United Kingdom Haemophilia Centre Doctors’ Organisation (UKHCDO). Incidence of factor VIII inhibitors throughout life in severe hemophilia A in the United Kingdom. Blood 2011, 117, 6367–6370. [Google Scholar] [CrossRef] [PubMed]
- Astermark, J. Inhibitor development: Patient-determined risk factors. Haemophilia 2010, 16, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Gouw, S.C.; Fijnvandraat, K. Identifying non genetic risk factors for inhibitor development in severe hemophilia A. Semin. Thromb. Hemost. 2013, 39, 740–751. [Google Scholar] [PubMed]
- Astermark, J.; Altisent, C.; Batorova, A.; Diniz, M.J.; Gringeri, A.; Holme, P.A.; Karafoulidou, A.; Lopez-Fernández, M.F.; Reipert, B.M.; Rocino, A.; et al. Non-genetic risk factors and the development of inhibitors in haemophilia: A comprehensive review and consensus report. Haemophilia 2010, 16, 747–766. [Google Scholar] [CrossRef] [PubMed]
- D’Oiron, R.; Pipe, S.W.; Jacquemin, M. Mild/moderate haemophilia A: New insights into molecular mechanisms and inhibitor development. Haemophilia 2008, 14, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Mauser-Bunschoten, E.P.; Den Ujil, I.E.; Schutgens, R.E.; Roosendaal, G.; Fischer, K. Risk of inhibitor development in mild haemophilia A increases with age. Haemophilia 2012, 18, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, C.L.; van Velzen, A.S.; Peters, M.; Astermark, J.; Brons, P.P.; Castaman, G.; Cnossen, M.H.; Dors, N.; Escuriola-Ettingshausen, C.; Hamulyak, K.; et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood 2013, 122, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, C.L.; Menke, L.A.; Van Ommen, C.H.; van der Lee, J.H.; Geskus, R.B.; Kamphuisen, P.W.; Peters, M.; Fijnvandraat, K. Intensive perioperative use of factor VIII and the Arg593-->Cys mutation are risk factors for inhibitor development in mild/moderate hemophilia A. J. Thromb. Haemost. 2009, 7, 930–937. [Google Scholar] [CrossRef] [PubMed]
- DiMichele, D. Inhibitor development in haemophilia B: An orphan disease in need of attention. Br. J. Haematol. 2007, 138, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Goodeve, A.C. Hemophilia B: Molecular pathogenesis and mutation analysis. J. Thromb. Haemost. 2015, 13, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- White, G.C., II; Rosendaal, F.; Aledort, L.M.; Lusher, J.M.; Rothschild, C.; Ingerslev, J.; Factor VIII and Factor IX Subcommittee. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the international society of thrombosis and haemostasis. Thromb. Haemost. 2001, 85, 560. [Google Scholar] [PubMed]
- Gringeri, A.; Mantovani, L.G.; Scalone, L.; Mannucci, P.M.; COCIS Study Group. Cost of care and quality of life for patients with hemophilia complicated by inhibitors: The COCIS Study Group. Blood 2003, 102, 2358–2363. [Google Scholar] [CrossRef] [PubMed]
- Morfini, M.; Haya, S.; Tagariello, G.; Pollmann, H.; Quintana, M.; Siegmund, B.; Stieltjes, N.; Dolan, G.; Tusell, J. European study on orthopaedic status of haemophilia patients with inhibitors. Haemophilia 2007, 13, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Scalone, L.; Mantovani, L.G.; Mannucci, P.M.; Gringeri, A. Quality of life is associated to the orthopaedic status in haemophilic patients with inhibitors. Haemophilia 2006, 12, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Brewer, A.K.; Mauser-Bunschoten, E.P.; Key, N.S.; Kitchen, S.; Llinas, A.; Ludlam, C.A.; Mahlangu, J.N.; Mulder, K.; Poon, M.C.; et al. Guidelines for the management of hemophilia. Haemophilia 2013, 19, e1–e47. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.W.; Chalmers, E.; Hart, D.P.; Liesner, R.; Rangarajan, S.; Talks, K.; Williams, M.; Hay, C.R. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: (4th edition). Br. J. Haematol. 2013, 160, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Rocino, A.; Coppola, A.; Franchini, M.; Castaman, G.; Santoro, C.; Zanon, E.; Santagostino, E.; Morfini, M.; Italian Association of Haemophilia Centres (AICE) Working Party. Principles of treatment and update of recommendations for the management of haemophilia and congenital bleeding disorders in Italy. Blood Transfus. 2014, 12, 575–598. [Google Scholar] [PubMed]
- Brackmann, H.H.; Lenk, H.; Scharrer, I.; Auerswald, G.; Kreuz, W. German recommendations for immune tolerance therapy in type A haemophiliacs with antibodies. Haemophilia 1999, 5, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Valentino, L.A.; Kempton, C.L.; Kruse-Jarres, R.; Mathew, P.; Meeks, S.L.; Reiss, U.M.; International Immune Tolerance Induction Study Investigators. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015, 21, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Colvin, B.T.; Astermark, J.; Fischer, K.; Gringeri, A.; Lassila, R.; Schramm, W.; Thomas, A.; Ingerslev, J.; Inter Disciplinary Working Group. European principles of haemophilia care. Haemophilia 2008, 14, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Astermark, J.; Morado, M.; Rocino, A.; van den Berg, H.M.; von Depka, M.; Gringeri, A.; Mantovani, L.; Garrido, R.P.; Schiavoni, M.; Villar, A.; et al. Current European practice in immune tolerance induction therapy in patients with haemophilia and inhibitors. Haemophilia 2006, 12, 363–371. [Google Scholar] [CrossRef] [PubMed]
- DiMichele, D.M.; Hoots, W.K.; Pipe, S.W.; Rivard, G.E.; Santagostino, E. International workshop on immune tolerance induction: Consensus recommendations. Haemophilia 2007, 13, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Benson, G.; Auerswald, G.; Elezović, I.; Lambert, T.; Ljung, R.; Morfini, M.; Remor, E.; Salek, S.Z. Immune tolerance induction in patients with severe hemophilia with inhibitors: Expert panel views and recommendations for clinical practice. Eur. J. Haematol. 2012, 88, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Colowick, A.B.; Bohn, R.L.; Avorn, J.; Ewenstein, B.M. Immune tolerance induction in hemophilia patients with inhibitors: Costly can be cheaper. Blood 2000, 96, 1698–1702. [Google Scholar] [PubMed]
- Rocino, A.; Cortesi, P.A.; Scalone, L.; Mantovani, L.G.; Crea, R.; Gringeri, A.; European Haemophilia Therapy Strategy Board (EHTSB). Immune tolerance induction in patients with haemophilia a and inhibitors: Effectiveness and cost analysis in an European Cohort (The ITER Study). Haemophilia 2016, 22, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Earnshaw, S.R.; Graham, C.N.; McDade, C.L.; Spears, J.B.; Kessler, C.M. Factor VIII alloantibody inhibitors: Cost analysis of immune tolerance induction vs. prophylaxis and on-demand with bypass treatment. Haemophilia 2015, 21, 310–319. [Google Scholar] [CrossRef] [PubMed]
- DiMichele, D. The North American Immune Tolerance Registry: Contributions to the thirty-year experience with immune tolerance therapy. Haemophilia 2009, 15, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Chitlur, M.; Warrier, I.; Rajpurkar, M.; Lusher, J.M. Inhibitors in factor IX deficiency a report of the ISTH-SSC international FIX inhibitor registry (1997–2006). Haemophilia 2009, 15, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Holstein, K.; Batorova, A.; Carvalho, M.; Fijnvandraat, K.; Holme, P.; Kavakli, K.; Lambert, T.; Rocino, A.; Jiménez-Yuste, V.; Astermark, J.; et al. Current view and outcome of ITI therapy—A change over time? Thromb. Res. 2016, 148, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Castaman, G.; Bonetti, E.; Messina, M.; Morfini, M.; Rocino, A.; Scaraggi, F.A.; Tagariello, G.; Italian Association of Hemophilia Centers. Inhibitors in haemophilia B: The Italian experience. Haemophilia 2013, 19, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Castaman, G.; Fijnvandraat, K. Molecular and clinical predictors of inhibitor risk and its prevention and treatment in mildhemophilia A. Blood 2014, 124, 2333–2336. [Google Scholar] [CrossRef] [PubMed]
- Kempton, C.L.; Allen, G.; Hord, J.; Kruse-Jarres, R.; Pruthi, R.K.; Walsh, C.; Young, G.; Soucie, J.M. Eradication of factor VIII inhibitors in patients with mild and moderate haemophilia A. Am. J. Hematol. 2012, 87, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Van Veltzen, A.S.; Eckhardt, C.L.; Hart, D.P.; Peters, M.; Rangarajan, S.; Mancuso, M.E.; Smiers, F.J.; Khair, K.; Petrini, P.; Jiménez-Yuste, V.; et al. Inhibitors in nonsevere haemophilia A: Outcome and eradication strategies. Thromb. Haemost. 2015, 114, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Gringeri, A.; Mannucci, P.M.; Italian Association of Haemophilia Centres. Italian guidelines for the diagnosis and treatment of patients with haemophilia and inhibitors. Haemophilia 2005, 11, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Kempton, C.L.; White, G.C., 2nd. How we treat a hemophilia A patient with a factor VIII inhibitor. Blood 2009, 113, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Lillicrap, D.; Schiviz, A.; Apostol, C.; Wojciechowski, P.; Horling, F.; Lai, C.K.; Piskernik, C.; Hoellriegl, W.; Lollar, P. Porcine recombinant factor VIII (Obizur; OBI-1; BAX801): Product characteristics and preclinical profile. Haemophilia 2016, 22, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.R.; Monroe, D.M.; White, G.C. The use of recombinant factor VIIa in the treatment of bleeding disorders. Blood 2004, 104, 3858–3864. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.; Dargaud, Y. Mechanisms and monitoring of bypassing agent therapy. J. Thromb. Haemost. 2012, 10, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Villar, A.; Aronis, S.; Morfini, M.; Santagostino, E.; Auerswald, G.; Thomsen, H.F.; Erhardtsen, E.; Giangrande, P.L. Pharmacokinetics of activated recombinant coagulation factor VII (NovoSeven) in children vs. adults with haemophilia A. Haemophilia 2004, 10, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Turecek, P.L.; Váradi, K.; Gritsch, H.; Schwarz, H.P. FEIBA: Mode of action. Haemophilia 2004, 10, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Coppola, A.; Tagliaferri, A.; Lippi, G. FEIBA versus NovoSeven in hemophilia patients with inhibitors. Semin. Thromb. Hemost. 2013, 39, 772–778. [Google Scholar] [PubMed]
- Iorio, A.; Matino, D.; D’Amico, R.; Makris, M. Recombinant Factor VIIa concentrate versus plasma derived concentrates for the treatment of acute bleeding episodes in people with haemophilia and inhibitors. Cochrane Database Syst. Rev. 2010, 8, CD004449. [Google Scholar]
- Negrier, C.; Gomperts, E.D.; Oldenburg, J. The history of FEIBA: A lifetime of success in the treatment of hemophilia complicated by an inhibitor. Haemophilia 2006, 12, 4–13. [Google Scholar] [CrossRef]
- Key, N.S.; Aledort, L.M.; Beardsley, D.; Cooper, H.A.; Davignon, G.; Ewenstein, B.M.; Gilchrist, G.S.; Gill, J.C.; Glader, B.; Hoots, W.K.; et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb. Haemost. 1998, 80, 912–918. [Google Scholar] [PubMed]
- Santagostino, E.; Gringeri, A.; Mannucci, P.M. Home treatment with recombinant activated factor VII in patients with factor VIII inhibitors: The advantages of early intervention. Br. J. Haematol. 1999, 104, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Kavakli, K.; Makris, M.; Zulfikar, B.; Erhardtsen, E.; Abrams, Z.S.; Kenet, G.; NovoSeven trial (F7HAEM-1510) investigators. Home treatment of haemarthroses using a single dose regimen of recombinant activated factor VII in patients with haemophilia and inhibitors. A multi-centre, randomised, double-blind, crossover trial. Thromb. Haemost. 2006, 95, 600–605. [Google Scholar] [PubMed]
- Santagostino, E.; Mancuso, M.E.; Rocino, A.; Mancuso, G.; Scaraggi, F.; Mannucci, P.M. A prospective randomised trial of high and standard dosages of recombinant factor VIIa for treatment of hemarthroses in hemophiliacs with inhibitors. J. Thromb. Haemost. 2006, 4, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Astermark, J.; Donfield, S.M.; DiMichele, D.M.; Gringeri, A.; Gilbert, S.A.; Waters, J.; Berntorp, E.; FENOC Study Group. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: The FEIBA NovoSeven Comparative (FENOC) Study. Blood 2007, 109, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Young, G.; Shafer, F.E.; Rojas, P.; Seremetis, S. Single 270 microg kg(-1)-dose rFVIIa vs. standard 90microg kg(-1)-dose rFVIIa and APCC for home treatment of joint bleeds in haemophilia patients with inhibitors: A randomized comparison. Haemophilia 2008, 14, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Negrier, C.; Goudemand, J.; Sultan, Y.; Bertrand, M.; Rothchild, C.; Lauroua, P.; the French FEIBA Study Group. Multicenter retrospective study on the utilization of FEIBA in France in patients with factor VIII and factor IX inhibitors. Thromb. Haemost. 1997, 77, 1113–1119. [Google Scholar] [PubMed]
- Escuriola-Ettingshausen, C.; Kreuz, W. Early long-term FEIBA prophylaxis in haemophilia A patients with inhibitor after failing immune tolerance induction: A prospective clinical case series. Haemophilia 2010, 16, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Teitel, J.; Berntorp, E.; Collins, P.; D’Oiron, R.; Ewenstein, B.; Gomperts, E.; Goudemand, J.; Gringeri, A.; Key, N.; Leissinger, C.; et al. A systematic approach to controlling problem bleeds in patients with severe congenital haemophilia A and high-titre inhibitors. Haemophilia 2007, 13, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Key, N.S.; Christie, B.; Henderson, N.; Nelsestuen, G.L. Possible synergy between recombinant factor VIIa and prothrombin complex concentrate in hemophilia therapy. Thromb. Haemost. 2002, 88, 60–65. [Google Scholar] [PubMed]
- Tomokiyo, K.; Nakatomi, Y.; Araki, T.; Teshima, K.; Nakano, H.; Nakagaki, T.; Miyamoto, S.; Funatsu, A.; Iwanaga, S. A novel therapeutic approach combining human plasma-derived Factors VIIa and X for haemophiliacs with inhibitors: Evidence of a higher thrombin generation rate in vitro and more sustained haemostatic activity in vivo than obtained with Factor VIIa alone. Vox Sang. 2003, 85, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Schneiderman, J.; Rubin, E.; Nugent, D.J.; Young, G. Sequential therapy with activated prothrombin complex concentrates and recombinant FVIIa in patients with severe haemophilia and inhibitors: Update of our previous experience. Haemophilia 2007, 13, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Gringeri., A.; Fischer, K.; Karafoulidou, A.; Klamroth, R.; Lopez-Fernandez, M.F.; Mancuso, E. Sequential combined bypassing therapy is safe and effective in the treatment of unresponsive bleeding in adults and children with haemophilia and inhibitors. Haemophilia 2011, 17, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Astermark, J.; Rocino, A.; Von Depka, M.; Van Den Berg, H.M.; Gringeri, A.; Mantovani, L.G.; Morado, M.; Garrido, R.P.; Schiavoni, M.; Villar, A.; et al. Current use of by-passing agents in Europe in the management of acute bleeds in patients with haemophilia and inhibitors. Haemophilia 2007, 13, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Hay, C.R.; Ludlam, C.A.; Colvin, B.T.; Hill, F.G.; Preston, F.E.; Wasseem, N.; Bagnall, R.; Peake, I.R.; Berntorp, E.; Mauser Bunschoten, E.P.; et al. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Thromb. Haemost. 1998, 79, 762–766. [Google Scholar] [PubMed]
- Ehrlich, H.J.; Henzl, M.J.; Gomperts, E.D. Safety of factor VIII inhibitor bypass activity (FEIBA): 10-Year compilation of thrombotic adverse events. Haemophilia 2002, 8, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Dimichele, D.; Négrier, C. A retrospective postlicensure survey of FEIBA efficacy and safety. Haemophilia 2006, 12, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Negrier, C.; Voisin, S.; Baghaei, F.; Numerof, R.; Novack, A.; Doralt, J.E.; Romanov, V.; Gringeri, A.; FEIBA PASS Study Group. Global Post-Authorization Safety Surveillance Study: Real-world data on prophylaxis and on-demand treatment using FEIBA (an activated prothrombin complex concentrate). Blood. Coagul. Fibrinolysis 2016, 27, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.R.; Monroe, D.M., 3rd; Hoffman, M. Safety profile of recombinant factor VIIa. Semin. Hematol. 2004, 41, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Aledort, L.M. Comparative thrombotic event incidence after infusion of recombinant factor VIIa versus FVIII bypassing activity. J. Thromb. Haemost. 2004, 2, 1700–1708. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.; Sørensen, B.; Rea, C.J. Bypassing agent therapy with and without tranexamic acid in haemophilia a patients with inhibitors—An in vivo prospective crossover study. Lancet 2013, 1, 1–10. [Google Scholar]
- Valentino, L.A.; Holme, P.A. Should anti-inhibitor coagulant complex and tranexamic acid be used concomitantly? Haemophilia 2015, 21, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Makris, M.; Hay, C.R.; Gringeri, A.; D’Oiron, R. How I treat inhibitors in haemophilia. Haemophilia 2012, 18, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Váradi, K.; Negrier, C.; Berntorp, E.; Astermark, J.; Bordet, J.C.; Morfini, M.; Linari, S.; Schwarz, H.P.; Turecek, P.L. Monitoring the bioavailability of FEIBA with a thrombin generation assay. J. Thromb. Haemost. 2003, 1, 2374–2380. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, B.; Ingerslev, J. Whole blood clot formation phenotypes in hemophilia A and rare coagulation disorders. Patterns of response to recombinant factor VIIa. J. Thromb. Haemost. 2004, 2, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Chitlur, M. Challenges in the laboratory analyses of bleeding disorders. Thromb. Res. 2012, 130, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.E.; Fasulo, M.R. Thrombin generation assay as a laboratory monitoring tool during bypassing therapy in patients with hemophilia and inhibitors. Semin. Thromb. Hemost. 2016, 42, 30–35. [Google Scholar] [PubMed]
- Berntorp, E.; Collins, P.; D’Oiron, R.; Ewing, N.; Gringeri, A.; Négrier, C.; Young, G. Identifying non-responsive bleeding episodes in patients with haemophilia and inhibitors: A consensus definition. Haemophilia. 2011, 17, e202–e210. [Google Scholar] [CrossRef] [PubMed]
- Gringeri, A.; Lambert, T.; Street, A.; Aledort, L.; Adolescent/Adult Prophylaxis Expert Working Group of the International Prophylaxis Study Group. Tertiary prophylaxis in adults: Is there a rationale? Haemophilia 2012, 18, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Valentino, L.A. Assessing the benefits of FEIBA prophylaxis in haemophilia patients with inhibitors. Haemophilia 2010, 16, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Young, G.; Auerswald, G.; Jimenez-Yuste, V.; Lambert, T.; Morfini, M.; Santagostino, E.; Blanchette, V. PRO-PACT: Retrospective observational study on the prophylactic use of recombinant factor VIIa in hemophilia patients with inhibitors. Thromb. Res. 2012, 130, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Konkle, B.A.; Ebbesen, L.S.; Erhardtsen, E.; Bianco, R.P.; Lissitchkov, T.; Rusen, L.; Serban, M.A. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J. Thromb. Haemost. 2007, 5, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Leissinger, C.; Gringeri, A.; Antmen, B.; Berntorp, E.; Biasoli, C.; Carpenter, S.; Cortesi, P.; Jo, H.; Kavakli, K.; Lassila, R.; et al. Anti-inhibitor coagulant complex prophylaxis in hemophilia with inhibitors. N. Engl. J. Med. 2011, 365, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Antunes, S.V.; Tangada, S.; Stasyshyn, O.; Mamonov, V.; Phillips, J.; Guzman-Becerra, N.; Grigorian, A.; Ewenstein, B.; Wong, W.Y. Randomized comparison of prophylaxis and on-demand regimens with FEIBA NF in the treatment of haemophilia A and B with inhibitors. Haemophilia 2014, 20, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Hoots, W.K.; Ebbesen, L.S.; Konkle, B.A.; Auerswald, G.K.; Roberts, H.R.; Weatherall, J.; Ferran, J.M.; Ljung, R.C.; Novoseven (F7HAEM-1505) Investigators. Secondary prophylaxis with recombinant activated factor VII improves health-related quality of life of haemophilia patients with inhibitors. Haemophilia 2008, 14, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Gringeri, A.; Leissinger, C.; Cortesi, P.A.; Jo, H.; Fusco, F.; Riva, S.; Antmen, B.; Berntorp, E.; Biasoli, C.; Carpenter, S.; et al. Health-related quality of life in patients with haemophilia and inhibitors on prophylaxis with anti-inhibitor complex concentrate: Results from the Pro-FEIBA study. Haemophilia 2013, 19, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Young, G.; Auerswald, G.; Jimenez-Yuste, V.; Konkle, B.A.; Lambert, T.; Morfini, M.; Santagostino, E.; Blanchette, V. When should prophylaxis therapy in inhibitor patients be considered? Haemophilia 2011, 17, e849–e857. [Google Scholar] [CrossRef] [PubMed]
- López-Fernández, M.F.; Altisent Roca, C.; Álvarez-Román, M.T.; Canaro Hirnyk, M.I.; Mingot-Castellano, M.E.; Jiménez-Yuste, V.; Cid Haro, A.R.; Pérez-Garrido, R.; Sedano Balbas, C. Spanish Consensus Guidelines on prophylaxis with bypassing agents in patients with haemophilia and inhibitors. Thromb. Haemost. 2016, 115, 872–895. [Google Scholar] [CrossRef] [PubMed]
- Brackmann, H.H.; Oldenburg, J.; Schwaab, R. Immune tolerance for the treatment of factor VIII inhibitors—Twenty years of the Bonn Protocol. Vox Sang. 1996, 70, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Yuste, V.; Alvarez, M.T.; Martin-Salces, M.; Quintana, M.; Rodriguez-Merchan, C.; Lopez-Cabarcos, C.; Velasco, F.; Hernández-Navarro, F. Prophylaxis in 10 patients with severe haemophilia A and inhibitor: Different approaches for different clinical situations. Haemophilia 2009, 15, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Manco-Johnson, M.J.; Abshire, T.C.; Shapiro, A.D.; Riske, B.; Hacker, M.R.; Kilcoyne, R.; Ingram, J.D.; Manco-Johnson, M.L.; Funk, S.; Jacobson, L.; et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N. Engl. J. Med. 2007, 357, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, M.; Kurnik, K.; Pfluger, T.; Bidlingmaier, C. Identification and long-term observation of early joint damage by magnetic resonance imaging in clinically asymptomatic joints in patients with haemophilia A or B despite prophylaxis. Haemophilia 2012, 18, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Kraft, J.; Blanchette, V.; Babyn, P.; Feldman, B.; Cloutier, S.; Israels, S.; Pai, M.; Rivard, G.E.; Gomer, S.; McLimont, M.; et al. Magnetic resonance imaging and joint outcomes in boys with severe hemophilia A treated with tailored primary prophylaxis in Canada. J. Thromb. Haemost. 2012, 10, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Roosendaal, G.; Vianen, M.E.; Marx, J.J.; van den Berg, H.M.; Lafeber, F.P.; Bijlsma, J.W. Blood-induced joint damage: A human in vitro study. Arthritis Rheum. 1999, 42, 1025–1032. [Google Scholar] [CrossRef]
- Jansen, N.W.; Roosendaal, G.; Bijlsma, JW.; Degroot, J.; Lafeber, F.P. Exposure of human cartilage tissue to low concentrations of blood for a short period of time leads to prolonged cartilage damage: An in vitro study. Arthritis Rheum. 2007, 56, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, M.N.; Ambrosino, P.; Quintavalle, G.; Coppola, A.; Tagliaferri, A.; Martinoli, C.; Rivolta, G.F. Assessment of hemophilic arthropathy by ultrasound: Where do we stand? Semin. Thromb. Hemost. 2016, 42, 541–549. [Google Scholar] [PubMed]
- Gringeri, A.; Ewenstein, B.; Reininger, A. The burden of bleeding in haemophilia: Is one bleed too many? Haemophilia 2014, 20, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Valentino, L.A.; Hakobyan, N.; Enockson, C.; Simpson, M.L.; Kakodkar, N.C.; Cong, L.; Song, X. Exploring the biological basis of haemophilic joint disease: Experimental studies. Haemophilia 2012, 18, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K. Prophylaxis for adults with haemophilia: One size does not fit all. Blood Transfus. 2012, 10, 169–173. [Google Scholar] [PubMed]
- Di Minno, M.N.; Di Minno, G.; Di Capua, M.; Cerbone, A.M.; Coppola, A. Cost of care of haemophilia with inhibitors. Haemophilia 2010, 16, e190–e201. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.W.; Zhou, Z.Y. Economical comparison of APCC vs. rFVIIa for mild-to-moderate bleeding episodes in haemophilia patients with inhibitors. Haemophilia 2011, 17, e969–e974. [Google Scholar] [CrossRef] [PubMed]
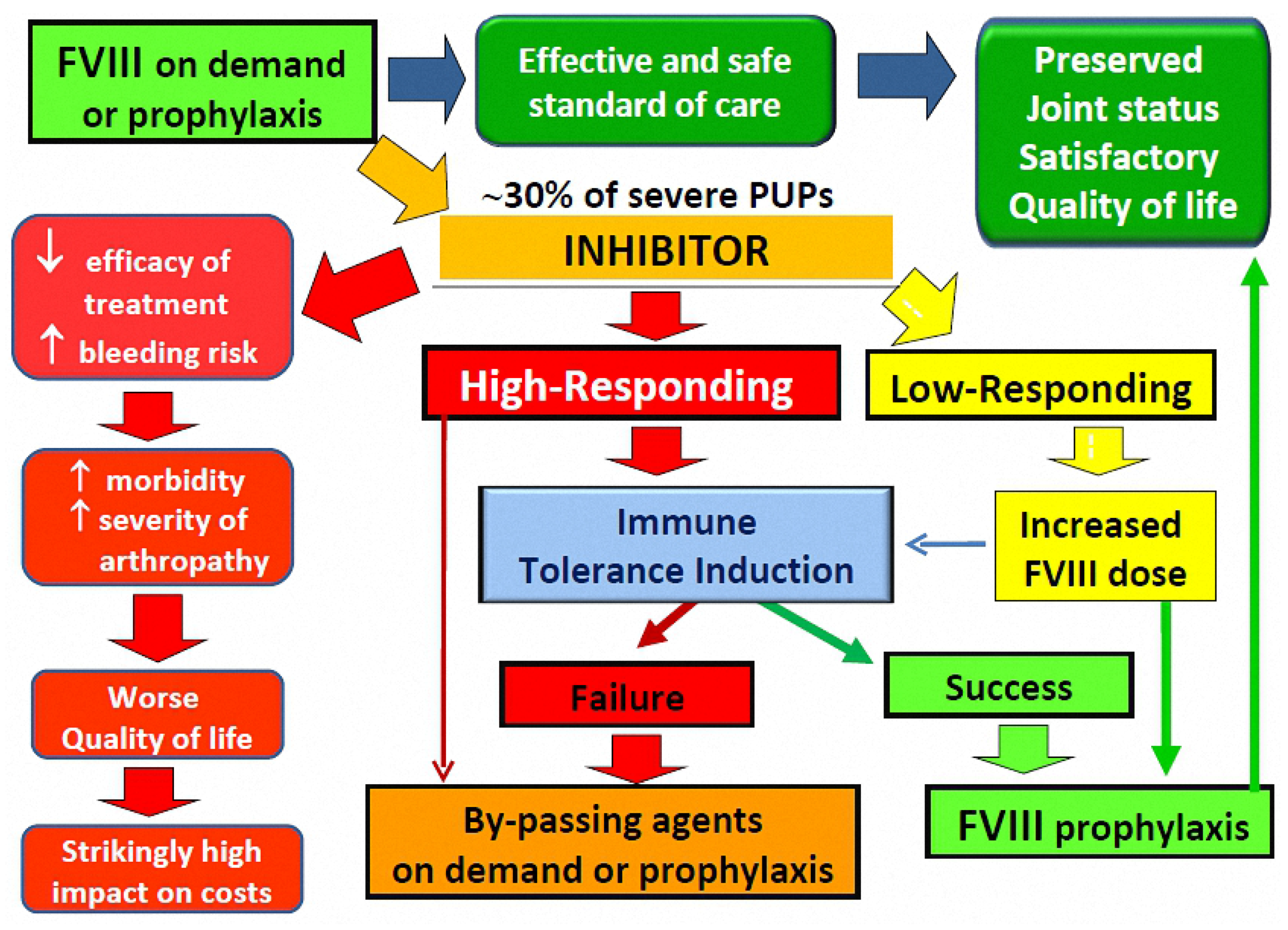

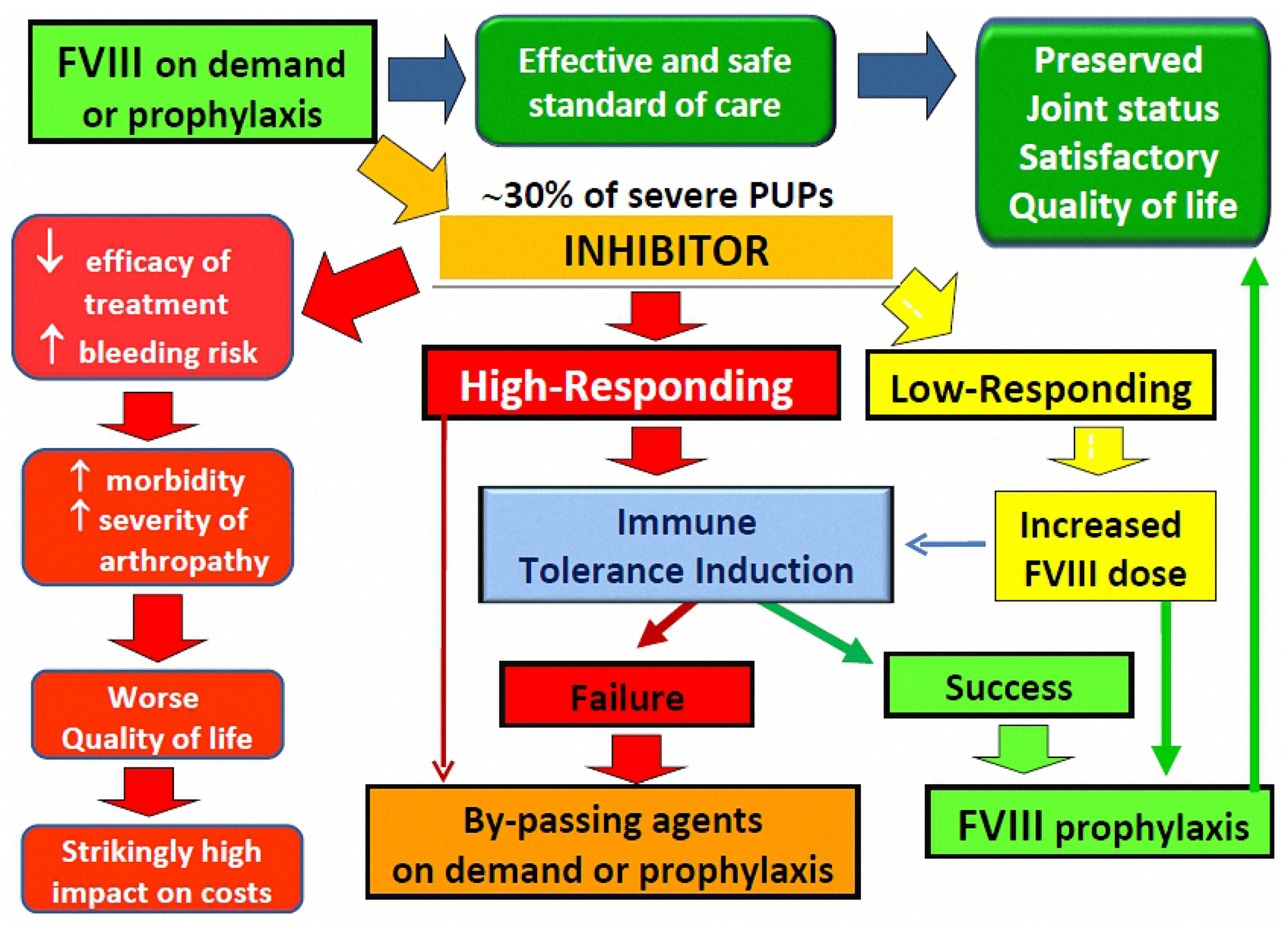
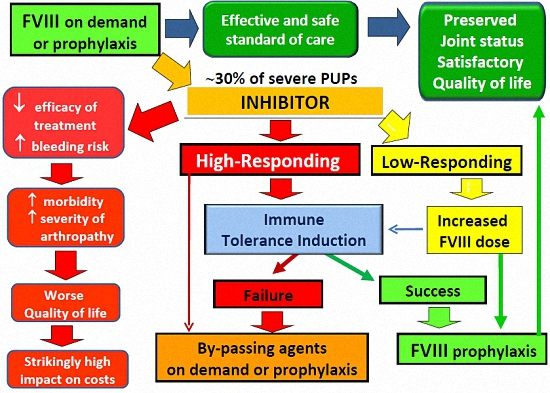{kind=link}
{kind=link}
| Bypassing Agent | Activated Prothrombin Complex Concentrate (aPCC) | Recombinant Activated Factor VII (rFVIIa) |
|---|---|---|
| Source | Pooled human plasma | Recombinant coagulation FVIIa produced in baby hamster kidney (BHK) cells by recombinant DNA technology (Eptacog alfa) |
| Therapeutic indications | Treatment of spontaneous bleeding and cover of surgical interventions in: haemophilia A patients with inhibitors to FVIII patients with acquired inhibitors to FVIII prophylaxis in haemophilia A patients with HR inhibitors and frequent joint bleeding. | Treatment of bleeding episodes and cover of surgical interventions in:
|
| Recommended regimen(s) | 50 to 100 U*/kg Dose interval: 6–12 h. A single dose of 100 U/kg and a daily dose of 200 U/kg should not be exceeded unless the severity of bleeding warrants and justifies the use of higher doses. For prevention of bleeding episodes during prophylaxis, 70 to 100 U/kg every other day with dose adjusted based on the patient’s clinical response. | Initial dose: 90 μg/kg Dose interval: 2–3 h until haemostasis is achieved. If continued therapy is needed, the dose interval can be increased successively to every 4, 6, 8 or 12 h for as long as treatment is judged as being indicated. For mild to moderate bleeding episodes (including home therapy) two dosing regimens can be recommended:
|
| Storage requirements | Room temperature (up to 25 °C) for up to 2 years. | Room temperature (up to 25 °C) for up to 2 years. |
| Volume | Diluent volume: 20 mL. | Diluent volume: 1.1 mL (1 mg); 2.1 mL (2 mg); 5.2 mL (5 mg). |
| Time of administration | The rate of intravenous administration should ensure the comfort of the patient and should not exceed a maximum of 2 U/kg per minute. | Intravenous bolus injection over 2–5 min |
| Half-life in plasma | Varying half-lives for the single components (FII, FIX, FX, FVIIa) | 2.3 h (range 1.7–2.7) |
| Inhibitor anamnesis | Possible in about 20%–30% of patients, since FVIII coagulant antigen (FVIII C:Ag) is present in a concentration of up to 0.1 U/1 U of aPCC. Upon continued administration, inhibitors may decrease over time. | No |
| Association with antifibrinolytic agents | Systemic antifibrinolytics should be administered at least 6 h apart. | Commonly used. |
| Laboratory monitoring | Not standardised | Not standardised |
| Thrombotic risk | Yes | Yes |
| Bypassing Agent | rFVIIa | aPCC | ||
|---|---|---|---|---|
| Study | Konkle et al. 2007 [76] | Lessinger et al. 2011 [77] PRO-FEIBA | Antunes et al. 2014 [78] PROOF | |
| Design | Double-blind, parallel group trial | Open-label, cross-over trial (intra-individual comparison) | Open-label, two arm, parallel trial (inter-individual comparison) | |
| Patients, n | 22 | 26 | 36 | |
| Treatment | 90 μg/kg/day vs. 270 μg/kg/day | 85 ± 15 U/kg three times per week vs. on-demand treatment | 85 ± 15 U/kg each other day (n = 17) vs. on-demand (n = 19) | |
| Follow-up | 3 months (vs. 3 months pre-study and 3 months post-prophylaxis) | 6 months in each treatment regimen (3 months wash-out) | 12 months | |
| Age, years (range) | 15.7 (5.1–56.5) | 28.7 (2.8–62.8) | 23.5 (7–56) | |
| Bleeding rate (as inclusion criterion) | ≥12 in 3 months | ≥6 in 6 months | ≥12 in 12 months | |
| 90 μg/kg/day | 270 μg/kg/day | |||
| Mean bleeding rate on demand | 5.6 * | 5.3 * | 13.1 ^ | 28.7 # |
| Bleeding rate on prophylaxis | 3.0 * | 2.2 * | 5.0 ^ | 7.9 # |
| Reduction (%) on prophylaxis | 45% | 59% | 62% | 72.5% |
| Reduction (%) of target joint bleeds on prophylaxis | 43% | 61% | 72% | 75% |
| Other findings | Benefits extended during the 3 months after prophylaxis | 62% good responders (bleeding rate reduction >50%, overall 84%). In 23% 0 bleeds | Lower development of new target joints on prophylaxis. 26% additional reduction of bleeding rate in the last 6 months vs. first 6 months | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocino, A.; Franchini, M.; Coppola, A. Treatment and Prevention of Bleeds in Haemophilia Patients with Inhibitors to Factor VIII/IX. J. Clin. Med. 2017, 6, 46. https://doi.org/10.3390/jcm6040046
Rocino A, Franchini M, Coppola A. Treatment and Prevention of Bleeds in Haemophilia Patients with Inhibitors to Factor VIII/IX. Journal of Clinical Medicine. 2017; 6(4):46. https://doi.org/10.3390/jcm6040046
Chicago/Turabian StyleRocino, Angiola, Massimo Franchini, and Antonio Coppola. 2017. "Treatment and Prevention of Bleeds in Haemophilia Patients with Inhibitors to Factor VIII/IX" Journal of Clinical Medicine 6, no. 4: 46. https://doi.org/10.3390/jcm6040046
APA StyleRocino, A., Franchini, M., & Coppola, A. (2017). Treatment and Prevention of Bleeds in Haemophilia Patients with Inhibitors to Factor VIII/IX. Journal of Clinical Medicine, 6(4), 46. https://doi.org/10.3390/jcm6040046