
Oxidative Stress: A New Target for Pancreatic Cancer Prognosis and Treatment



Abstract
:1. Introduction
2. The Oxidative Stress in Cancer
2.1. ROS
2.2. ROS and microRNAs Regulation
2.3. Polimorphisms Associated to Oxidative Stress
2.4. Inflammatory Cytokines and ROS Accumulation in PDAC
3. Therapies against Oxidative Stress
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ARE | Anti-oxidant Response Elements |
| BCL-XL | B-cell lymphoma-extra large |
| BRCA | Breast cancer susceptibility genes |
| CAT | Catalase |
| CCNE1 | Cyclin E1 |
| CI | Coefficient interval |
| CK | Cytokeratin |
| CoQ | Coenzyme Q10 |
| COX2 | Cyclooxygenase-2 |
| EMT | Epithelial-mesenchymal transition |
| ERK | Extracellular-regulated kinase |
| FGF-2 | Fibroblast growth factor 2 |
| GLUT-1 | Glucose transporter 1 |
| GPXs | Glutathione peroxidases |
| GSH | Glutation |
| HIF | Hypoxia-inducible factor |
| 4-HNE | 4-Hydroxynonenal |
| HR | Hazard ratio |
| IGF1 | Insulin-like growth factor I |
| IL | Interleukin |
| KEAP1 | Kelch-like protein 1 |
| LDH | lActate dehydrogenase |
| MAPK | Mitogen activated protein kinase |
| M1-dG | Pyrimido 1,2-a purin-10 3H- one |
| MDA | Malondialdehyde |
| MEK | Mitogen-activated protein kinase kinase |
| MMP | Metalloproteinases |
| MTHFR | Methylenetetrahydrofolate reductase |
| mTOR | Mammalian target of rapamicin |
| NADPH | Nicotinamide adenine dinucleotide |
| NQO1 | Nicotinamide adenine dinucleotide phosphate quinone oxidoreductase |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PIK3 | Phosphoinositide 3-kinase |
| PKC | Protein kinase C |
| PKM2 | Pyruvate kinase isozymes M2 |
| PON1 | Human paraoxonase 1 |
| PRXs | Peroxiredoxins |
| RB | Retinoblastoma |
| RCS | Reactive chloride species |
| RHO-RAC | Ras homolog gene/Ras-related C3 |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RSS | Reactive sulfur species |
| SNPs | Single nucleotide polymorphisms |
| SOD | Superoxide dismutase |
| TNFα | Tumor necrosis factor alpha |
| TRXs | Thioredoxins |
| UTR | Untranslated region |
| VEGF | Vascular endothelial growth factor |
| XIAP | X-linked inhibitor of apoptosis proteins |
| XRCC1 | X-ray repair cross-complementing group 1 |
References
- Raimondi, S.; Maisonneuve, P.; Lowenfels, A.B. Epidemiology of pancreatic cancer: An overview. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Egawa, S.; Takeda, K.; Fukuyama, S.; Motoi, F.; Sunamura, M.; Matsuno, S. Clinicopathological aspects of small pancreatic cancer. Pancreas 2004, 28, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Ariyama, J.; Suyama, M.; Satoh, K.; Sai, J. Imaging of small pancreatic ductal adenocarcinoma. Pancreas 1998, 16, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Kelsen, D.P.; Portenoy, R.; Thaler, H.; Tao, Y.; Brennan, M. Pain as a predictor of outcome in patients with operable pancreatic carcinoma. Surgery 1997, 122, 53–59. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andren-Sandberg, A.; Domellof, L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Chari, S.T.; Leibson, C.L.; Rabe, K.G.; Timmons, L.J.; Ransom, J.; de Andrade, M.; Petersen, G.M. Pancreatic cancer-associated diabetes mellitus: Prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008, 134, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Lowenfels, A.B. Risk factors for pancreatic cancer: A summary review of meta-analytical studies. Int. J. Epidemiol. 2015, 44, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Yeo, T.P. Demographics, epidemiology, and inheritance of pancreatic ductal adenocarcinoma. Semin. Oncol. 2015, 42, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.N.A.; Krapcho, M.; Garshell, J.; Miller, D.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; et al. SEER Cancer Statistics Review, 1975–2012; National Cancer Institute: Bethesda, MD, USA, 2015.
- Ghadirian, P.; Baillargeon, J.; Simard, A.; Perret, C. Food habits and pancreatic cancer: A case-control study of the Francophone community in Montreal, Canada. Cancer Epidemiol. Biomark. Prev. 1995, 4, 895–899. [Google Scholar]
- Amaral, A.F.; Porta, M.; Silverman, D.T.; Milne, R.L.; Kogevinas, M.; Rothman, N.; Cantor, K.P.; Jackson, B.P.; Pumarega, J.A.; Lopez, T.; et al. Pancreatic cancer risk and levels of trace elements. Gut 2012, 61, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Water Quality and Health Strategy 2013–2020. Geneva, WHO, 2013. Available online: http://www.who.int/water_sanitation_health/publications/2013/water_quality_strategy/en/ (accessed on 3 November 2016).
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A. Red and processed meat consumption and risk of pancreatic cancer: Meta-analysis of prospective studies. Br. J. Cancer 2012, 106, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Zarkovic, N. 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Mol. Asp. Med. 2003, 24, 281–291. [Google Scholar] [CrossRef]
- Wang, M.; Dhingra, K.; Hittelman, W.N.; Liehr, J.G.; de Andrade, M.; Li, D. Lipid peroxidation-induced putative malondialdehyde-DNA adducts in human breast tissues. Cancer Epidemiol. Biomark. Prev. 1996, 5, 705–710. [Google Scholar]
- Sram, R.J.; Farmer, P.; Singh, R.; Garte, S.; Kalina, I.; Popov, T.A.; Binkova, B.; Ragin, C.; Taioli, E. Effect of vitamin levels on biomarkers of exposure and oxidative damage-the EXPAH study. Mutat. Res. 2009, 672, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Sosa, V.; Moline, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Galeotti, T.; Chiarugi, P. Metastasis: Cancer cell’s escape from oxidative stress. Cancer Metastasis Rev. 2010, 29, 351–378. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, E.C.; Edderkaoui, M.; Pandol, S.J.; Gukovsky, I.; Gukovskaya, A.S. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J. Biol. Chem. 2004, 279, 34643–34654. [Google Scholar] [CrossRef] [PubMed]
- Narendhirakannan, R.T.; Hannah, M.A. Oxidative stress and skin cancer: An overview. Indian J. Clin. Biochem. 2013, 28, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Antony, S.; Juhasz, A.; Lu, J.; Ge, Y.; Jiang, G.; Roy, K.; Doroshow, J.H. Up-regulation and sustained activation of Stat1 are essential for interferon-γ (IFN-γ)-induced dual oxidase 2 (Duox2) and dual oxidase A2 (DuoxA2) expression in human pancreatic cancer cell lines. J. Biol. Chem. 2011, 286, 12245–12256. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zou, P.; Zou, J.; Wang, J.; Zhou, D.; Liu, L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. Exp. Gerontol. 2011, 46, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Han, X.D.; Kan, Y.W. An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA 2001, 98, 4611–4616. [Google Scholar] [CrossRef] [PubMed]
- Chiera, F.; Meccia, E.; Degan, P.; Aquilina, G.; Pietraforte, D.; Minetti, M.; Lambeth, D.; Bignami, M. Overexpression of human NOX1 complex induces genome instability in mammalian cells. Free Radic. Biol. Med. 2008, 44, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Miron, N.; Miron, M.M.; Milea, V.G.; Cristea, V. Proinflammatory cytokines: An insight into pancreatic oncogenesis. Roum. Arch. Microbiol. Immunol. 2010, 69, 183–189. [Google Scholar] [PubMed]
- Lee, J.K.; Edderkaoui, M.; Truong, P.; Ohno, I.; Jang, K.T.; Berti, A.; Pandol, S.J.; Gukovskaya, A.S. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology 2007, 133, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Ju, K.D.; Lim, J.W.; Kim, K.H.; Kim, H. Potential role of NADPH oxidase-mediated activation of Jak2/Stat3 and mitogen-activated protein kinases and expression of TGF-β1 in the pathophysiology of acute pancreatitis. Inflamm. Res. 2011, 60, 791–800. [Google Scholar] [CrossRef]
- Calvert, P.; Yao, K.S.; Hamilton, T.C.; O’Dwyer, P.J. Clinical studies of reversal of drug resistance based on glutathione. Chem. Biol. Interact. 1998, 111–112, 213–224. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Khor, T.O.; Huang, M.T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.N. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lei, W.; Mandlekar, S.; Weber, M.J.; Der, C.J.; Wu, J.; Kong, A.N. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J. Biol. Chem. 1999, 274, 27545–27552. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Lee, S.J.; Park, J.W.; Kim, S.G. Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2-related factor 2 through actin rearrangement in response to oxidative stress. Mol. Pharmacol. 2002, 62, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor suppressor genes and ROS: Complex networks of interactions. Free Radic. Biol. Med. 2012, 52, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, K.; Khuda-Bukhsh, A.R.; Huh, S.O. PLGA-Loaded Gold-Nanoparticles Precipitated with Quercetin Downregulate HDAC-Akt Activities Controlling Proliferation and Activate p53-ROS Crosstalk to Induce Apoptosis in Hepatocarcinoma Cells. Mol. Cells 2015, 38, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, R.; Kato, M.; Miyagawa, S.; Kamata, T. Nox4-derived ROS signaling contributes to TGF-β induced epithelial-mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2003, 33, 4431–4438. [Google Scholar]
- Zhang, C.; Cao, S.; Toole, B.P.; Xu, Y. Cancer may be a pathway to cell survival under persistent hypoxia and elevated ROS: A model for solid-cancer initiation and early development. Int. J. Cancer 2015, 136, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Afanas’ev, I. Reactive oxygen species signaling in cancer: Comparison with aging. Aging Dis. 2011, 2, 219–230. [Google Scholar] [PubMed]
- Donadelli, M.; Dando, I.; Zaniboni, T.; Costanzo, C.; Dalla Pozza, E.; Scupoli, M.T.; Scarpa, A.; Zappavigna, S.; Marra, M.; Abbruzzese, A.; et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011, 2, e152. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, B.; Jan, K.Y.; Chen, C.H.; Hour, T.C.; Yu, H.J.; Pu, Y.S. Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res. 2005, 65, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, C.; Cordani, M.; Gotte, G.; Picone, D.; Donadelli, M. Onconase induces autophagy sensitizing pancreatic cancer cells to gemcitabine and activates Akt/mTOR pathway in a ROS-dependent manner. Biochim. Biophys. Acta 2015, 1853, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Simone, N.L.; Soule, B.P.; Ly, D.; Saleh, A.D.; Savage, J.E.; Degraff, W.; Cook, J.; Harris, C.C.; Gius, D.; Mitchell, J.B. Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS ONE 2009, 4, e6377. [Google Scholar] [CrossRef] [PubMed]
- Favaro, E.; Ramachandran, A.; McCormick, R.; Gee, H.; Blancher, C.; Crosby, M.; Devlin, C.; Blick, C.; Buffa, F.; Li, J.L.; et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS ONE 2010, 5, e10345. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhu, C.F.; Ma, M.Z.; Chen, G.; Song, M.; Zeng, Z.L.; Lu, W.H.; Yang, J.; Wen, S.; Chiao, P.J.; et al. Micro-RNA-155 is induced by K-Ras oncogenic signal and promotes ROS stress in pancreatic cancer. Oncotarget 2015, 6, 21148–21158. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, S.; Alimova, I.; Fan, R.; Harris, P.; Foreman, N.; Vibhakar, R. MicroRNA 128a increases intracellular ROS level by targeting Bmi-1 and inhibits medulloblastoma cancer cell growth by promoting senescence. PLoS ONE 2010, 5, e10748. [Google Scholar] [CrossRef] [PubMed]
- Mateescu, B.; Batista, L.; Cardon, M.; Gruosso, T.; de Feraudy, Y.; Mariani, O.; Nicolas, A.; Meyniel, J.P.; Cottu, P.; Sastre-Garau, X.; et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat. Med. 2011, 17, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Jajoo, S.; Mukherjea, D.; Kaur, T.; Sheehan, K.E.; Sheth, S.; Borse, V.; Rybak, L.P.; Ramkumar, V. Essential role of NADPH oxidase-dependent reactive oxygen species generation in regulating microRNA-21 expression and function in prostate cancer. Antioxid. Redox Signal. 2013, 19, 1863–1876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, X.; Dhakal, I.B.; Gross, M.D.; Kadlubar, F.F.; Anderson, K.E. Sequence variants in antioxidant defense and DNA repair genes, dietary antioxidants, and pancreatic cancer risk. Int. J. Mol. Epidemiol. Genet. 2011, 2, 236–244. [Google Scholar] [PubMed]
- Mohelnikova-Duchonova, B.; Marsakova, L.; Vrana, D.; Holcatova, I.; Ryska, M.; Smerhovsky, Z.; Slamova, A.; Schejbalova, M.; Soucek, P. Superoxide dismutase and nicotinamide adenine dinucleotide phosphate: Quinone oxidoreductase polymorphisms and pancreatic cancer risk. Pancreas 2011, 40, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Choudhuri, G.; Kumar, R.; Agarwal, S. Association of 5, 10-methylenetetrahydrofolate reductase C677T polymorphism in susceptibility to tropical chronic pancreatitis in north Indian population. Cell Mol. Biol. 2012, 58, 122–127. [Google Scholar] [PubMed]
- Vecka, M.; Jachymova, M.; Vavrova, L.; Kodydkova, J.; Macasek, J.; Urbanek, M.; Krechler, T.; Slaby, A.; Duskova, J.; Muravska, A.; et al. Paraoxonase-1 (PON1) status in pancreatic cancer: Relation to clinical parameters. Folia Biol. 2012, 58, 231–237. [Google Scholar]
- Jung, S.H.; Kim, S.M.; Lee, C.E. Mechanism of suppressors of cytokine signaling 1 inhibition of epithelial-mesenchymal transition signaling through ROS regulation in colon cancer cells: Suppression of Src leading to thioredoxin up-regulation. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, D.M.; Asaithamby, A.; Bailey, S.M.; Costes, S.V.; Doetsch, P.W.; Dynan, W.S.; Kronenberg, A.; Rithidech, K.N.; Saha, J.; Snijders, A.M.; et al. Understanding cancer development processes after HZE-particle exposure: Roles of ROS, DNA damage repair and inflammation. Radiat. Res. 2015, 183, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Lee, S.; Lee, E.K. ROS and energy metabolism in cancer cells: Alliance for fast growth. Arch. Pharm. Res. 2015, 38, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/IL-8 and CXCL12/SDF-1α co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Wigmore, S.J.; Fearon, K.C.; Sangster, K.; Maingay, J.P.; Garden, O.J.; Ross, J.A. Cytokine regulation of constitutive production of interleukin-8 and -6 by human pancreatic cancer cell lines and serum cytokine concentrations in patients with pancreatic cancer. Int. J. Oncol. 2002, 21, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Bellone, G.; Smirne, C.; Mauri, F.A.; Tonel, E.; Carbone, A.; Buffolino, A.; Dughera, L.; Robecchi, A.; Pirisi, M.; Emanuelli, G. Cytokine expression profile in human pancreatic carcinoma cells and in surgical specimens: Implications for survival. Cancer Immunol. Immunother. 2006, 55, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.; Gansauge, F.; Beger, H.; Dolara, P.; Winde, G.; Bartsch, H. Increased etheno-DNA adducts in affected tissues of patients suffering from Crohn’s disease, ulcerative colitis, and chronic pancreatitis. Antioxid. Redox Signal. 2006, 8, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Tas, F.; Aykan, F.; Alici, S.; Kaytan, E.; Aydiner, A.; Topuz, E. Prognostic factors in pancreatic carcinoma: Serum LDH levels predict survival in metastatic disease. Am. J. Clin. Oncol. 2001, 24, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Hocevar, B.A.; Kamendulis, L.M.; Pu, X.; Perkins, S.M.; Wang, Z.Y.; Johnston, E.L.; DeWitt, J.M.; Li, L.; Loehrer, P.J.; Klaunig, J.E.; et al. Contribution of environment and genetics to pancreatic cancer susceptibility. PLoS ONE 2014, 9, e90052. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.F.; Wang, S.X.; Zhang, F.R.; Peng, L.; Xiao, Y.; Zhang, M. Interleukin-1α, 6 regulate the secretion of vascular endothelial growth factor, A, C in pancreatic cancer. Hepatobiliary Pancreat. Dis. Int. 2005, 4, 460–463. [Google Scholar] [PubMed]
- Kang, R.; Tang, D.; Lotze, M.T.; Zeh, H.J., 3rd. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy 2012, 8, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Vorndamm, J.; Muerkoster, S.; Yu, H.; Schmidt, W.E.; Folsch, U.R.; Schafer, H. Autocrine production of interleukin 1beta confers constitutive nuclear factor kappaB activity and chemoresistance in pancreatic carcinoma cell lines. Cancer Res. 2002, 62, 910–916. [Google Scholar] [PubMed]
- Kuwada, Y.; Sasaki, T.; Morinaka, K.; Kitadai, Y.; Mukaida, N.; Chayama, K. Potential involvement of IL-8 and its receptors in the invasiveness of pancreatic cancer cells. Int. J. Oncol. 2003, 22, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Egberts, J.H.; Cloosters, V.; Noack, A.; Schniewind, B.; Thon, L.; Klose, S.; Kettler, B.; von Forstner, C.; Kneitz, C.; Tepel, J.; et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008, 68, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Ellenrieder, V.; Hendler, S.F.; Ruhland, C.; Boeck, W.; Adler, G.; Gress, T.M. TGF-β-induced invasiveness of pancreatic cancer cells is mediated by matrix metalloproteinase-2 and the urokinase plasminogen activator system. Int. J. Cancer 2001, 93, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Truty, M.J.; Urrutia, R. Basics of TGF-β and pancreatic cancer. Pancreatology 2007, 7, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Xiong, Y.; Lei, Q.Y.; Guan, K.L. LDH-A Acetylation: Implication in pancreatic cancer initiation and diagnosis. Oncotarget 2013. [Google Scholar] [CrossRef] [PubMed]
- Tas, F.; Karabulut, S.; Ciftci, R.; Sen, F.; Sakar, B.; Disci, R.; Duranyildiz, D. Serum levels of LDH, CEA, and CA19–9 have prognostic roles on survival in patients with metastatic pancreatic cancer receiving gemcitabine-based chemotherapy. Cancer Chemother. Pharmacol. 2014, 73, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Heinemann, V.; Kullmann, F.; Laubender, R.P.; Klose, C.; Bruns, C.J.; Holdenrieder, S.; Modest, D.P.; Schulz, C.; Boeck, S. Prognostic value of CA 19–9, CEA, CRP, LDH and bilirubin levels in locally advanced and metastatic pancreatic cancer: Results from a multicenter, pooled analysis of patients receiving palliative chemotherapy. J. Cancer Res. Clin. Oncol. 2013, 139, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Useros, J.; Garcia-Foncillas, J. Obesity and colorectal cancer: Molecular features of adipose tissue. J. Transl. Med. 2016, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Hirai, F.; Motoori, S.; Kakinuma, S.; Tomita, K.; Indo, H.P.; Kato, H.; Yamaguchi, T.; Yen, H.C.; Clair, D.K.; Nagano, T.; et al. Mitochondrial signal lacking manganese superoxide dismutase failed to prevent cell death by reoxygenation following hypoxia in a human pancreatic cancer cell line, KP4. Antioxid. Redox Signal. 2004, 6, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Otani, K.; Shimizu, S.; Chijiiwa, K.; Yamaguchi, K.; Noshiro, H.; Tanaka, M. Immunohistochemical detection of 8-hydroxy-2′-deoxyguanosine in gallbladder epithelium of patients with pancreaticobiliary maljunction. Eur. J. Gastroenterol. Hepatol. 2001, 13, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lei, X.G.; Wang, J. Malondialdehyde regulates glucose-stimulated insulin secretion in murine islets via TCF7L2-dependent Wnt signaling pathway. Mol. Cell Endocrinol. 2014, 382, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Blair, I.A. DNA adducts with lipid peroxidation products. J. Biol. Chem. 2008, 283, 15545–15549. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, L.Y.; Song, T.S.; Ni, L.; Yang, L.; Hu, X.Y.; Hu, J.S.; Song, L.P.; Luo, Y.; Si, L.S. Apoptosis of pancreatic cancer BXPC-3 cells induced by indole-3-acetic acid in combination with horseradish peroxidase. World J. Gastroenterol. 2005, 11, 4519–4523. [Google Scholar] [CrossRef] [PubMed]
- Schiavon, C.C.; Vieira, F.G.; Ceccatto, V.; de Liz, S.; Cardoso, A.L.; Sabel, C.; Gonzalez-Chica, D.A.; da Silva, E.L.; Galvan, D.; Crippa, C.G.; et al. Nutrition education intervention for women with breast cancer: Effect on nutritional factors and oxidative stress. J. Nutr. Educ. Behav. 2015, 47, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Centeno, B.A.; Chen, D.T.; Fulp, W.J.; Perez, M.; Lee, G.Z.; Luetteke, N.; Hingorani, S.R.; Sebti, S.M.; Malafa, M.P. Prolonged survival and delayed progression of pancreatic intraepithelial neoplasia in LSL-KrasG12D/+;Pdx-1-Cre mice by vitamin E delta-tocotrienol. Carcinogenesis 2013, 34, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Springett, G.M.; Husain, K.; Neuger, A.; Centeno, B.; Chen, D.T.; Hutchinson, T.Z.; Lush, R.M.; Sebti, S.; Malafa, M.P. A Phase I Safety, Pharmacokinetic, and Pharmacodynamic Presurgical Trial of Vitamin E delta-tocotrienol in Patients with Pancreatic Ductal Neoplasia. EBioMedicine 2015, 2, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Patacsil, D.; Osayi, S.; Tran, A.T.; Saenz, F.; Yimer, L.; Shajahan, A.N.; Gokhale, P.C.; Verma, M.; Clarke, R.; Chauhan, S.C.; et al. Vitamin E succinate inhibits survivin and induces apoptosis in pancreatic cancer cells. Genes Nutr. 2012, 7, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Lawson, S. Vitamin C and cancer revisited. Proc. Natl. Acad. Sci. USA 2008, 105, 11037–11038. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Vitamin C: Poison, prophylactic or panacea? Trends Biochem. Sci. 1999, 24, 255–259. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, O.; Munoz-Sagastibelza, M.; Torrejon, B.; Borrero-Palacios, A.; Del Puerto-Nevado, L.; Martinez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; Garcia, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Martin, S.M.; Levine, M.; Wagner, B.A.; Buettner, G.R.; Wang, S.H.; Taghiyev, A.F.; Du, C.; Knudson, C.M.; Cullen, J.J. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin. Cancer Res. 2010, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Calderon, P.B. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic. Biol. Med. 2009, 47, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.A.; Mitchell, E.; Bazzan, A.J.; Littman, S.; Zabrecky, G.; Yeo, C.J.; Pillai, M.V.; Newberg, A.B.; Deshmukh, S.; Levine, M. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS ONE 2012, 7, e29794. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Roberts, L.J.; et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra218. [Google Scholar] [CrossRef] [PubMed]
- Bimonte, S.; Barbieri, A.; Leongito, M.; Piccirillo, M.; Giudice, A.; Pivonello, C.; de Angelis, C.; Granata, V.; Palaia, R.; Izzo, F. Curcumin AntiCancer Studies in Pancreatic Cancer. Nutrients 2016, 8, 433. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Imaizumi, A.; Otsuka, Y.; Sasaki, H.; Hashiguchi, M.; Tsujiko, K.; Matsumoto, S.; Ishiguro, H.; Chiba, T. Dose-escalation and pharmacokinetic study of nanoparticle curcumin, a potential anticancer agent with improved bioavailability, in healthy human volunteers. Cancer Chemother. Pharmacol. 2012, 69, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.J.; Vazquez Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochim. Biophys. Acta 2016, 1857, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Hertz, N.; Lister, R.E. Improved survival in patients with end-stage cancer treated with coenzyme Q(10) and other antioxidants: A pilot study. J. Int. Med. Res. 2009, 37, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef]
- Shin, S.H.; Kim, S.C.; Hong, S.M.; Kim, Y.H.; Song, K.B.; Park, K.M.; Lee, Y.J. Genetic alterations of K-ras, p53, c-erbB-2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas 2013, 42, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Deramaudt, T.; Rustgi, A.K. Mutant KRAS in the initiation of pancreatic cancer. Biochim. Biophys. Acta 2005, 25, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Nothlings, U.; Wilkens, L.R.; Murphy, S.P.; Hankin, J.H.; Henderson, B.E.; Kolonel, L.N. Meat and fat intake as risk factors for pancreatic cancer: The multiethnic cohort study. J. Natl. Cancer Inst. 2005, 97, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
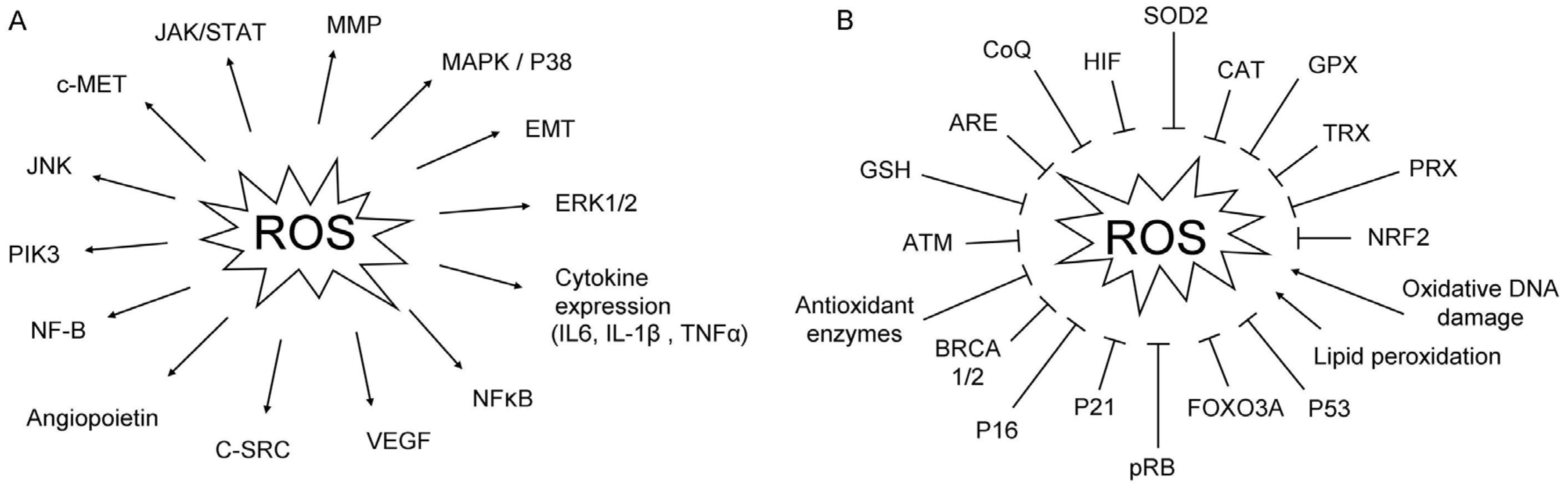

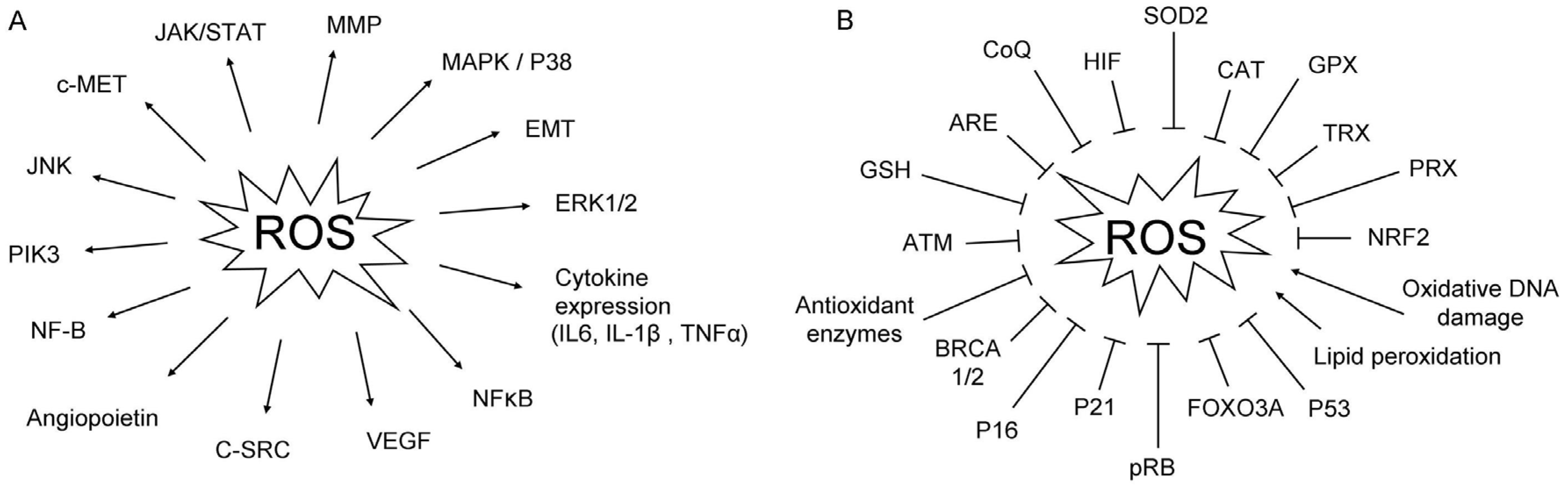{kind=link}
| Factor | Target | Role in Tumorogenesis | References |
|---|---|---|---|
| NADPH | H2O2, -OH, O2-radicals | loss of biochemical homeostasis | [66] |
| IGF1 | Increased ROS production and NAD(P)H oxidase activity | anti-apoptosis and agresiveness | [25] |
| FGF2 | Increased ROS production | anti-apoptosis | [25] |
| IL-2 | VEGF | angiogenesis | [73] |
| IL-6 | STAT3 | proliferation | [74] |
| IL-1β | NF-κB, COX2 | invasiveness, chemoresistance | [75] |
| IL-8 | VEGF, VEGFR, Neuropilin-2, MAPK, MMP2 | proliferation, invasiveness, survival angiogenesis | [67,76] |
| TNF-α | NF-κB, AP1 | invasiveness | [77] |
| TNF-β | MMP2, urokinase | proliferation, invasiveness | [78,79] |
| LDH | Regulated by c-Myc and HIF1 | predictive biomarker of gemcitabine response, prognosis | [80,81,82] |
| 4-HNE | GSH | inflammation, pancreatic maljunction | [70,84,85,86] |
| MDA | DNA, WNT pathway | inflammation, apoptotic biomarker | [72,87,88,89] |
| Molecule | Dose | Study | n | Parameters | Results | Reference |
|---|---|---|---|---|---|---|
| Vitamin E | 200 mg/kg twice a day, for 12 months | In vivo | 92 mice | Survival, progression | Increased survival (p < 0.025). Induced BAX and Caspase 3 | [91] |
| Vitamin E | 200–3200 mg daily for 13 days | Phase I | 25 patients | Safety, pharmacokinetics, apoptosis | Apoptosis induction (p = 0.044) | [92] |
| Vitamin E | 25.1 to 51.3 μM | In vitro | PANC-1, COLO-357, and ASPC-1 cell lines | Cell viability, apoptosis, cell cycle | Inhibition of proliferation. Apoptosis induction (p < 0.01). | [93] |
| Curcumin | 8 g orally daily | Phase II | 25 patients | Tumor volume and interleukin levels | Decreased pSTAT3 (p = 0.009), COX2 (p = 0.029), and L-6, IL-8, IL-10, and IL-1RA (- to 35-fold) | [105] |
| Ascorbate | Ascorbate dose of 15 g was infused with subsequent dose escalation of 25 to 100 g over 50 min/0–20 mM for 1 h | In vivo | 194 mice | Tumor volume and ascorbate levels | Ascorbate decreased growth of ovarian (p < 0.005), pancreatic (p < 0.05), and glioblastoma (p < 0.001) mice tumors | [96] |
| Ascorbate | 50.75 and 100 g three infusions per week, for eight weeks | Phase I | 9 patients (stage IV) | Safety and progression | Null toxicity. Seven patients with stable disease, 2 patients with progression disease | [101] |
| Ascorbate | 4 g/kg for two weeks 0.5–10 mmol/L for 1 h | In vivo | 28 mice | Tumor growth | Ascorbate inhibited tumor growth (p = 0.001) | [99] |
| Ascorbate | 15–125 g twice weekly | Phase I | 9 patients | Safety and progression | Ascorbate combined with gemcitabine should be safe and well tolerated | [102] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Useros, J.; Li, W.; Cabeza-Morales, M.; Garcia-Foncillas, J. Oxidative Stress: A New Target for Pancreatic Cancer Prognosis and Treatment. J. Clin. Med. 2017, 6, 29. https://doi.org/10.3390/jcm6030029
Martinez-Useros J, Li W, Cabeza-Morales M, Garcia-Foncillas J. Oxidative Stress: A New Target for Pancreatic Cancer Prognosis and Treatment. Journal of Clinical Medicine. 2017; 6(3):29. https://doi.org/10.3390/jcm6030029
Chicago/Turabian StyleMartinez-Useros, Javier, Weiyao Li, Marticela Cabeza-Morales, and Jesus Garcia-Foncillas. 2017. "Oxidative Stress: A New Target for Pancreatic Cancer Prognosis and Treatment" Journal of Clinical Medicine 6, no. 3: 29. https://doi.org/10.3390/jcm6030029
APA StyleMartinez-Useros, J., Li, W., Cabeza-Morales, M., & Garcia-Foncillas, J. (2017). Oxidative Stress: A New Target for Pancreatic Cancer Prognosis and Treatment. Journal of Clinical Medicine, 6(3), 29. https://doi.org/10.3390/jcm6030029