
Protein Kinase D Enzymes as Regulators of EMT and Cancer Cell Invasion

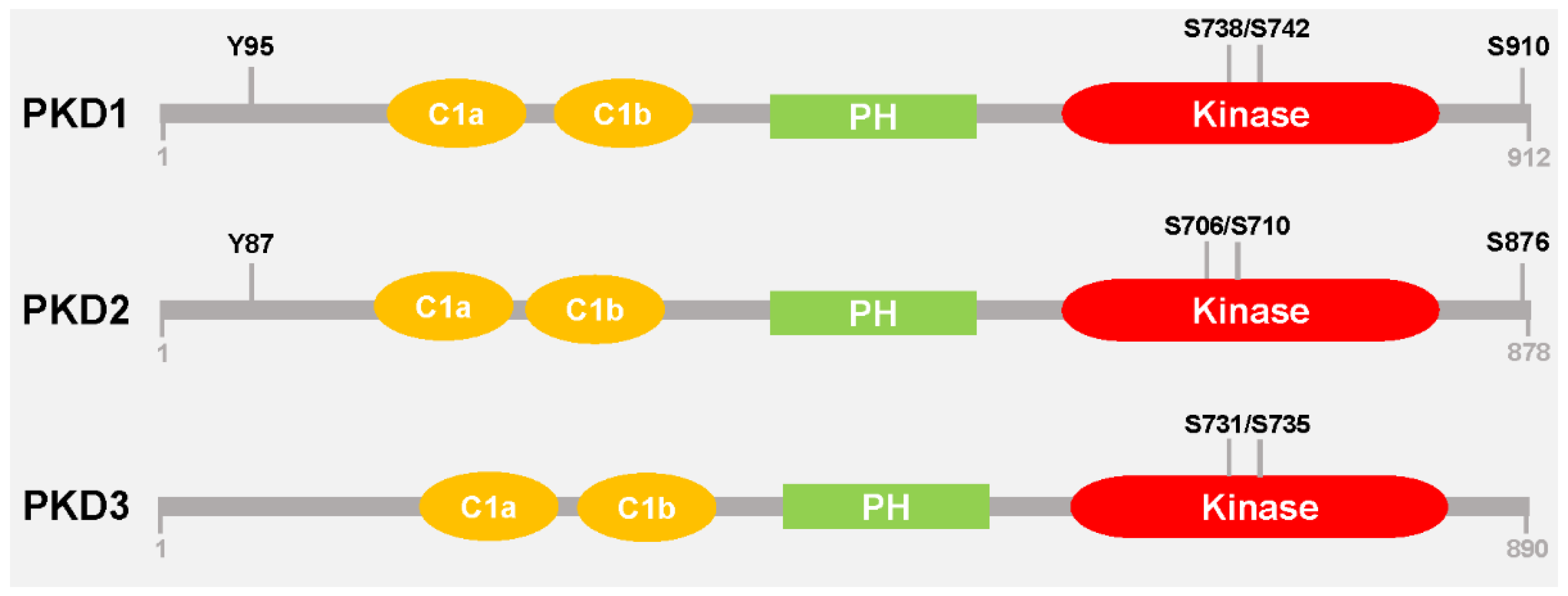{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. EMT and Cell Migration
3. PKD1 Maintains the Epithelial Phenotype and Inhibits Cell Migration
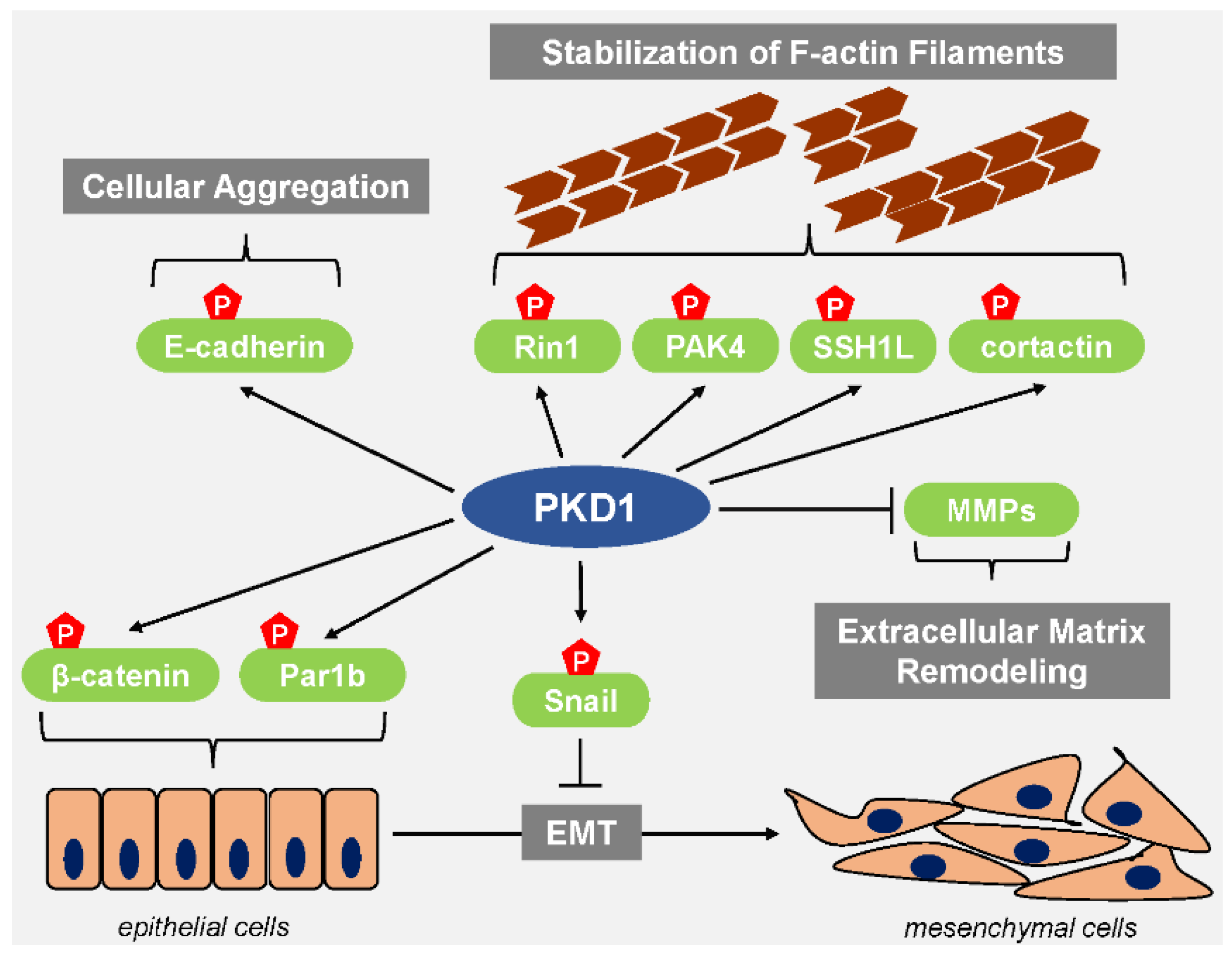
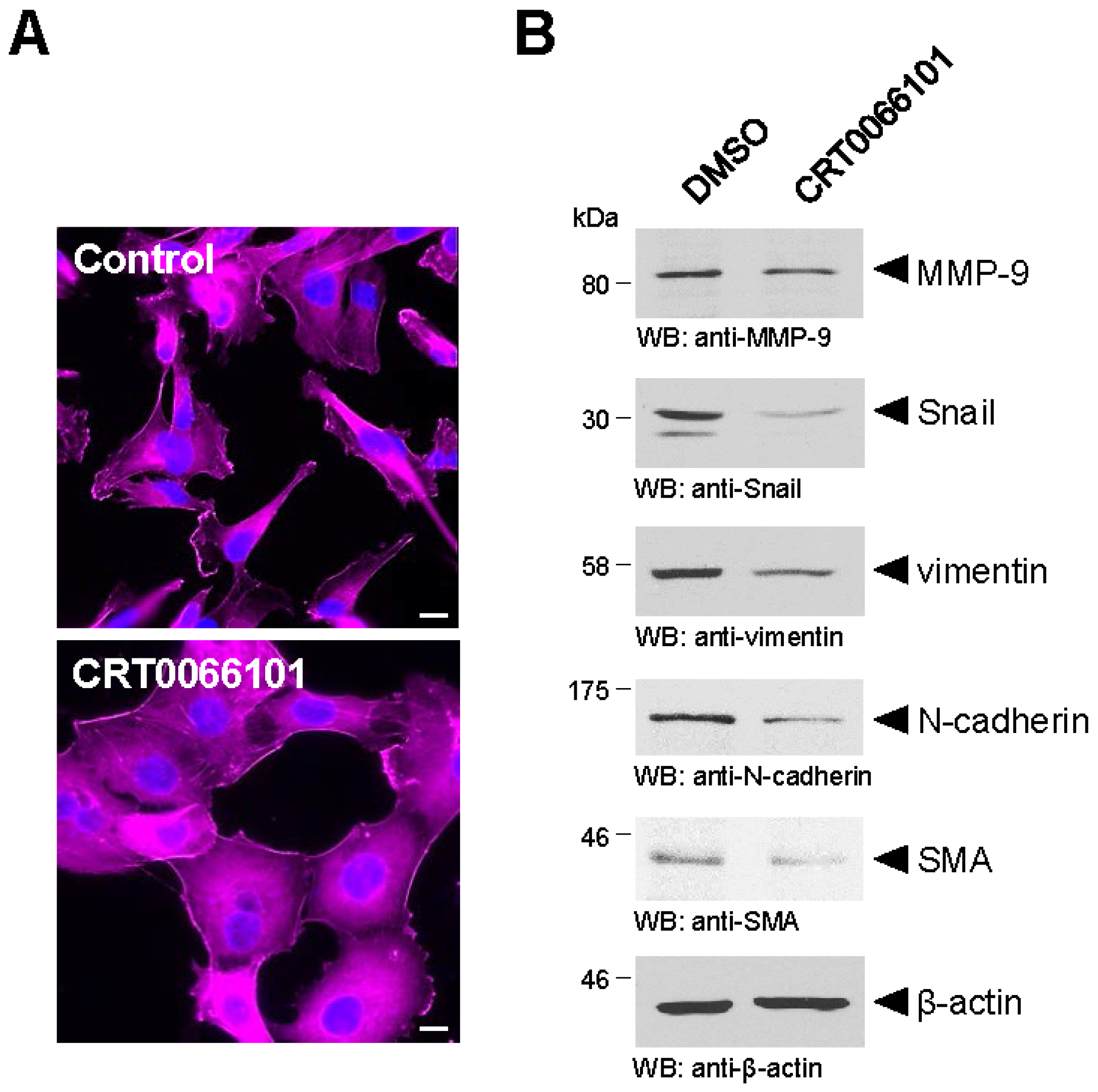
4. PKD1 in Cancer
5. Contribution of PKD2 and PKD3 to EMT and Cell Migration
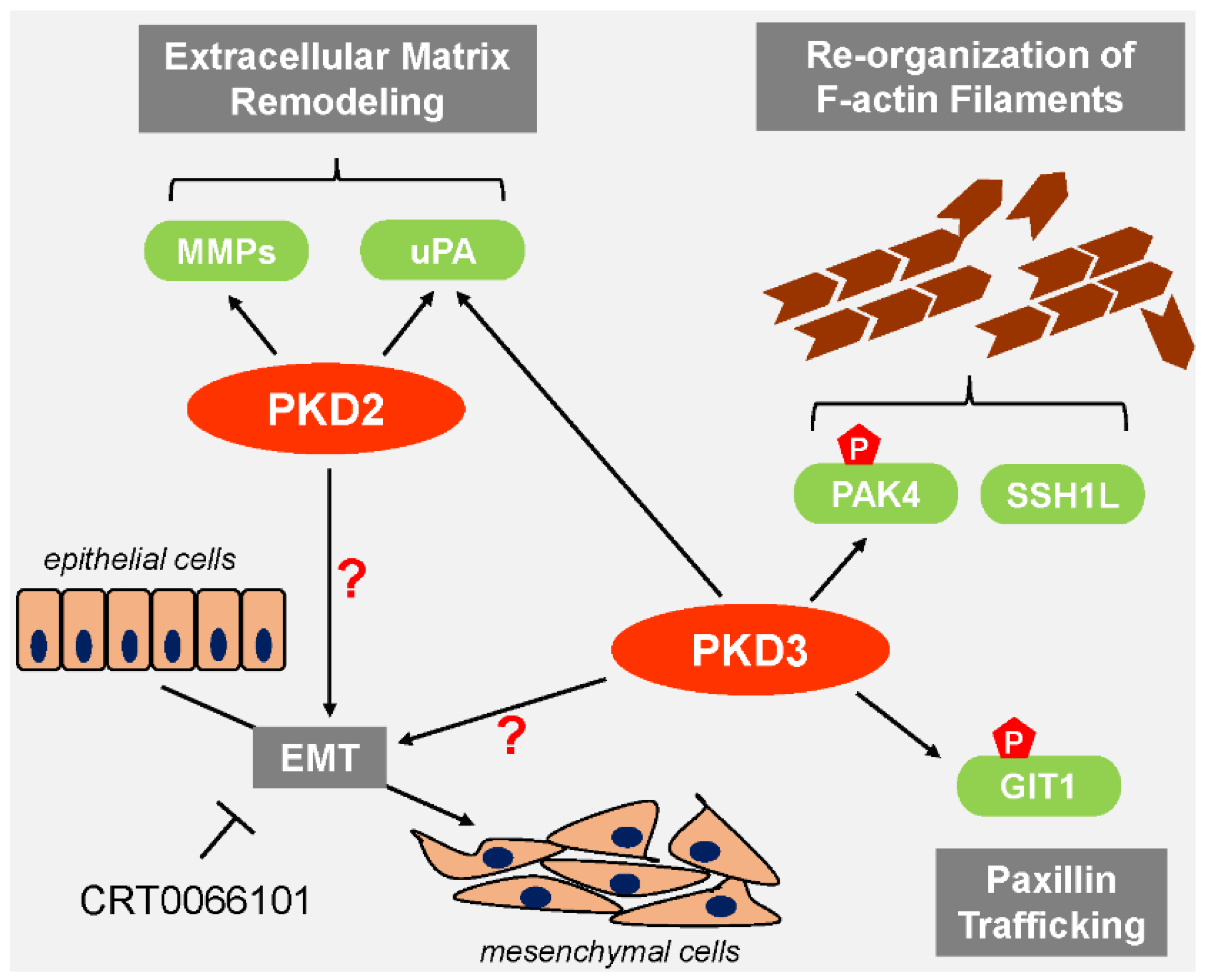
6. PKD2 and PKD3 in Cancer
7. PKD as a Potential Therapeutic Target
8. Summary and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fu, Y.; Rubin, C.S. Protein kinase d: Coupling Extracellular Stimuli to the Regulation of Cell Physiology. EMBO Rep. 2011, 12, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Johannes, F.J.; Prestle, J.; Eis, S.; Oberhagemann, P.; Pfizenmaier, K. Pkcu is a novel, atypical member of the protein kinase c family. J. Biol. Chem. 1994, 269, 6140–6148. [Google Scholar] [PubMed]
- Valverde, A.M.; Sinnett-Smith, J.; van Lint, J.; Rozengurt, E. Molecular cloning and characterization of protein kinase d: A Target for Diacylglycerol and Phorbol Esters with a Distinctive Catalytic Domain. Proc. Natl Acad. Sci. USA 1994, 91, 8572–8576. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E.; Rey, O.; Waldron, R.T. Protein kinase d signaling. J. Biol. Chem. 2005, 280, 13205–13208. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Beznoussenko, G.V.; van Lint, J.; Mironov, A.A.; Malhotra, V. Recruitment of protein kinase d to the trans-golgi network via the first cysteine-rich domain. EMBO J. 2001, 20, 5982–5990. [Google Scholar] [CrossRef] [PubMed]
- Rey, O.; Sinnett-Smith, J.; Zhukova, E.; Rozengurt, E. Regulated nucleocytoplasmic transport of protein kinase d in response to g protein-coupled receptor activation. J. Biol. Chem. 2001, 276, 49228–49235. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, T.; Rozengurt, E. Protein kinase d activation by mutations within its pleckstrin homology domain. J. Biol. Chem. 1998, 273, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E.; Sinnett-Smith, J.; van Lint, J.; Valverde, A.M. Protein kinase d (pkd): A Novel Target for Diacylglycerol and Phorbol Esters. Mutat. Res. 1995, 333, 153–160. [Google Scholar] [CrossRef]
- Steinberg, S.F. Regulation of protein kinase d1 activity. Mol. Pharmacol. 2012, 81, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Toker, A. Protein kinase d mediates a stress-induced nf-kappab activation and survival pathway. EMBO J. 2003, 22, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Waldron, R.T.; Rozengurt, E. Oxidative stress induces protein kinase d activation in intact cells. Involvement of src and dependence on protein kinase c. J. Biol. Chem. 2000, 275, 17114–17121. [Google Scholar] [CrossRef] [PubMed]
- Doppler, H.; Storz, P. A novel tyrosine phosphorylation site in protein kinase d contributes to oxidative stress-mediated activation. J. Biol. Chem. 2007, 282, 31873–31881. [Google Scholar] [CrossRef] [PubMed]
- Jamora, C.; Yamanouye, N.; van Lint, J.; Laudenslager, J.; Vandenheede, J.R.; Faulkner, D.J.; Malhotra, V. Gbetagamma-mediated regulation of golgi organization is through the direct activation of protein kinase D. Cell 1999, 98, 59–68. [Google Scholar] [CrossRef]
- Endo, K.; Oki, E.; Biedermann, V.; Kojima, H.; Yoshida, K.; Johannes, F.J.; Kufe, D.; Datta, R. Proteolytic cleavage and activation of protein kinase c [micro] by caspase-3 in the apoptotic response of cells to 1-beta -d-arabinofuranosylcytosine and other genotoxic agents. J. Biol. Chem. 2000, 275, 18476–18481. [Google Scholar] [CrossRef] [PubMed]
- Eiseler, T.; Doppler, H.; Yan, I.K.; Kitatani, K.; Mizuno, K.; Storz, P. Protein kinase d1 regulates cofilin-mediated f-actin reorganization and cell motility through slingshot. Nat. Cell Biol. 2009, 11, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Peterburs, P.; Heering, J.; Link, G.; Pfizenmaier, K.; Olayioye, M.A.; Hausser, A. Protein kinase d regulates cell migration by direct phosphorylation of the cofilin phosphatase slingshot 1 like. Cancer Res. 2009, 69, 5634–5638. [Google Scholar] [CrossRef] [PubMed]
- Spratley, S.J.; Bastea, L.I.; Doppler, H.; Mizuno, K.; Storz, P. Protein kinase d regulates cofilin activity through p21-activated kinase 4. J. Biol. Chem. 2011, 286, 34254–34261. [Google Scholar] [CrossRef] [PubMed]
- Bastea, L.I.; Doppler, H.; Balogun, B.; Storz, P. Protein kinase d1 maintains the epithelial phenotype by inducing a DNA-bound, inactive snai1 transcriptional repressor complex. PLoS ONE 2012, 7, e30459. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Zhang, C.; Hassan, S.; Biswas, M.H.; Balaji, K.C. Protein kinase d1 suppresses epithelial-to-mesenchymal transition through phosphorylation of snail. Cancer Res. 2010, 70, 7810–7819. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Shen, M.; Zha, Y.L.; Li, W.; Wei, Y.; Blanco, M.A.; Ren, G.; Zhou, T.; Storz, P.; Wang, H.Y.; et al. Pkd1 phosphorylation-dependent degradation of snail by scf-fbxo11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell 2014, 26, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Baron, C.L.; Malhotra, V. Role of diacylglycerol in pkd recruitment to the tgn and protein transport to the plasma membrane. Science 2002, 295, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Bossard, C.; Bresson, D.; Polishchuk, R.S.; Malhotra, V. Dimeric pkd regulates membrane fission to form transport carriers at the tgn. J. Cell Biol. 2007, 179, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Hausser, A.; Storz, P.; Martens, S.; Link, G.; Toker, A.; Pfizenmaier, K. Protein kinase d regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase iiibeta at the golgi complex. Nat. Cell Biol. 2005, 7, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.H.; Wang, W.; Jhun, B.S.; Wong, C.; Hausser, A.; Pfizenmaier, K.; McKinsey, T.A.; Olson, E.N.; Jin, Z.G. Protein kinase d-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J. Biol. Chem. 2008, 283, 14590–14599. [Google Scholar] [CrossRef] [PubMed]
- Dequiedt, F.; van Lint, J.; Lecomte, E.; van Duppen, V.; Seufferlein, T.; Vandenheede, J.R.; Wattiez, R.; Kettmann, R. Phosphorylation of histone deacetylase 7 by protein kinase d mediates t cell receptor-induced nur77 expression and apoptosis. J. Exp. Med. 2005, 201, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Feng, H.; Fu, Y.; Land, M.; Rubin, C.S. Protein kinase d is an essential regulator of c. Elegans innate immunity. Immunity 2009, 30, 521–532. [Google Scholar] [PubMed]
- Sumara, G.; Formentini, I.; Collins, S.; Sumara, I.; Windak, R.; Bodenmiller, B.; Ramracheya, R.; Caille, D.; Jiang, H.; Platt, K.A.; et al. Regulation of pkd by the mapk p38delta in insulin secretion and glucose homeostasis. Cell 2009, 136, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, E.; Cazorla, O.; Schwenk, R.W.; Lorenzen-Schmidt, I.; Sadayappan, S.; van Lint, J.; Carrier, L.; van Eys, G.J.; Glatz, J.F.; Luiken, J.J. Protein kinase d increases maximal ca2+-activated tension of cardiomyocyte contraction by phosphorylation of cmybp-c-ser315. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H323–H331. [Google Scholar] [CrossRef] [PubMed]
- Borges, S.; Doppler, H.; Perez, E.A.; Andorfer, C.A.; Sun, Z.; Anastasiadis, P.Z.; Thompson, E.; Geiger, X.J.; Storz, P. Pharmacologic reversion of epigenetic silencing of the prkd1 promoter blocks breast tumor cell invasion and metastasis. Breast Cancer Res. 2013, 15, R66. [Google Scholar] [CrossRef] [PubMed]
- Borges, S.; Perez, E.A.; Thompson, E.A.; Radisky, D.C.; Geiger, X.J.; Storz, P. Effective targeting of estrogen receptor-negative breast cancers with the protein kinase d inhibitor crt0066101. Mol. Cancer Ther. 2015, 14, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lu, L.; Feng, Y.; Wang, H.; Dai, L.; Li, Y.; Zhang, P. Pkd2 mediates multi-drug resistance in breast cancer cells through modulation of p-glycoprotein expression. Cancer Lett. 2011, 300, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; McKenzie, R.; Gan, H.; Tang, H. Protein kinases d2 and d3 are novel growth regulators in hcc1806 triple-negative breast cancer cells. Anticancer Res. 2013, 33, 393–399. [Google Scholar] [PubMed]
- Huck, B.; Duss, S.; Hausser, A.; Olayioye, M.A. Elevated protein kinase d3 (pkd3) expression supports proliferation of triple-negative breast cancer cells and contributes to mtorc1-s6k1 pathway activation. J. Biol. Chem. 2014, 289, 3138–3147. [Google Scholar] [CrossRef] [PubMed]
- Wille, C.; Kohler, C.; Armacki, M.; Jamali, A.; Gossele, U.; Pfizenmaier, K.; Seufferlein, T.; Eiseler, T. Protein kinase d2 induces invasion of pancreatic cancer cells by regulating matrix metalloproteinases. Mol. Biol. Cell 2014, 25, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Tanasanvimon, S.; Sinnett-Smith, J.; Rozengurt, E. Role of protein kinase d signaling in pancreatic cancer. Biochem. Pharmacol. 2010, 80, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ochi, N.; Tong, Z.; Deorukhkar, A.; Sung, B.; Kelland, L.; Jamieson, S.; Sutherland, R.; Raynham, T.; et al. A novel small-molecule inhibitor of protein kinase d blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Ochi, N.; Tanasanvimon, S.; Matsuo, Y.; Tong, Z.; Sung, B.; Aggarwal, B.B.; Sinnett-Smith, J.; Rozengurt, E.; Guha, S. Protein kinase d1 promotes anchorage-independent growth, invasion, and angiogenesis by human pancreatic cancer cells. J. Cell. Physiol. 2011, 226, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Rozengurt, E. Pkd, pkd2, and p38 mapk mediate hsp27 serine-82 phosphorylation induced by neurotensin in pancreatic cancer panc-1 cells. J. Cell. Biochem. 2008, 103, 648–662. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Deng, F.; Singh, S.V.; Wang, Q.J. Protein kinase d3 (pkd3) contributes to prostate cancer cell growth and survival through a pkcepsilon/pkd3 pathway downstream of akt and erk 1/2. Cancer Res. 2008, 68, 3844–3853. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, M.; Rao, P.S.; Smith, D.J.; Hemstreet, G.P.; Balaji, K.C. Protein kinase c mu is down-regulated in androgen-independent prostate cancer. Biochem. Biophys. Res. Commun. 2003, 307, 254–260. [Google Scholar] [CrossRef]
- Jaggi, M.; Rao, P.S.; Smith, D.J.; Wheelock, M.J.; Johnson, K.R.; Hemstreet, G.P.; Balaji, K.C. E-cadherin phosphorylation by protein kinase d1/protein kinase c{mu} is associated with altered cellular aggregation and motility in prostate cancer. Cancer Res. 2005, 65, 483–492. [Google Scholar] [PubMed]
- LaValle, C.R.; Zhang, L.; Xu, S.; Eiseman, J.L.; Wang, Q.J. Inducible silencing of protein kinase d3 inhibits secretion of tumor-promoting factors in prostate cancer. Mol. Cancer Ther. 2012, 11, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Zeng, F.; Xu, W.; Wang, C.; Ke, Z.; Wang, Q.J.; Deng, F. Pkd2 and pkd3 promote prostate cancer cell invasion by modulating nf-kappab- and hdac1-mediated expression and activation of upa. J. Cell Sci. 2012, 125, 4800–4811. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Schroer, J.; Azoitei, N.; Eiseler, T.; Bergmann, W.; Kohntop, R.; Lin, Q.; Costa, I.G.; Zenke, M.; Genze, F.; et al. A time frame permissive for protein kinase d2 activity to direct angiogenesis in mouse embryonic stem cells. Sci. Rep. 2015, 5, 11742. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhu, X.; Zhu, Y.; Liu, J.; Hu, X.; Wang, Y.; Peng, S.; Chen, Y.; Chen, R.; Ding, F.; et al. Protein kinase d3 is essential for prostratin-activated transcription of integrated hiv-1 provirus promoter via nf-kappab signaling pathway. Biomed. Res. Int. 2014, 2014, 968027. [Google Scholar] [PubMed]
- Huck, B.; Kemkemer, R.; Franz-Wachtel, M.; Macek, B.; Hausser, A.; Olayioye, M.A. Git1 phosphorylation on serine 46 by pkd3 regulates paxillin trafficking and cellular protrusive activity. J. Biol. Chem. 2012, 287, 34604–34613. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.N.; Feijoo-Carnero, C.; Arandilla, A.G.; Trost, M.; Cantrell, D.A. Protein kinase d2 is a digital amplifier of t cell receptor-stimulated diacylglycerol signaling in naive cd8(+) t cells. Sci. Signal. 2014, 7, ra99. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.; Borges, S.; Storz, P. Functional and therapeutic significance of protein kinase d enzymes in invasive breast cancer. Cell. Mol. Life Sci. 2015, 72, 4369–4382. [Google Scholar] [CrossRef] [PubMed]
- Eiseler, T.; Doppler, H.; Yan, I.K.; Goodison, S.; Storz, P. Protein kinase d1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 2009, 11, R13. [Google Scholar] [CrossRef] [PubMed]
- Alpsoy, A.; Gunduz, U. Protein kinase d2 silencing reduced motility of doxorubicin-resistant mcf7 cells. Tumour Biol. 2015, 36, 4417–4426. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F.; Berx, G. The cell-cell adhesion molecule e-cadherin. Cell. Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, A.; Perotti, A.; Sessa, C.; Ruegg, C. N-cadherin as a therapeutic target in cancer. Expert Opin. Investig. Drugs 2007, 16, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. Tgf-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Sun, Y.; Shimokado, A.; Muragaki, Y. The roles of mitogen-activated protein kinase pathways in tgf-beta-induced epithelial-mesenchymal transition. J. Signal. Transduct. 2012, 2012, 289243. [Google Scholar] [CrossRef] [PubMed]
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A Physically Integrated Molecular Process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; Magalhaes, M.A.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell. Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Stossel, T.P.; Fenteany, G.; Hartwig, J.H. Cell surface actin remodeling. J. Cell Sci. 2006, 119, 3261–3264. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Mizushima, T.; Saeki, Y. The proteasome: Molecular Machinery and Pathophysiological Roles. Biol. Chem. 2012, 393, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in emt and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef] [PubMed]
- Martin-Belmonte, F.; Perez-Moreno, M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 2012, 12, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.L.; Lewandowski, K.T.; Meek, S.E.; Storz, P.; Toker, A.; Piwnica-Worms, H. Phosphorylation of the par-1 polarity kinase by protein kinase d regulates 14-3-3 binding and membrane association. Proc. Natl. Acad. Sci. USA 2008, 105, 18378–18383. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Jaggi, M.; Zhang, C.; Balaji, K.C. Protein kinase d1-mediated phosphorylation and subcellular localization of beta-catenin. Cancer Res. 2009, 69, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.; Condeelis, J. The cofilin activity cycle in lamellipodia and invadopodia. J. Cell. Biochem. 2009, 108, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A.; Barisic, S.; Hausser, A. Multi-level control of actin dynamics by protein kinase d. Cell. Signal. 2013, 25, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.; Eiseler, T.; Scholz, R.P.; Beck, A.; Link, G.; Hausser, A. A novel protein kinase d phosphorylation site in the tumor suppressor rab interactor 1 is critical for coordination of cell migration. Mol. Biol. Cell 2011, 22, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Eiseler, T.; Hausser, A.; De Kimpe, L.; van Lint, J.; Pfizenmaier, K. Protein kinase d controls actin polymerization and cell motility through phosphorylation of cortactin. J. Biol. Chem. 2010, 285, 18672–18683. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Nagase, H. Matrix metalloproteinases in cancer. Essays Biochem. 2002, 38, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Sundram, V.; Chauhan, S.C.; Jaggi, M. Emerging roles of protein kinase d1 in cancer. Mol. Cancer Res. 2011, 9, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jang, H.R.; Kim, J.H.; Noh, S.M.; Song, K.S.; Cho, J.S.; Jeong, H.Y.; Norman, J.C.; Caswell, P.T.; Kang, G.H.; et al. Epigenetic inactivation of protein kinase d1 in gastric cancer and its role in gastric cancer cell migration and invasion. Carcinogenesis 2008, 29, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Chu, E.; Wipf, P.; Schmitz, J.C. Protein kinase d as a potential chemotherapeutic target for colorectal cancer. Mol. Cancer Ther. 2014, 13, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Doppler, H.; Bastea, L.I.; Borges, S.; Spratley, S.J.; Pearce, S.E.; Storz, P. Protein kinase d isoforms differentially modulate cofilin-driven directed cell migration. PLoS ONE 2014, 9, e98090. [Google Scholar] [CrossRef] [PubMed]
- Azoitei, N.; Kleger, A.; Schoo, N.; Thal, D.R.; Brunner, C.; Pusapati, G.V.; Filatova, A.; Genze, F.; Moller, P.; Acker, T.; et al. Protein kinase d2 is a novel regulator of glioblastoma growth and tumor formation. Neuro. Oncol. 2011, 13, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Kates, R.E.; Gauger, K.; Willems, A.; Kiechle, M.; Magdolen, V.; Schmitt, M. Urokinase-type plasminogen activator (upa) and its inhibitor pai-i: Novel Tumor-Derived Factors with a High Prognostic and Predictive Impact in Breast Cancer. Thromb. Haemost 2004, 91, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P. Myelodysplastic syndromes: Diagnosis and Treatment. Mayo Clin. Proc. 2015, 90, 969–983. [Google Scholar] [CrossRef] [PubMed]
- Borges, S.; Storz, P. Protein kinase d isoforms: New Targets for Therapy in Invasive Breast Cancers? Expert Rev. Anticancer Ther. 2013, 13, 895–898. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durand, N.; Borges, S.; Storz, P. Protein Kinase D Enzymes as Regulators of EMT and Cancer Cell Invasion. J. Clin. Med. 2016, 5, 20. https://doi.org/10.3390/jcm5020020
Durand N, Borges S, Storz P. Protein Kinase D Enzymes as Regulators of EMT and Cancer Cell Invasion. Journal of Clinical Medicine. 2016; 5(2):20. https://doi.org/10.3390/jcm5020020
Chicago/Turabian StyleDurand, Nisha, Sahra Borges, and Peter Storz. 2016. "Protein Kinase D Enzymes as Regulators of EMT and Cancer Cell Invasion" Journal of Clinical Medicine 5, no. 2: 20. https://doi.org/10.3390/jcm5020020
APA StyleDurand, N., Borges, S., & Storz, P. (2016). Protein Kinase D Enzymes as Regulators of EMT and Cancer Cell Invasion. Journal of Clinical Medicine, 5(2), 20. https://doi.org/10.3390/jcm5020020