
Epithelial-Mesenchymal Transition (EMT) and Regulation of EMT Factors by Steroid Nuclear Receptors in Breast Cancer: A Review and in Silico Investigation


Abstract
1. Introduction
2. Epithelial to Mesenchymal Transition (EMT) and Mesenchymal to Epithelial Transition (MET) in Breast Cancer
3. SNRs and EMT in Breast Cancer
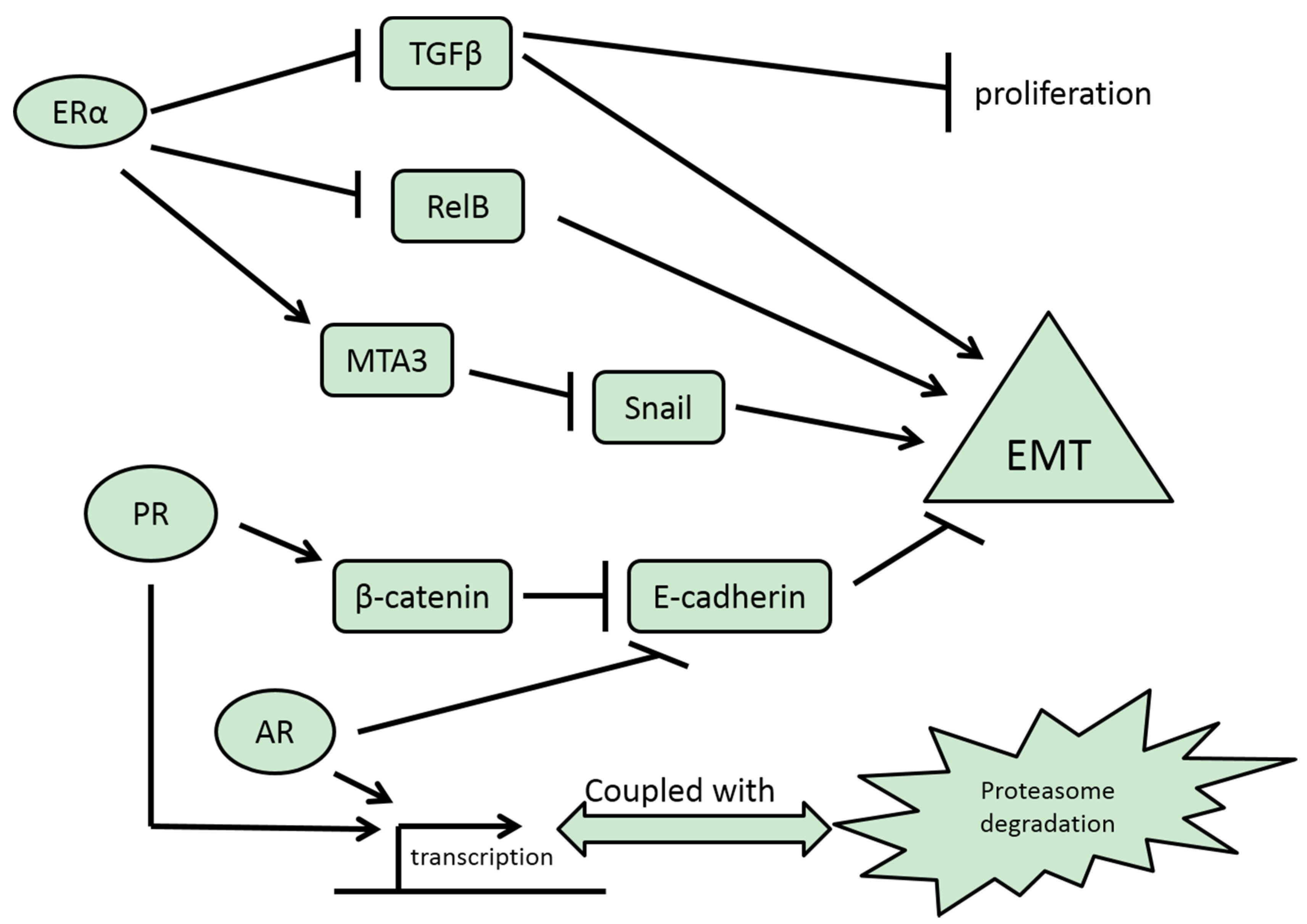
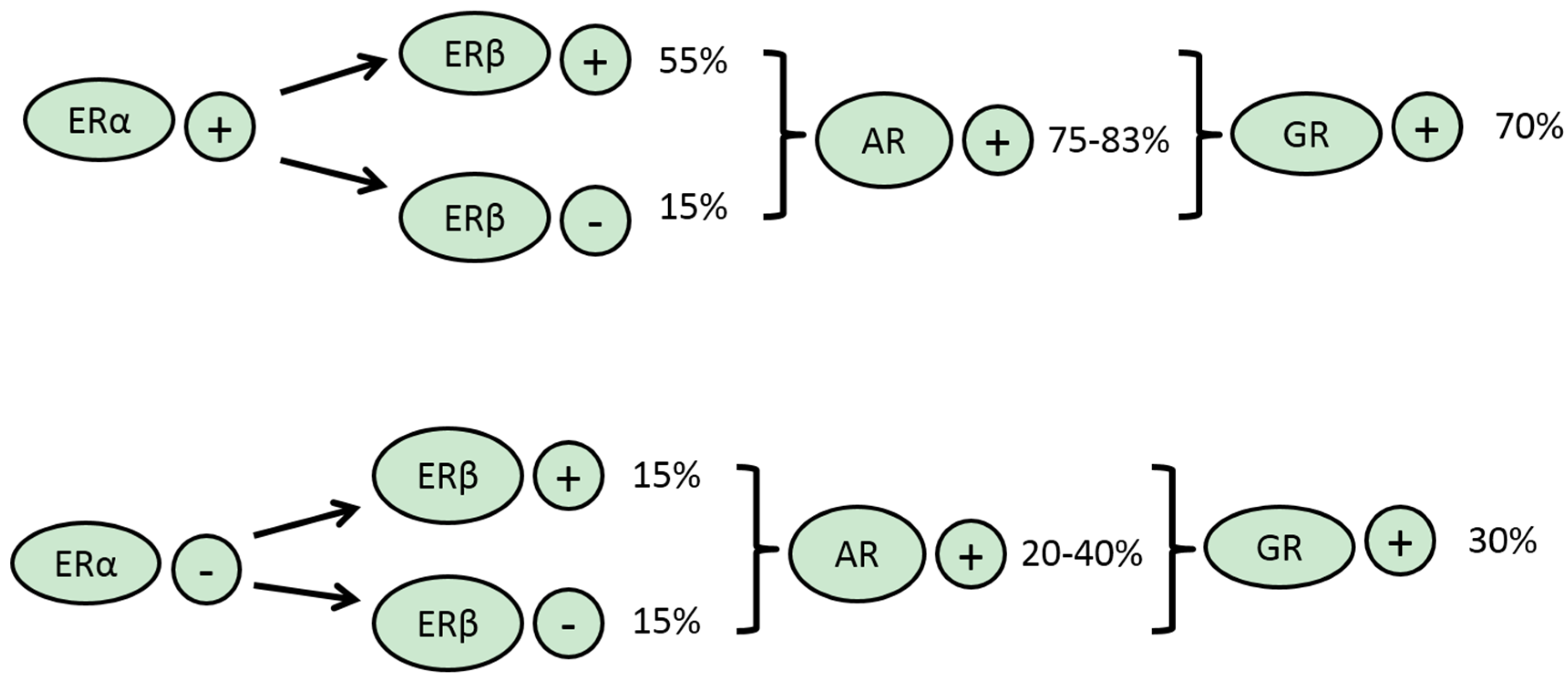
3.1. ERα
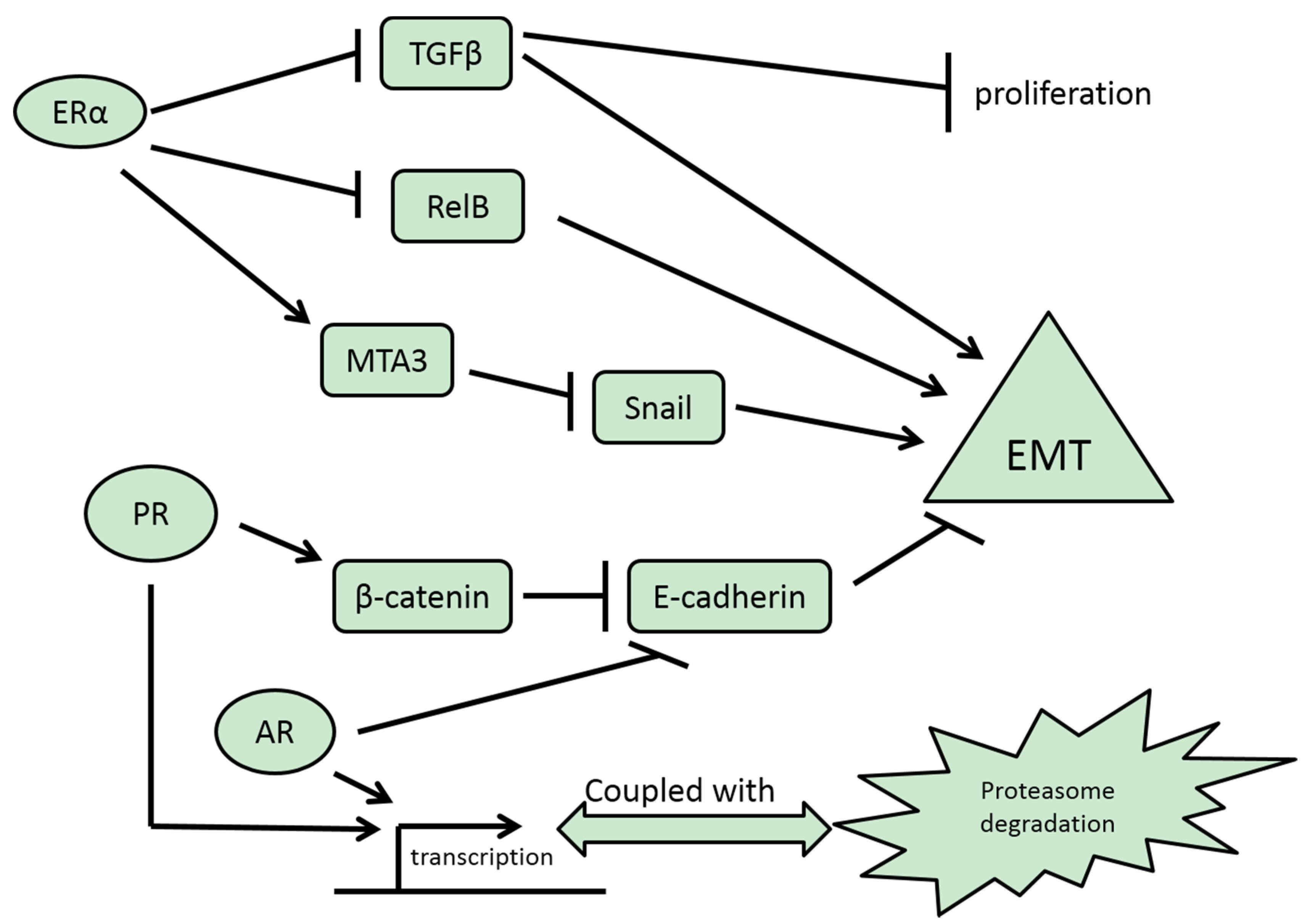
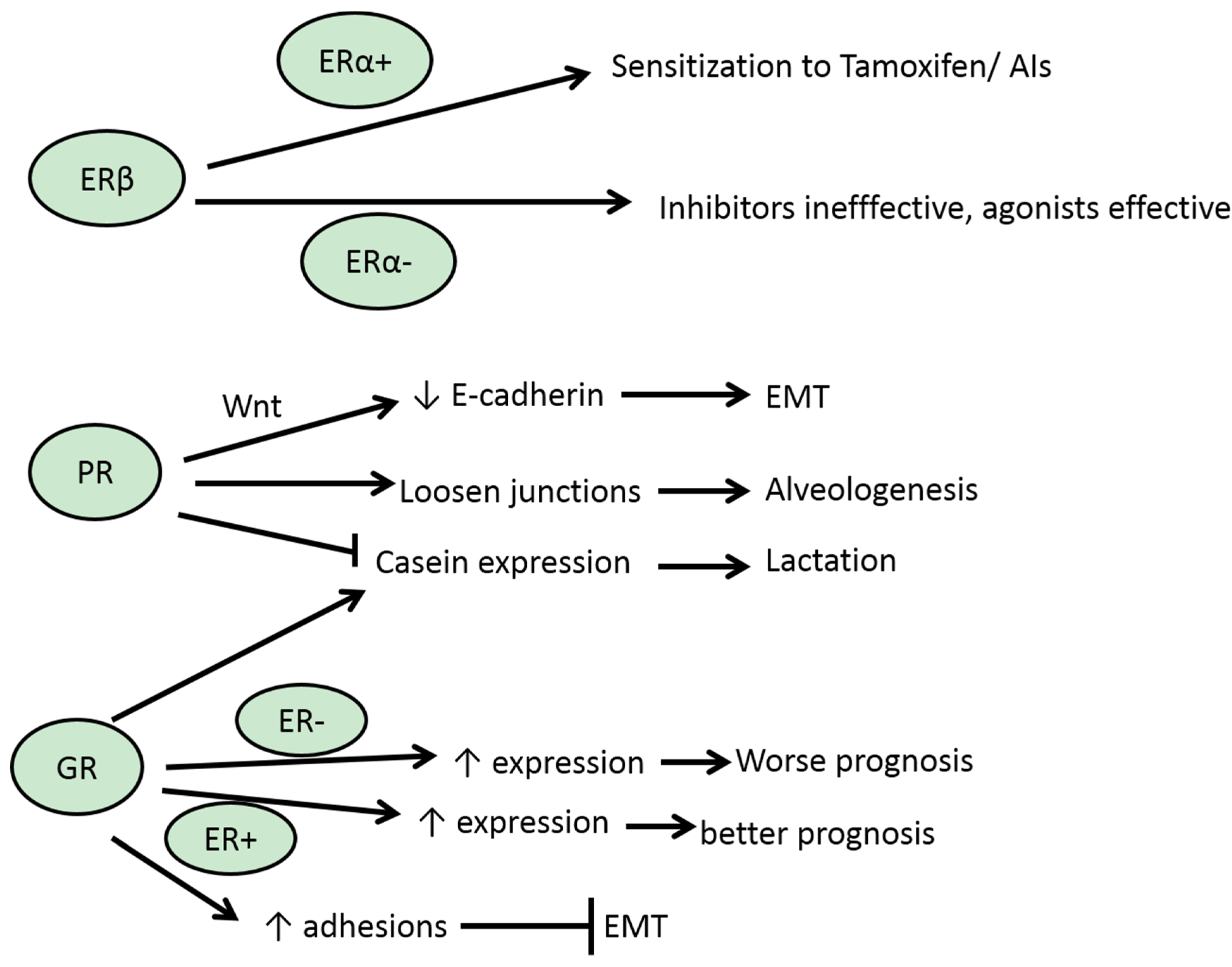
3.2. ERβ
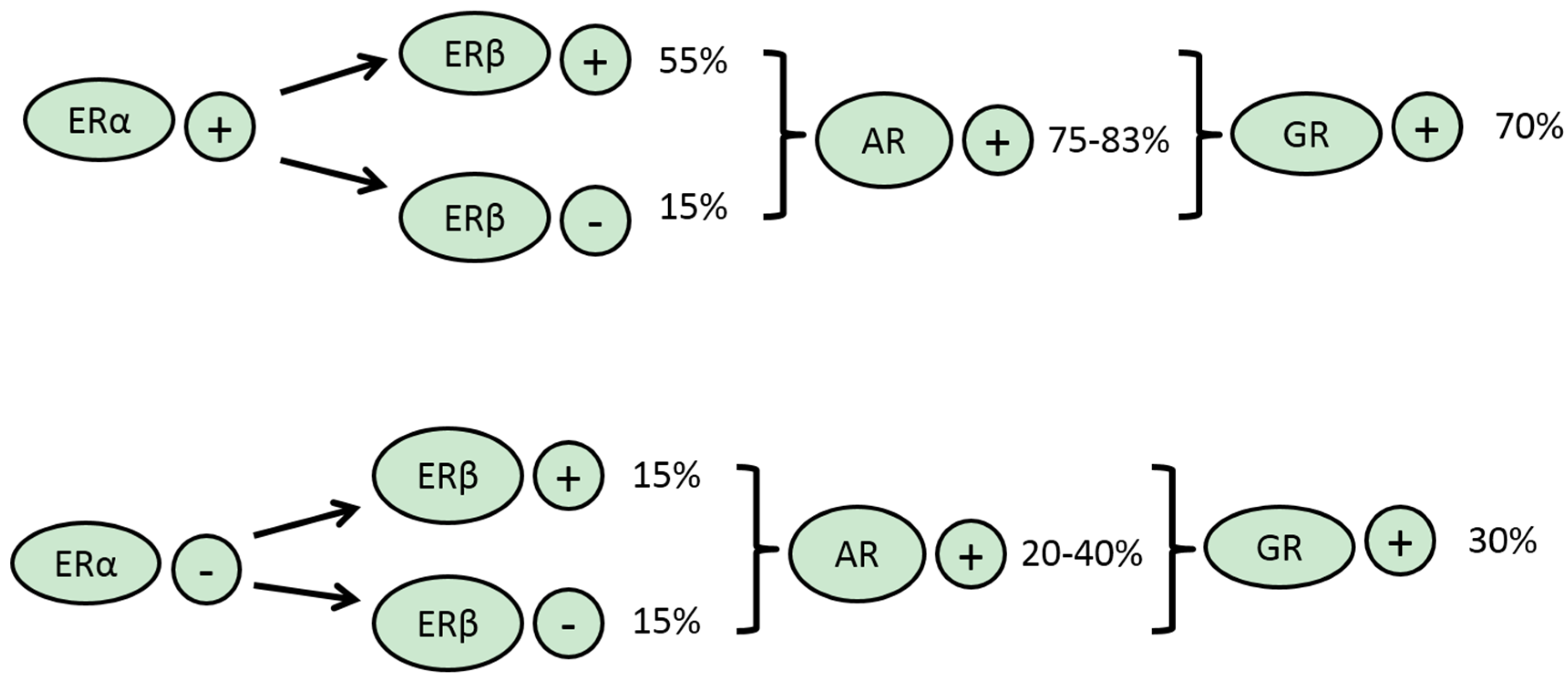
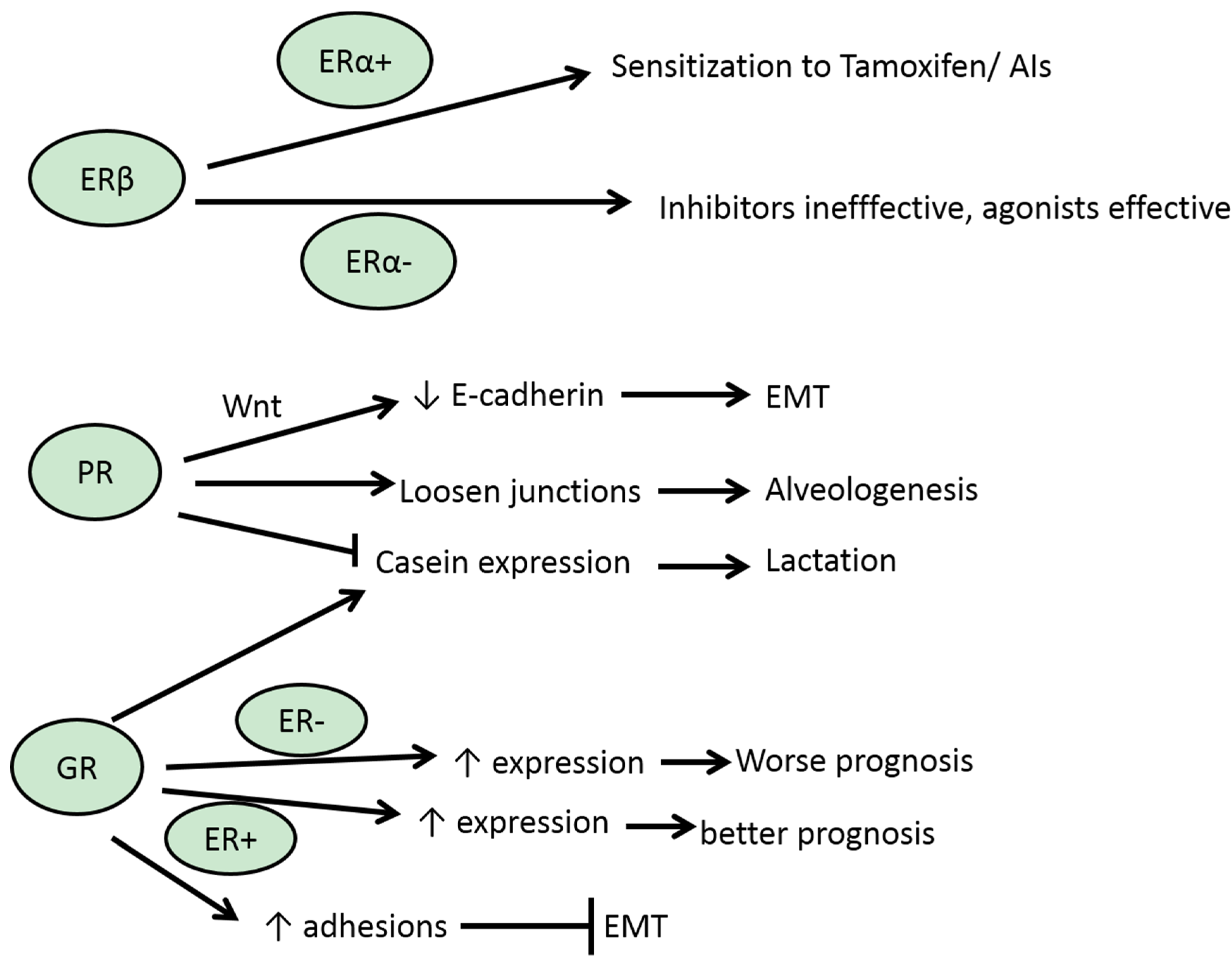
3.3. PR
3.4. AR
3.5. GR
3.6. MR
4. Snail1 and Slug and Regulation by SNRs

{kind=link}
{kind=link}
{kind=link}
| Factor | Database | Promoter | ERα | ERβ | AR (PR) | GR (MR) | Clusters |
|---|---|---|---|---|---|---|---|
| Snail | TRED | 1 | 0 | 3 | 2 | 4 | 0/3/0/2 |
| 2 | 1 | 7 | 1 | 3 | |||
| 3 | 0 | 3 | 1 | 2 | |||
| Slug | TRED | 1 | 0 | 7 | 4 | 4 | 0/2/2/3 |
| 2 | 0 | 6 | 5 | 4 | |||
| 3 | 0 | 0 | 2 | 7 | |||
| ZEB1 | TRED | 1 | 1 | 7 | 2 | 6 | 0/4/1//5 |
| 2 | 0 | 4 | 3 | 8 | |||
| 3 | 1 | 7 | 2 | 6 | |||
| EPD | 1 | 0 | 4 | 1 | 5 | ||
| 2 | 0 | 1 | 0 | 3 | |||
| ZEB2 | TRED | 1 | 0 | 2 | 2 | 4 | 0/1/3/4 |
| 2 | 0 | 2 | 3 | 3 | |||
| 3 | 0 | 3 | 4 | 7 | |||
| EPD | 1 | 0 | 1 | 1 | 2 | ||
| 2 | 0 | 1 | 7 | 4 |
5. ZEB1 and ZEB2 and miR200 Family
6. Twist
7. TCF3 and Id Proteins
| Factor | Database | Promoter | ERα | ERβ | AR (PR) | GR (MR) | Clusters |
|---|---|---|---|---|---|---|---|
| Twist | EPD | 1 | 0 | 0 | 2 | 2 | 0/0/0/0 |
| TCF3 | TRED | 1 | 0 | 1 | 1 | 2 | 0/1/1/2 |
| 2 | 1 | 3 | 0 | 6 | |||
| 3 | 0 | 1 | 3 | 8 | |||
| FOXC2 | TRED | 1 | 0 | 1 | 3 | 1 | 0/1/1/1 |
| 2 | 0 | 3 | 2 | 3 | |||
| Goosecoid | EPD | 1 | 0 | 0 | 1 | 1 | 0/0/0/0 |
| LBX1 | TRED | 1 | 0 | 2 | 4 | 1 | 0/1/3/2 |
| 2 | 0 | 3 | 1 | 0 | |||
| 3 | 0 | 2 | 3 | 3 | |||
| 4 | 0 | 0 | 4 | 3 |
8. FoxC2
9. Goosecoid
10. LBX1
11. Pioneer Factors
12. Perspective: EMT and Therapeutic Resistance Development and Reversal
Conflicts of Interest
References
- Le Du, F.; Eckhardt, B.L.; Lim, B.; Litton, J.K.; Moulder, S.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ueno, N.T. Is the future of personalized therapy in triple-negative breast cancer based on molecular subtype? Oncotarget 2015, 6, 12890–12908. [Google Scholar] [PubMed]
- Voutsadakis, I.A. Peroxisome proliferator activated receptor-γ and the ubiquitin-proteasome system in colorectal cancer. World J. Gastrointest. Oncol. 2010, 2, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Anbalagan, M.; Huderson, B.; Murphy, L.; Rowan, B.G. Post-translational modifications of nuclear receptors and human disease. Nuclear Recept. Signal. 2012, 10, e001. [Google Scholar]
- Helsen, C.; Claessens, F. Looking at nuclear receptors from a new ankle. Mol. Cell. Endocrinol. 2014, 382, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Beato, M.; Vicent, G.P. Impact of chromatin structure and dynamics on PR signaling. The initial steps in hormonal gene regulation. Mol. Cell. Endocrinol. 2012, 357, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Grøntved, L.; Hager, G.L. Impact of chromatin structure on PR signaling: Transition from local to global analysis. Mol. Cell. Endocrinol. 2012, 357, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.J.; Archer, T.K. Chromatin architecture defines the glucocorticoid response. Mol. Cell. Endocrinol. 2013, 380, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. The ubiquitin-proteasome system and signal transduction pathways regulating Epithelial Mesenchymal transition of cancer. J. Biomed. Sci. 2012, 19, 67. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. Ubiquitination and the Ubiquitin-Proteasome System as regulators of transcription and transcription factors in epithelial masenchymal transition of cancer. Tumour Biol. 2012, 33, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Parker Kerrigan, B.C.; Yang, D.; Hu, L.; Shmulevich, L.; Sood, A.K.; Xue, F.; Zhang, W. Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J. Hematol. Oncol. 2014, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. The network of pluripotency, epithelial-mesenchymal transition, and prognosis of breast cancer. Breast Cancer 2015, 7, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Cubillo, E.; Sarrió, D.; Peinado, H.; Rodríguez-Pinilla, S.M.; Villa, S.; Bolós, V.; Jordá, M.; Fabra, A.; Portillo, F.; et al. Genetic profiling of epithelial cells expressing E-Cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in Epithelial-Mesenchymal Transition. Cancer Res. 2006, 66, 9543–9556. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.D.; Corsa, C.A.S.; Biswas, H.; Aft, R.L. Longmore GD. Temporal and spatial cooperation of Snail1 and Twist1 during Epithelial-Mesenchymal Transition predicts for human breast cancer recurrence. Mol. Cancer Res. 2011, 9, 1644–1657. [Google Scholar] [CrossRef] [PubMed]
- De Herreros, A.G.; Peiró, S.; Nassour, M.; Savagner, P. Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011, 8, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Mitre-Aguilar, I.B.; Cabrera-Quintero, A.J.; Zentella-Dehesa, A. Genomic and non-genomic effects of glucocorticoids: Implications for breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 1–10. [Google Scholar] [PubMed]
- Denayer, S.; Helsen, C.; Thorrez, L.; Haelens, A.; Claessens, F. The rules of DNA recognition by the Androgen Receptor. Mol. Endocrinol. 2010, 24, 898–913. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Dahlman-Wright, K.; Gustafsson, J.-Å. Estrogen signalling via Estrogen Receptor β. J. Biol. Chem. 2010, 285, 39575–39579. [Google Scholar] [CrossRef] [PubMed]
- Ratman, D.; Vande Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; de Bosscher, K. How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Lupien, M.; Brown, M. Cistromics of hormone-dependent cancer. Endocr. Relat. Cancer 2009, 16, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, K.B.; Dye, W.W.; Chuck Harrell, J.; Kabos, P.; Sartorius, C.A. Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc. Natl. Acad. Sci. USA 2008, 105, 5774–5779. [Google Scholar] [CrossRef] [PubMed]
- Kabos, P.; Haughian, J.M.; Wang, X.; Dye, W.W.; Finlayson, C.; Elias, A.; Horwitz, K.B.; Sartorius, C.A. Cytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Res. Treat. 2011, 128, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Harrison, H.; Simões, B.M.; Rogerson, L.; Howell, S.J.; Landberg, G.; Clarke, R.B. Oestrogen increases the activity of oestrogen receptor negative breast cancer stem cells through paracrine EGFR and Notch signalling. Breast Cancer Res. 2013, 15, R21. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Nah, H.Y.; Lee, Y.J.; Lee, J.W.; Park, J.H.; Kim, S.J.; Lee, J.B.; Yoon, H.S.; Kim, C.H. Expression of Estrogen Receptor-α and –β, Glucocorticoid Receptor, and Progesterone Receptor genes in human embryonic stem cells and embryoid bodies. Mol. Cells 2005, 18, 320–325. [Google Scholar]
- Guttilla, I.K.; Adams, B.D.; White, B.A. ERα, microRNAs, and the epithelial-mesenchymal transition in breast cancer. Trends Endocrinol. Metable 2012, 23, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Al Saleh, S.; Al Mulla, F.; Luqmani, Y.A. Estrogen Receptor silencing induces Epithelial to Mesenchymal Transition in human breast cancer cells. PLoS ONE 2011, 6, e20610. [Google Scholar] [CrossRef] [PubMed]
- Bouris, P.; Skandalis, S.S.; Piperigkou, Z.; Afratis, N.; Karamanou, K.; Aletras, A.J.; Moustakas, A.; Theocharis, A.D.; Karamanos, N.K. Estrogen receptor alpha mediates epithelial to mesenchymal transition, expression of specific matrix effectors and functional properties of breast cancer cells. Matrix Biol. 2015, 43, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Ito, I.; Hanyu, A.; Wayama, M.; Goto, N.; Katsuno, Y.; Kawasaki, S.; Nakajima, Y.; Kajiro, M.; Komatsu, Y.; Fujimura, A.; et al. Estrogen inhibits Transforming Growth Factor β signalling by promoting Smad2/3 degradation. J. Biol. Chem. 2010, 285, 14747–14755. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.L.; Rosen, J.M. Stop! In the name of transforming growth factor-β; keeping estrogen receptor-α-positive mammary epithelial cells from proliferating. Breast Cancer Res. 2006, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Belguise, K.; Kersual, N.; Kirsch, K.H.; Mineva, N.D.; Galtier, F.; Chalbos, D.; Sonenshein, G.E. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 2007, 9, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Belguise, K.; O’Neill, C.F.; Sánchez-Morgan, N.; Romagnoli, M.; Eddy, S.F.; Mineva, N.D.; Yu, Z.; Min, C.; Trinkaus-Randall, V.; et al. RelB NF-κB represses Estrogen Receptor α expression via induction of the zinc finger protein Blimp1. Mol. Cell. Biol. 2009, 29, 3832–3844. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Li, Y.; Tong, Q.; Gu, F.; Zhu, T.; Fu, L.; Yang, S. δEF1 down-regulates ER-α expression and confers tamoxifen resistance in breast cancer. PLoS ONE 2012, 7, e52380. [Google Scholar] [CrossRef] [PubMed]
- Scherbakov, A.M.; Andreeva, O.E.; Shatskaya, V.A.; Krasil’nikov, M.A. The relationships between Snail1 and estrogen receptor signalling in breast cancer cells. J. Cell. Biochem. 2012, 113, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Talukder, A.H.; Gururaj, A.E.; Yang, Z.; Singh, R.R.; Mahoney, M.G.; Franci, C.; Vadlamudi, R.K.; Kumar, R. Upstream determinants of Estrogen Receptor-α regulation of Metastatic Tumor Antigen 3 pathway. J. Biol. Chem. 2004, 279, 32709–32715. [Google Scholar] [CrossRef] [PubMed]
- Haldosén, L.-A.; Zhao, C.; Dahlman-Wright, K. Estrogen receptor beta in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Speirs, V.; Skliris, G.P.; Burdall, S.E.; Carder, P.J. Distinct expression patterns of ER alpha and ER beta in normal human mammary gland. J. Clin. Pathol. 2002, 55, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.M.; Davis, R.J.; Shupnik, M.A. ERβ in breast cancer- Onlooker, passive player, or active protector? Steroids 2008, 73, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Meng, J.; Yilamu, D.; Jakulin, A.; Fu, M.; Wang, B.; Abulajiang, G. Significance of ERβ expression in different molecular subtypes of breast cancer. Diagn. Pathol. 2014, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.M.; Suman, V.J.; Subramaniam, M.; Wu, X.; Negron, V.; Gingery, A.; Pitel, K.S.; Shah, S.S.; Cunliffe, H.E.; McCullough, A.E.; et al. ERβ1: Characterization, prognosis, and evaluation of treatment strategies in ERα-positive and -negative cancer. BMC Cancer 2014, 14, 749. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.C.; Frasor, J.; Komm, B.; Katzenellenbogen, B.S. Impact of Estrogen Receptor β on gene networks regulated by Estrogen Receptor α in breast cancer cells. Endocrinology 2006, 147, 4831–4842. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Edvardsson, K.; Lewandowski, S.A.; Ström, A.; Gustafsson, J.-Å. A genome-wide study of the repressive effects of estrogen receptor beta on estrogen receptor alpha signaling in breast cancer cells. Oncogene 2008, 27, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Hefti, M.M.; Hu, R.; Knoblauch, N.W.; Collins, L.C.; Haibe-Kains, B.; Tamimi, R.M.; Beck, A.H. Estrogen receptor negative/progesterone receptor positive breast cancer is not a reproducible subtype. Breast Cancer Res. 2013, 15, R68. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Anderson, G.L.; Gass, M.; Lane, D.S.; Aragaki, A.K.; Kuller, L.H.; Manson, J.E.; Stefanick, M.L.; Ockene, J.; Sarto, G.E.; et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010, 304, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Cancello, G.; Maisonneuve, P.; Rotmensz, N.; Viale, G.; Mastropasqua, M.G.; Pruneri, G.; Montagna, E.; Iorfida, M.; Mazza, M.; Balduzzi, A.; et al. Progesterone receptor loss identifies luminal B breast cancer subgroups at higher risk of relapse. Ann. Oncol. 2013, 24, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Diep, C.H.; Daniel, A.R.; Mauro, L.J.; Knutson, T.P.; Lange, C.A. Progesterone action in breast, uterine, and ovarian cancers. J. Mol. Endocrinol. 2015, 54, R31–R53. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Hardy, D.B.; Mendelson, C.R. Progesterone Receptor inhibits proliferation of human breast cancer cells via induction of MAPK Phosphatase 1 (MKP-1/DUSP1). J. Biol. Chem. 2011, 286, 43091–43102. [Google Scholar] [CrossRef] [PubMed]
- Knutson, T.P.; Lange, C.A. Tracking progesterone receptor-mediated actions in breast cancer. Pharmacol. Ther. 2014, 142, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Parlow, A.F.; Neville, M.C. Hormonal regulation of tight junction closure in the mouse mammary epithelium during the transition from pregnancy to lactation. J. Endocrinol. 2001, 170, 347–356. [Google Scholar] [CrossRef]
- Obr, A.E.; Edwards, D.P. The biology of progesterone receptor in the normal mammary gland and in breast cancer. Mol. Cell. Endocrinol. 2012, 357, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Brisken, C.; Heineman, A.; Chavarria, T.; Elenbaas, B.; Tan, J.; Dey, S.K.; McMahon, J.A.; McMahon, A.P.; Weinberg, R.A. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev. 2000, 14, 650–654. [Google Scholar] [PubMed]
- Fernandez-Valdivia, R.; Mukherjee, A.; Creighton, C.J.; Buser, A.C.; DeMayo, F.J.; Edwards, D.P.; Lydon, J.P. Transcriptional response of the murine mammary gland to acute progesterone exposure. Endocrinology 2008, 149, 6236–6250. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Valdivia, R.; Mukherjee, A.; Ying, Y.; Li, J.; Paquet, M.; DeMayo, F.J.; Lydon, J.P. The RANKL signaling axis is sufficient to elicit ductal side-branching and alveologenesis in the mammary gland of the virgin mouse. Dev. Biol. 2009, 328, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Kariagina, A.; Xie, J.; Langohr, I.M.; Opreanu, R.C.; Basson, M.D.; Haslam, S.Z. Progesterone decreases levels of the adhesion protein E-cadherin and promotes invasiveness of steroid receptor positive breast cancers. Horm. Cancer 2013, 4, 381–390. [Google Scholar]
- Conneely, O.M.; Mulac-Jericevic, B.; Lydon, J.P. Progesterone-dependent regulation of female reproductive activity by two distinct progesterone receptor isoforms. Steroids 2003, 68, 771–778. [Google Scholar] [CrossRef]
- Pistelli, M.; Caramanti, M.; Biscotti, T.; Santinelli, A.; Pagliacci, A.; de Lisa, M.; Ballatore, Z.; Ridolfi, F.; Maccaroni, E.; Bracci, R.; et al. Androgen Receptor expression in early triple-negative breast cancer: Clinical significance and prognostic associations. Cancers 2014, 6, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, Y. Apocrine carcinoma as triple-negative breast cancer: Novel definition of apocrine-type carcinoma as Estrogen/Progesterone Receptor-negative and Androgen Receptor-positive invasive ductal carcinoma. Jpn. J. Clin. Oncol. 2012, 42, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Koo, J.; Park, H.S.; Kim, J.H.; Choi, S.Y.; Lee, J.H.; Park, B.W.; Lee, K.S. Expression of androgen receptors in primary breast cancer. Ann. Oncol. 2010, 21, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Vera-Badillo, F.E.; Templeton, A.J.; de Gouveia, P.; Diaz-Padilla, I.; Bedard, P.L.; Al-Mubarak, M.; Seruga, B.; Tannock, I.F.; Ocana, A.; Amir, E. Androgen Receptor expression and outcomes in early breast cancer: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2014, 106, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-N.; Lee, H.-J.; Hsu, Y.-H.; Chen, J.-H. Activated Androgen Receptor downregulates E-cadherin gene expression and promotes tumor metastasis. Mol. Cell. Biol. 2008, 28, 7096–7108. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A.; Papandreou, C.N. The ubiquitin-proteasome system in prostate cancer and its transition to castration resistance. Urol. Oncol. 2012, 30, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Hundertmark, S.; Buhler, H.; Rudolf, M.; Weitzel, H.K.; Ragosch, V. Inhibition of 11 beta-hydroxysteroid gehydrogenase activity enhances the antiproliferative effect of glucocorticosteroids on MCF-7 and ZR-75–1 breast cancer cells. J. Endocrinol. 1997, 155, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Shen, H.; Maki, C.G. Glucocorticoid receptor activation inhibits p53-induced apoptosis of MCF10Amyc cells via induction of protein kinase Cε. J. Biol. Chem. 2012, 287, 29825–29836. [Google Scholar] [CrossRef] [PubMed]
- Abduljabbar, R.; Negm, O.H.; Lai, C.F.; Jerjees, D.A.; Al-Kaabi, M.; Hamed, M.R.; Tighe, P.J.; Buluwela, L.; Mukherjee, A.; Green, A.R.; et al. Clinical and biological significance of glucocorticoid receptor (GR) expression in breast cancer. Breast Cancer Res. Treat. 2015, 150, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Buxant, F.; Engohan-Aloghe, C.; Noël, J.-C. Estrogen Receptor, Progesterone Receptor, and Glucoccorticoid Receptor expression in normal breast tissue, breast in situ carcinoma, and invasive breast cancer. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Vilasco, M.; Communal, L.; Mourra, N.; Courtin, A.; Forgez, P.; Gompel, A. Glucocorticoid receptor and breast cancer. Breast Cancer Res. Treat. 2011, 130, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Jin, Y.; Nagaich, A.K. Interaction of Glucoccorticoid Receptor (GR) with Estrogen Receptor (ER) α and Activator Protein 1 (AP1) in dexamethasone-mediated interference of ERα activity. J. Biol. Chem. 2013, 288, 24020–24034. [Google Scholar] [CrossRef] [PubMed]
- Bolt, M.J.; Stossi, F.; Newberg, J.Y.; Orjalo, A.; Johansson, H.E.; Mancini, M.A. Coactivators enable glucocorticoid receptor recruitment to fine-tune estrogen receptor transcriptional responses. Nucleic Acids Res. 2013, 41, 4036–4048. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Voss, T.C.; Sung, M.-H.; Baek, S.; John, S.; Hawkins, M.; Grøntved, L.; Schiltz, R.L.; Hager, G.L. Reprogramming the chromatin landscape: Interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res. 2013, 73, 5130–5139. [Google Scholar] [CrossRef] [PubMed]
- Vilasco, M.; Communal, L.; Hugon-Rodin, J.; Penault-Llorca, F.; Mourra, N.; Wu, Z.; Forgez, P.; Gompel, A.; BRACAPS. Loss of glucocorticoid receptor activation is a hallmark of BRCA1-mutated breast tissue. Breast Cancer Res. Treat. 2013, 142, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Chan, H.L.; Scott, A.; Smith, M.D.; Fan, C.; Herschkowitz, J.I.; Perou, C.M.; Livingstone, A.S.; Robbins, D.J.; Capobianco, A.J.; et al. BRCA1 suppresses epithelial-to-mesenchymal transition and stem cell dedifferentiation during mammary and tumor development. Cancer Res. 2014, 74, 6161–6172. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, G.L.; Visvader, J.E. Cell fate takes a slug in BRCA1-associated breast cancer. Breast Cancer Res. 2011, 13, 306. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Kocherginsky, M.; Conzen, S.D. Activation of the Glucocorticoid Receptor is associated with poor prognosis in Estrogen Receptor-negative breast cancer. Cancer Res. 2011, 71, 6360–6370. [Google Scholar] [CrossRef] [PubMed]
- Wyszomierski, S.L.; Rosen, J.M. Cooperative effects of STAT5 (signal transducer and activator of transcription 5) and C/EBPbeta (CCAAT/ Enhancer-binding protein-beta) on beta-casein gene transcription are mediated by the glucocorticoid receptor. Mol. Endocrinol. 2001, 15, 228–240. [Google Scholar] [PubMed]
- Leo, J.C.L.; Guo, C.; Woon, C.T.; Aw, S.E.; Lin, V.C.L. Glucocorticoid and Mineralocorticoid cross-talk with Progesterone Receptor to induce focal adhesion and growth inhibition in breast cancer cells. Endocrinology 2004, 145, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Skor, M.N.; Wonder, E.L.; Kocherginsky, M.; Goyal, A.; Hall, B.A.; Cai, Y.; Conzen, S.D. Glucocorticoid Receptor antagonism as a novel therapy for triple-negative breast cancer. Clin. Cancer Res. 2013, 19, 6163–6172. [Google Scholar] [CrossRef] [PubMed]
- García de Herreros, A.; Baulida, J. Cooperation, amplification, and feed-back in epithelial-mesenchymal transition. Biochim. Biophys. Acta 2012, 1825, 223–228. [Google Scholar]
- Kingsley-Kallesen, M.; Mukhopadhyay, S.S.; Wyszomierski, S.L.; Schanler, S.; Schutz, G.; Rosen, J.M. The mineralocorticoid receptor may compensate for the loss of the glucocorticoid receptor at specific stages of mammary gland development. Mol. Endocrinol. 2002, 16, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Diah, S.; Zhang, G.-X.; Nagai, Y.; Zhang, W.; Gang, L.; Kimura, S.; Abdul Hamid, M.R.W.; Tamiya, T.; Nishiyama, A.; Hitomi, H. Aldosterone induces myofibroblastic transdifferentiation and collagen gene expression through the Rho-kinase dependent signaling pathway in rat mesangial cells. Exp. Cell Res. 2008, 314, 3654–3662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Jia, Z.; Guo, X.; Yang, T. Aldosterone induces epithelial-mesenchymal transition via ROS of mitochondrial origin. Am. J. Physiol. Renal Physiol. 2007, 293, F723–F731. [Google Scholar] [CrossRef] [PubMed]
- Foubert, E.; De Craene, B.; Berx, G. Key signaling nodes in mammary gland development and cancer. The Snail1-Twist1 conspiracy in malignant breast cancer progression. Breast Cancer Res. 2010, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Gjorevski, N.; Boghaert, E.; Radisky, D.C.; Nelson, C.M. Snail1, Snail2, and E47 promote mammary epithelial branching morphogenesis. EMBO J. 2011, 30, 2662–2674. [Google Scholar] [CrossRef] [PubMed]
- Gras, B.; Jacqueroud, L.; Wierinckx, A.; Lamblot, C.; Fauvet, F.; Lachuer, J.; Puisieux, A.; Ansieau, S. Snail family members unequally trigger EMT and thereby differ in their ability to promote the neoplastic transformation of mammary epithelial cells. PLoS ONE 2014, 9, e92254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, Q.; Han, Z.; Hu, W.; Xi, L.; Gao, Q.; Wang, S.; Zhou, J.; Xu, G.; Meng, L.; et al. Reduced expression of Snail decreases breast cancer cell motility by downregulating the expression and inhibiting the activity of RhoA GTPase. Oncol. Lett. 2013, 6, 339–346. [Google Scholar] [PubMed]
- Okubo, T.; Truong, T.K.; Yu, B.; Itoh, T.; Zhao, J.; Grube, B.; Zhou, D.; Chen, S. Down-regulation of promoter I.3 activity of the human aromatase gene in breast tissue by zinc-finger protein, Snail (SnaH). Cancer Res. 2001, 61, 1338–1346. [Google Scholar] [PubMed]
- Martin, T.A.; Goyal, A.; Watkins, G.; Jiang, W.G. Expression of the transcription factor Snail, Slug, and Twist and their clinical significance in human breast cancer. Ann. Surg. Oncol. 2005, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Wang, W.; Yearsley, K.; Gao, J.-X.; Barsky, S.H. ERα suppresses slug expression directly by translational repression. Biochem. J. 2008, 416, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Xiao, Y.; Wang, W.; Yearsley, K.; Gao, J.-X.; Shetuni, B.; Barsky, S.H. ERα signalling through slug regulates E-cadherin and EMT. Oncogene 2010, 29, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Jaye, D.L.; Kajita, M.; Gelgerman, C.; Moreno, C.S.; Wade, P.A. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003, 113, 207–219. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, F.; Li, S.; Chang, A.K.; Jia, Z.; Chen, Y.; Xu, F.; Pan, H.; Wu, H. AIB1 cooperates with ERα to promote epithelial mesenchymal transition in breast cancer through SNAI1 activation. PLoS ONE 2013, 8, e65556. [Google Scholar] [CrossRef] [PubMed]
- Chimge, N.-O.; Baniwal, S.K.; Little, G.H.; Chen, Y.; Kahn, M.; Tripathy, D.; Borok, Z.; Frenkel, B. Regulation of breast cancer metastasis by Runx2 and estrogen signalling: The role of SNAI2. Breast Cancer Res. 2011, 13, R127. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Lin, W.-J.; Izumi, K.; Wang, X.; Xu, D.; Fang, L.-Y.; Li, L.; Jiang, Q.; Jin, J.; Chang, C. Targeting Androgen Receptor to suppress macrophage-induced EMT and benign prostatic hyperplasia (BPH) development. Mol. Endocrinol. 2012, 26, 1707–1715. [Google Scholar]
- Wu, K.; Gore, C.; Yang, L.; Fazli, L.; Gleave, M.; Pong, R.-C.; Xiao, G.; Zhang, L.; Yun, E.-J.; Tseng, S.-F.; et al. Slug, a unique Androgen-regulated transcription factor, coordinates Androgen Receptor to facilitate castration resistance in prostate cancer. Mol. Endocrinol. 2012, 26, 1496–1507. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Lee, S.O.; Yeh, S.; Chang, T.M. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene 2014, 33, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Kim, K.; Charn, T.H.; Komm, B.; Chang, K.C.N.; Kraus, W.L.; Benner, C.; Glass, C.K.; Katzenellenbogen, B.S. Genome-wide analysis of Estrogen Receptor α DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol. Cell. Biol. 2010, 30, 3943–3955. [Google Scholar] [CrossRef] [PubMed]
- Hill, L.; Browne, G.; Tulchinsky, E. ZEB/miR-200 feedback loop: At the crossroads of signal transduction in cancer. Int. J. Cancer 2013, 132, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Hurteau, G.J.; Carlson, J.A.; Spivack, S.D.; Brock, G.J. Overexpression of the MicroRNA has-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res. 2007, 67, 7972–7976. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, Z.; Filmore, R.; Xi, Y. miR-200, a new star miRNA in human cancer. Cancer Lett. 2014, 344, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Hilmarsdottir, B.; Briem, E.; Bergthorsson, J.T.; Magnusson, M.K.; Gudjonsson, T. Functional role of the microRNA-200 family in breast morphogenesis and neoplasia. Genes 2014, 5, 804–820. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Li, X.; Wright, J.A.; Lawrence, D.M.; Pillman, K.A.; Salmanidis, M.; Anderson, M.A.; Dredge, B.K.; Gregory, P.A.; Tsykin, A.; et al. Genome-wide identification of miR-200 targets reveals a regulatory network controlling cell invasion. EMBO J. 2014, 33, 2040–2056. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Brabletz, T. The ZEB/ miR-200 feedback loop- a motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, E.M.; Sanders, M.M. Identification of the novel player δEF1 in estrogen transcriptional cascades. Mol. Cell. Biol. 1999, 19, 3600–3606. [Google Scholar] [CrossRef] [PubMed]
- Spoeltra, N.S.; Manning, N.G.; Higashi, Y.; Darling, D.; Singh, M.; Shroyer, K.R.; Broaddus, R.R.; Horwitz, K.B.; Richer, J.K. The transcription factor ZEB1 is aberrantly expressed in aggressive uterine cancers. Cancer Res. 2006, 66, 3893–3902. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Saykally, J.N.; Anose, B.M.; Kalli, K.R.; Sanders, M.M. Expression of the ZEB1 (δEF1) transcription factor in human: Additional insights. Mol. Cell. Biochem. 2008, 318, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Tuomarila, M.; Luostari, K.; Soini, Y.; Kataja, V.; Kosma, V.-M.; Mannermaa, A. Overexpression of micro-RNA-200c predicts poor outcome in patients with PR-negative breast cancer. PLoS ONE 2014, 9, e109508. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, T.T.; Teng, Y.; Litchfield, L.M.; Muluhngwi, P.; Al-Rayyan, N.; Klinge, C.M. Reduced expression of miR-200 family members contributes to antiestrogen resistance in LY2 human breast cancer cells. PLoS ONE 2013, 8, e62334. [Google Scholar] [CrossRef] [PubMed]
- Richer, J.K.; Jacobsen, B.M.; Manning, N.G.; Abel, M.G.; Wolf, D.M.; Horwitz, K.B. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J. Biol. Chem. 2002, 277, 5209–5218. [Google Scholar] [CrossRef] [PubMed]
- Anose, B.M.; Sanders, M.M. Androgen receptor regulates transcription of the ZEB1 transcription factor. Int. J. Endocrinol. 2011, 2011, 903918. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.R.; Yacoub, R.; Taliaferro-Smith, L.; Osunkoya, A.O.; Odero-Marah, V.A.; Liu, T.; Kimbro, K.S.; Sharma, D.; O’Reagan, R.M. Reciprocal regulation of ZEB1 and AR in triple negative breast cancer cells. Breast Cancer Res. Treat. 2010, 123, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Kasimir-Bauer, S.; Hoffmann, O.; Wallwiener, D.; Kimmig, R.; Fehm, T. Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res. 2012, 14, R15. [Google Scholar] [CrossRef] [PubMed]
- Van Nes, J.G.; de Kruijf, E.M.; Putter, H.; Faratian, D.; Munro, A.; Campbell, F.; Smit, V.T.H.B.M.; Liefers, G.-J.; Kuppen, P.J.K; van de Velde, C.J.H.; et al. Co-expression of SNAIL and TWIST determines prognosis in estrogen receptor-positive early breast cancer patients. Breast Cancer Res Treat. 2012, 133, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, S.L.; Townley, I.; Zhong, Q.; Carriere, P.P.; Zou, J.; Llopis, S.D.; Preyan, L.C.; Williams, C.C.; Skripnikova, E.; Bratton, M.R.; et al. Proteomic signatures of acquired letrozole resistance in breast cancer: Suppresssed estrogen signalling and increased cell motility and invasiveness. Mol. Cell. Proteom. 2013, 12, 2440–2455. [Google Scholar] [CrossRef] [PubMed]
- Vesuna, F.; Lisok, A.; Kimble, B.; Domek, J.; Kato, Y.; van der Groep, P.; Artemov, D.; Kowalski, J.; Carraway, H.; van Diest, P.; et al. Twist contributes to hormone resistance in breast cancer by down-regulating estrogen receptor alpha. Oncogene 2012, 31, 3223–3234. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhang, L.; He, T.; Xiao, X.; Liu, X; Wang, L.; Yang, L.; Yang, M.; Zhang, T.; Chen, R.; et al. TWIST represses Estrogen Receptor-alpha expression by recruiting the NuRD protein complex in breast cancer cells. Int. J. Biol. Sci. 2012, 8, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, R.G.; Ferguson, A.T.; Ottaviano, Y.L.; Parl, F.F.; Smith, H.S.; Weitzman, S.A.; Baylin, S.B.; Issa, J.P.; Davidson, N.E. Methylation of estrogen receptor gene 5’ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin. Cancer Res. 1996, 2, 805–810. [Google Scholar] [PubMed]
- Cubillo, E.; Diaz-Lopez, A.; Cuevas, E.P.; Moreno-Bueno, G.; Peinado, H.; Montes, A.; Santos, V.; Portillo, F.; Cano, A. E47 and Id1 interplay in epithelial-mesenchymal transition. PLoS ONE 2013, 8, e59948. [Google Scholar] [CrossRef] [PubMed]
- Perk, J.; Gil-Bazo, I.; Chin, Y.; de Candia, P.; Chen, J.J.; Zhao, Y.; Chao, S.; Cheong, W.; Ke, Y.; Al-Ahmadie, H.; et al. Reassessement of Id1 protein expression in human mammary, prostate, and bladder cancers using a monospecific rabbit monoclonal anti-id1 antibody. Cancer Res. 2006, 66, 10870–10877. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Yang, J.; Brooks, M.; Schwaninger, G.; Zhou, A.; Miura, N.; Kutok, J.L.; Hartwell, K.; Richardson, A.L.; Weinberg, R.A. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2007, 104, 10069–10074. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.B.L.; Lam, M.S.; Aitken, S. A bioinformatics approach for identifying candidate transcriptional regulators of Mesenchyme-to-Epithelium Transitions in mouse embryos. Dev. Dyn. 2008, 237, 2748–2754. [Google Scholar] [CrossRef] [PubMed]
- Hader, C.; Marlier, A.; Cantley, L. Mesenchymal-epithelial transition in epithelial response to injury: The role of Foxc2. Oncogene 2010, 29, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Grønning, L.M.; Cederberg, A.; Miura, N.; Evenbäck, S.; Taskén, K. Insulin and TNFα induce expression of the Forkhead transcription factor gene Foxc2 in 3T3-L1 adipocytes via PI3K and ERK 1/2-dependent pathways. Mol. Endocrinol. 2002, 16, 873–883. [Google Scholar] [PubMed]
- Golden, D.; Cantley, L.G. Casen kinase 2 prevents mesenchymal transformation by maintaining Foxc2 in the cytoplasm. Oncogene 2015, 34, 4702–4712. [Google Scholar] [CrossRef] [PubMed]
- Hollier, B.G.; Tinnirello, A.A.; Werden, S.J.; Evans, K.W.; Taube, J.H.; Roy Sarkar, T.; Sphyris, N.; Shariati, M.; Kumar, S.V.; Battula, V.L.; et al. FOXC2 expression links Epithelial-Mesenchymal Transition and stem cell properties in breast cancer. Cancer Res. 2012, 73, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, K.A.; Muir, B.; Reinhardt, F.; Carpenter, A.E.; Sgroi, D.C.; Weinberg, R.A. The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 18969–18974. [Google Scholar] [CrossRef] [PubMed]
- Watabe, T.; Kim, S.; Candia, A.; Rothbächer, U.; Hashimoto, C.; Inoue, K.; Cho, K.W.Y. Molecular mechanisms of Spemann’s organizer formation: Conserved growth factor synergy between Xenopus and mouse. Genes Dev. 1995, 9, 3038–3050. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.-C.; Ge, N.-L.; Zhang, L.; Cui, J.-F.; Chen, R.-X.; You, Y.; Ye, S.-L.; Ren, Z.-G. Goosecoid promotes the metastasis of hepatocellular carcinoma by modulating the epithelial-mesenchymal transition. PLoS ONE 2014, 9, e109695. [Google Scholar] [CrossRef] [PubMed]
- Ell, B.; Kang, Y. Transcriptional control of cancer metastasis. Trends Cell Biol. 2013, 23, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.K.; Moran-Rivard, L.; Velasquez, T.; Nakatsu, M.N.; Jagla, K.; Goulding, M. Lbx1 is required for muscle precursor migration along a lateral pathway into the limb. Development 2000, 127, 413–424. [Google Scholar] [PubMed]
- Yu, M.; Smolen, G.A.; Zhang, J.; Wittner, B.; Schott, B.J.; Brachtel, E.; Ramaswamy, S.; Maheswaran, S.; Haber, D.A. A developmentally regulated inducer of EMT, LBX1, contributes to breast cancer progression. Genes Dev. 2009, 23, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Jozwik, K.M.; Carroll, J.S. Pioneer factors in hormone-dependent cancers. Nat. Rev. Cancer 2012, 12, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Carroll, J.S. FoxA1 is a key mediator of hormonal response in breast and prostate cancer. Front. Endocrinol. 2012, 3, 68. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.L.; MacArthur, S.; Ross-Innes, C.S.; Tilley, W.D.; Neal, D.E.; Mills, I.G.; Carroll, J.S. Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. EMBO J. 2011, 30, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Guiu, S.; Charon-Barra, C.; Vernerey, D.; Fumoleau, P.; Campone, M.; Spielmann, M.; Roché, H.; Mesleard, C.; Arnould, L.; Lemonnier, J.; et al. Coexpression of androgen receptor and FOXA1 in nonmetastatic triple-negative breast cancer: Ancillary study from PACS08 trial. Future Oncol. 2015, 11, 2283–2297. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Lee, W.W.; Wang, C.Y.; Chao, T.H.; Chen, Y.; Chen, J.H. Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene 2005, 24, 8277–8290. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Preca, B.-T.; Bajdak, K.; Mock, K.; Sundararajan, V.; Pfannstiel, J.; Maurer, J.; Wellner, U.; Hopt, U.T.; Brummer, T.; Brabletz, S.; et al. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int. J. Cancer 2015, 137, 2566–2577. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; He, J. Epithelial mesenchymal transition and lung cancer. J. Thorac. Dis. 2010, 2, 154–159. [Google Scholar] [PubMed]
- Huang, J.; Li, H.; Ren, G. Epithelial-mesenchymal transition and drug resistance in breast cancer. Int. J. Oncol. 2015, 47, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voutsadakis, I.A. Epithelial-Mesenchymal Transition (EMT) and Regulation of EMT Factors by Steroid Nuclear Receptors in Breast Cancer: A Review and in Silico Investigation. J. Clin. Med. 2016, 5, 11. https://doi.org/10.3390/jcm5010011
Voutsadakis IA. Epithelial-Mesenchymal Transition (EMT) and Regulation of EMT Factors by Steroid Nuclear Receptors in Breast Cancer: A Review and in Silico Investigation. Journal of Clinical Medicine. 2016; 5(1):11. https://doi.org/10.3390/jcm5010011
Chicago/Turabian StyleVoutsadakis, Ioannis A. 2016. "Epithelial-Mesenchymal Transition (EMT) and Regulation of EMT Factors by Steroid Nuclear Receptors in Breast Cancer: A Review and in Silico Investigation" Journal of Clinical Medicine 5, no. 1: 11. https://doi.org/10.3390/jcm5010011
APA StyleVoutsadakis, I. A. (2016). Epithelial-Mesenchymal Transition (EMT) and Regulation of EMT Factors by Steroid Nuclear Receptors in Breast Cancer: A Review and in Silico Investigation. Journal of Clinical Medicine, 5(1), 11. https://doi.org/10.3390/jcm5010011