
Human Papillomavirus: Current and Future RNAi Therapeutic Strategies for Cervical Cancer



Abstract
:1. Introduction
2. HPV and Cervical Cancer
The Role of E6 and E7 in Carcinogenesis
3. TP53 Mutations in Human Cancer
4. Therapeutics against HPVs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | References | Note | |
|---|---|---|---|
| HPV E7 antagonist | Peptide | [65] | |
| E6-binding aptamer | Peptide | [58] | |
| E6-AP mimetic epitope | Helical peptides | [60] | |
| Organic disulfides containing dithiobisamine moiety | Organic compound | [55] | |
| Anti-E6 ribozyme | RNA molecule | [63] | |
| GS-9191 | Nucleotide analog prodrug | [66] | |
| 5-aza-2′-deoxycytidine (DAC) | Nucleotide analog | [69] | Combination with therapeutic HPV DNA vaccine |
| 5,6-dimethylxanthenone-4-acetic acid (DMXAA) | Small molecule | [70] | |
| Cisplatin | Platinum compound | [61] | Alone or combination treatment |
| 4,4′-dithiodimorpholine | Zinc-ejecting inhibitor | [55] | |
| Glycosaminoglycans (GAGs) | Heparin-like | [68] | |
| Chitosan hydrogel | Natural biopolymer | [67] | Combination treatment |
| Methyl jasmonate | Plant hormone | [72] | Plant-originated products |
| Epigallocatechingallate (EGCG) | Plant-derived natural compound | [78] | |
| Nordihydroguaiaretic acid (NDGA) | Plant lignan derivative | [80] | |
| Silymarin | Plant flavonoid | [76] | |
| Jaceosidin (4′,5,7-trihydroxy-3′,6-dimethoxyflavone) | Plant-derived natural compound | [79] | |
| Withaferin A | Plant-derived natural compound | [77] | |
| Praneem tablet | Plant extract | [71] | |
| Curcumin | Plant extract | [75] | |
| Phytoglycoprotein | Plant-originated glycoprotein | [74] | |
| Soyasaponins | Plant-derived natural compound | [73] | |
| Carrageenan | Compound from red algae | [81] | |
| 17-N-allylyamino-17-demethoxygeldanamycin (17-AAG) | A derivative of the antibiotic geldanamycin | [64] | Combination treatment with GRP78 inhibitor |
4.1. RNAi-based Therapeutics against HPVs
| References | Cell line | Target Transcripts | Note | ||
|---|---|---|---|---|---|
| Cun et al., 2013 | [149] | OCM1, OM431, VUP and SP6.5 | Synthesized siRNA | E6 & E7 mRNA | |
| Li et al., 2013 | [150] | SiHa | Plasmid-based E6-specific siRNA | E6 mRNA | Dual coexpressed-E6-specific siRNA and wild type TP53 |
| Zhou et al., 2012 | [93] | SiHa | Synthesized siRNA | E6/E7 mRNA | BALB/C nude mice, intratumoral injections every other dayduring a 12-day period |
| Jung et al., 2012 | [94] | HeLa, CaSki, SiHa | Synthesized siRNA | E6/E7 mRNA | BALB/C nude mice, intravenous injections in combination with Cisplatin |
| Chang et al., 2010 | [95] | CaSki, HeLa | siRNA Plasmid | E6 only or E6/E7 mRNA | BALB/C nude mice, intratumoral injections twice a week for two weeks |
| Hong et al., 2009 | [96] | SiHa | Synthesized siRNA | E6/E7 mRNA | |
| Jonson et al., 2008 | [97] | CaSki | Synthesized siRNA | E6/E7 mRNA | nu/nu mice, intratumoral injections every three days for 35 days |
| Sima et al., 2008 | [88] | SiHa, CaSki | shRNA | E6/E7 mRNA | |
| Lea et al., 2007 | [98] | HeLa | Synthesized siRNA | E6 only or E6/E7 mRNA | |
| Courtete et al., 2007 | [99] | CaSki, SiHa, HeLa | Synthesized siRNA | E6 mRNA | |
| Fujii et al., 2006 | [100] | SKG-IIIa, SKG-II, HeLa | Synthesized siRNA | E6 & E7 mRNA | Nude mice, Intratumoral infections for 10 days |
| Tang et al., 2006 | [20] | CaSki and SiHa, HeLa | Synthesized siRNA | E6 or E6*I mRNA and E7 mRNA | |
| Yamato et al., 2008 | [89] | CaSki and SiHa, | Synthesized siRNA | E6 mRNA | |
| Koivusalo et al.,2005 | [101] | HeLa | Synthesized siRNA | E6 mRNA | siRNA combination with different drugs |
| Putral et al., 2005 | [102] | SiHa, CaSki | Synthesized siRNA | E6* mRNA and full length E6 mRNA | |
| Yoshinouchi et al., 2003 | [62] | SiHa | Synthesized siRNA | E6* mRNA and full length E6 mRNA | |
| Butz et al., 2003 | [58] | HeLa | Vector and Synthesized siRNA | full length E6 mRNA | |
| Hall and Alexander, 2003 | [91] | HeLa | Synthesized siRNA | E7 mRNA | |
| Jiang and Milner, 2002 | [59] | CaSki, SiHa | Synthesized siRNA | E6 and E7 mRNA |
4.2. Anticancer Therapeutic Strategies Targeting Activation of the TP53 Pathway
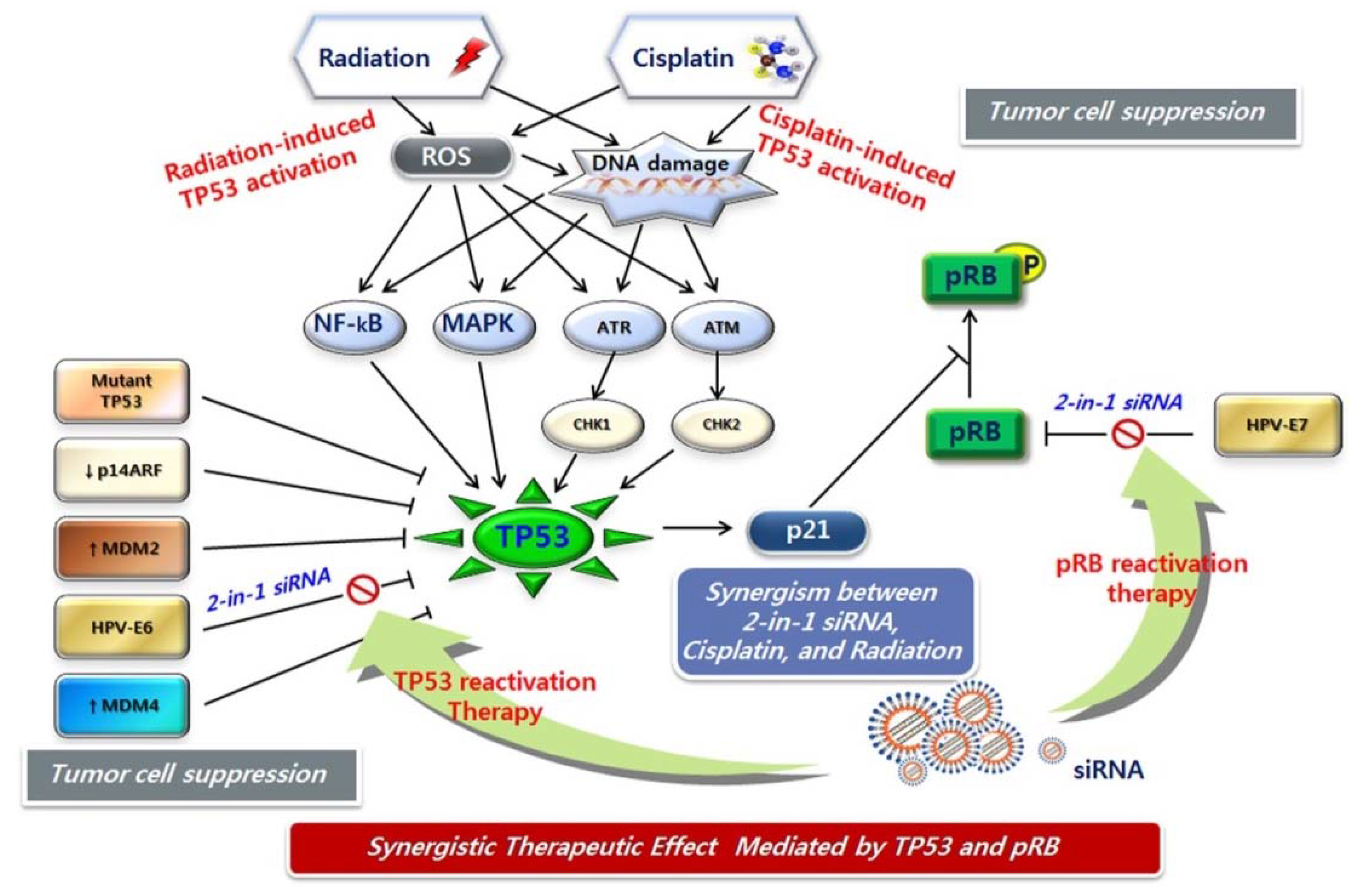
5. RNAi-based Combination Therapeutics against HPV
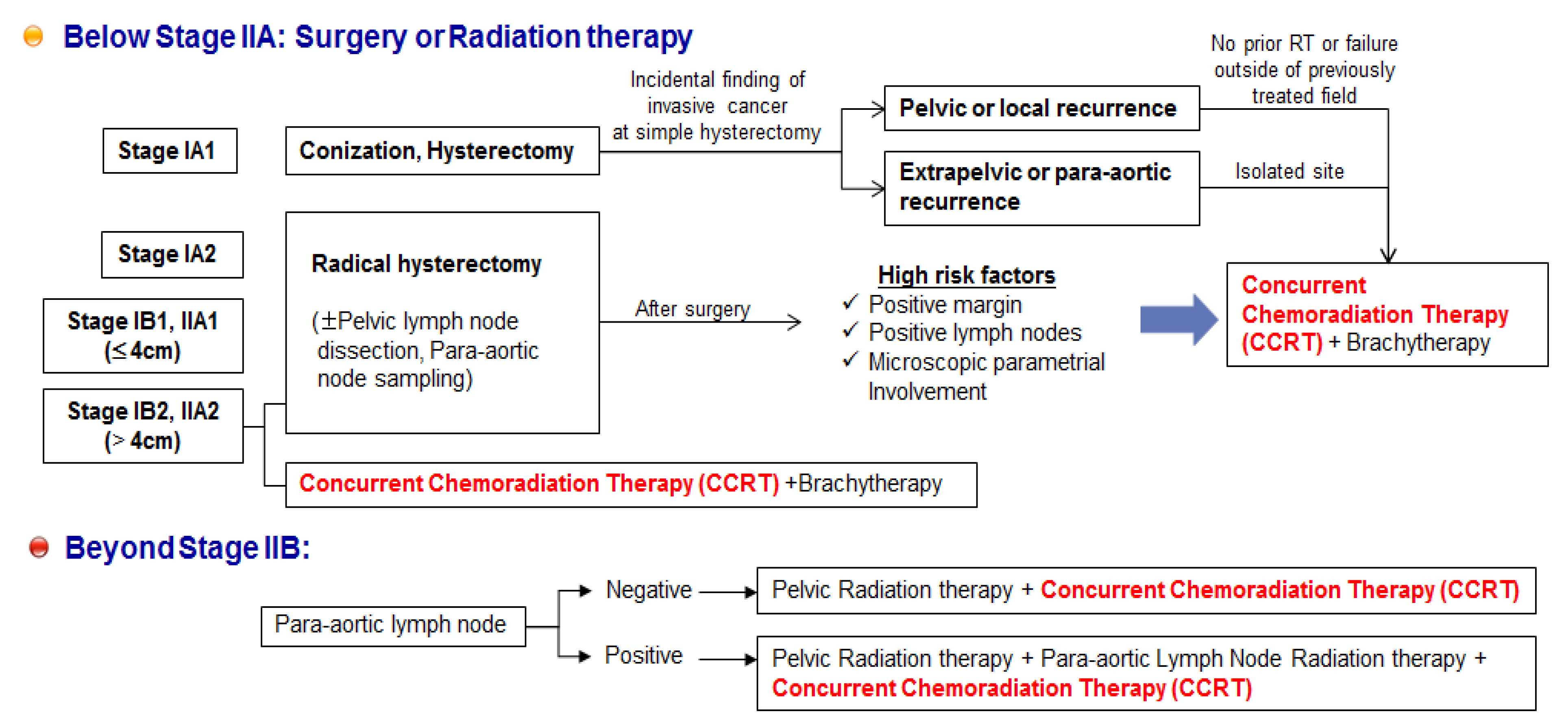
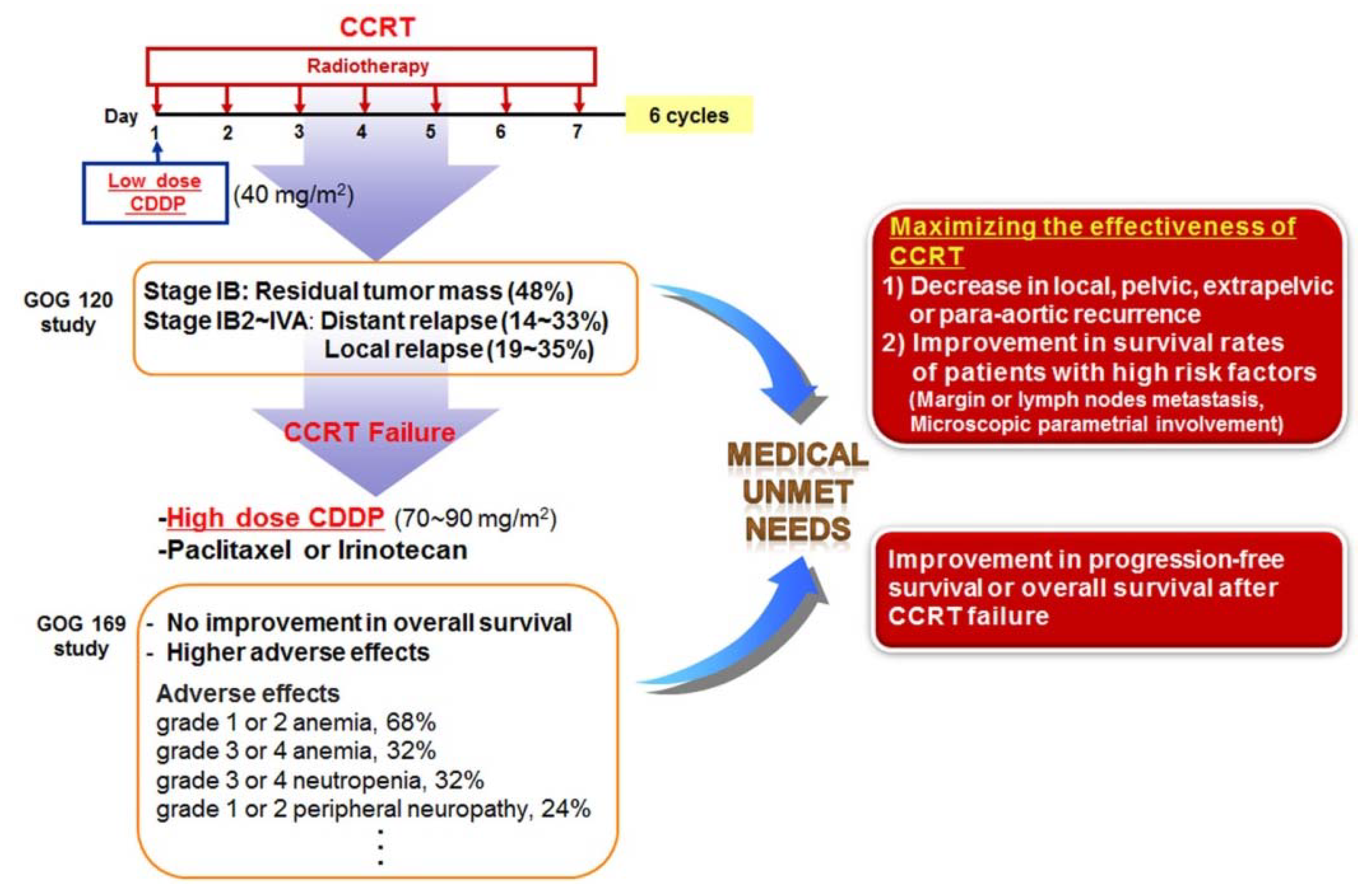
6. Unmet Medical Needs of Cervical Cancer
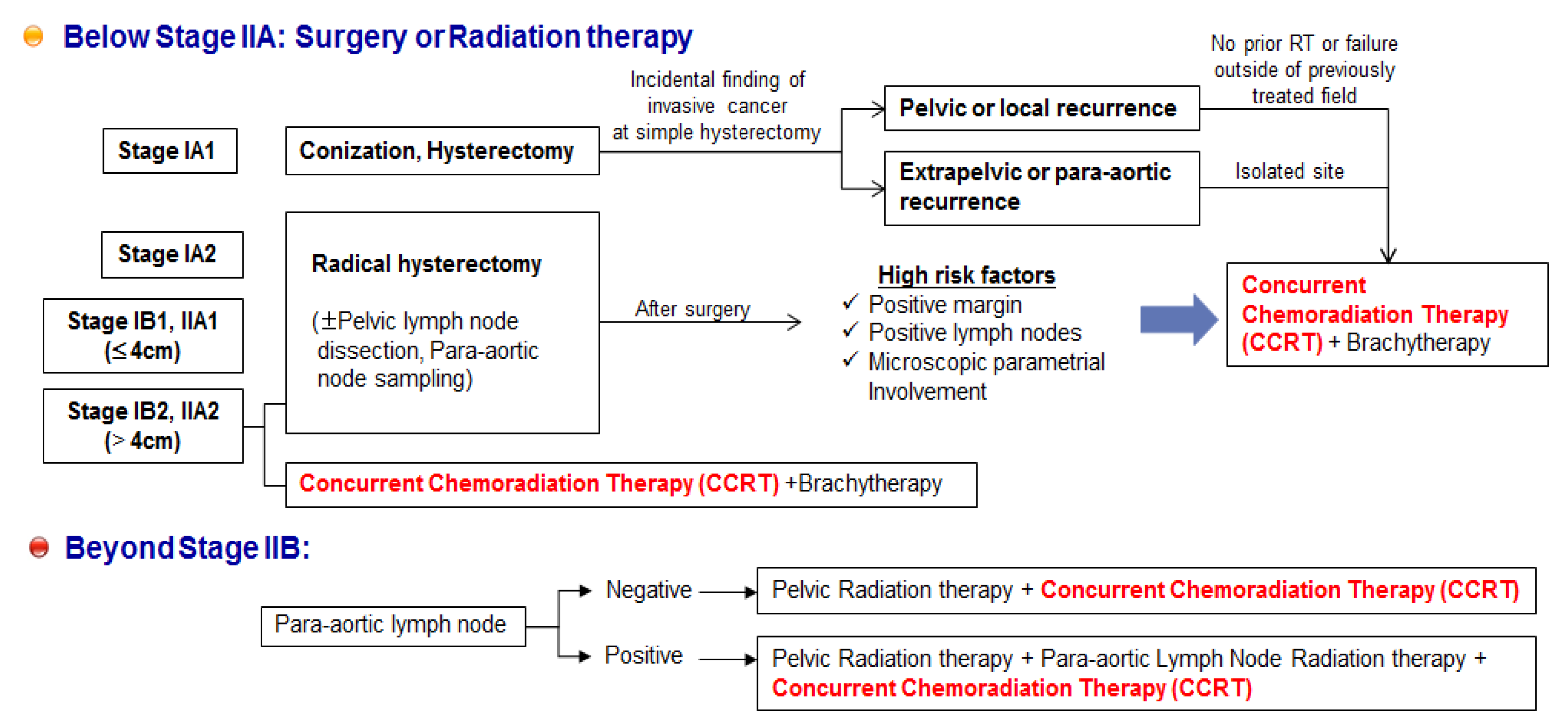
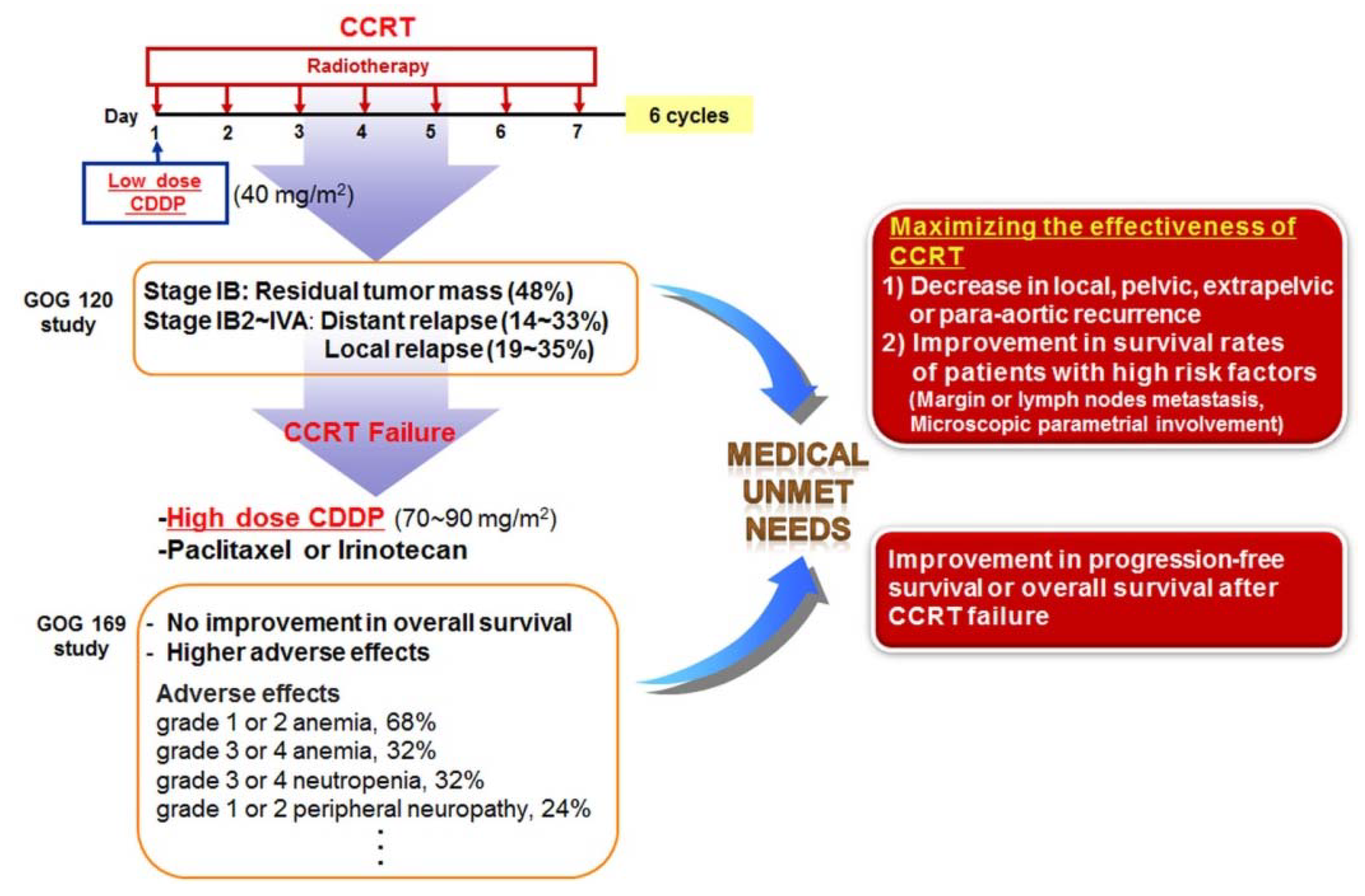
7. Potential Combination Therapies for Cervical Cancer
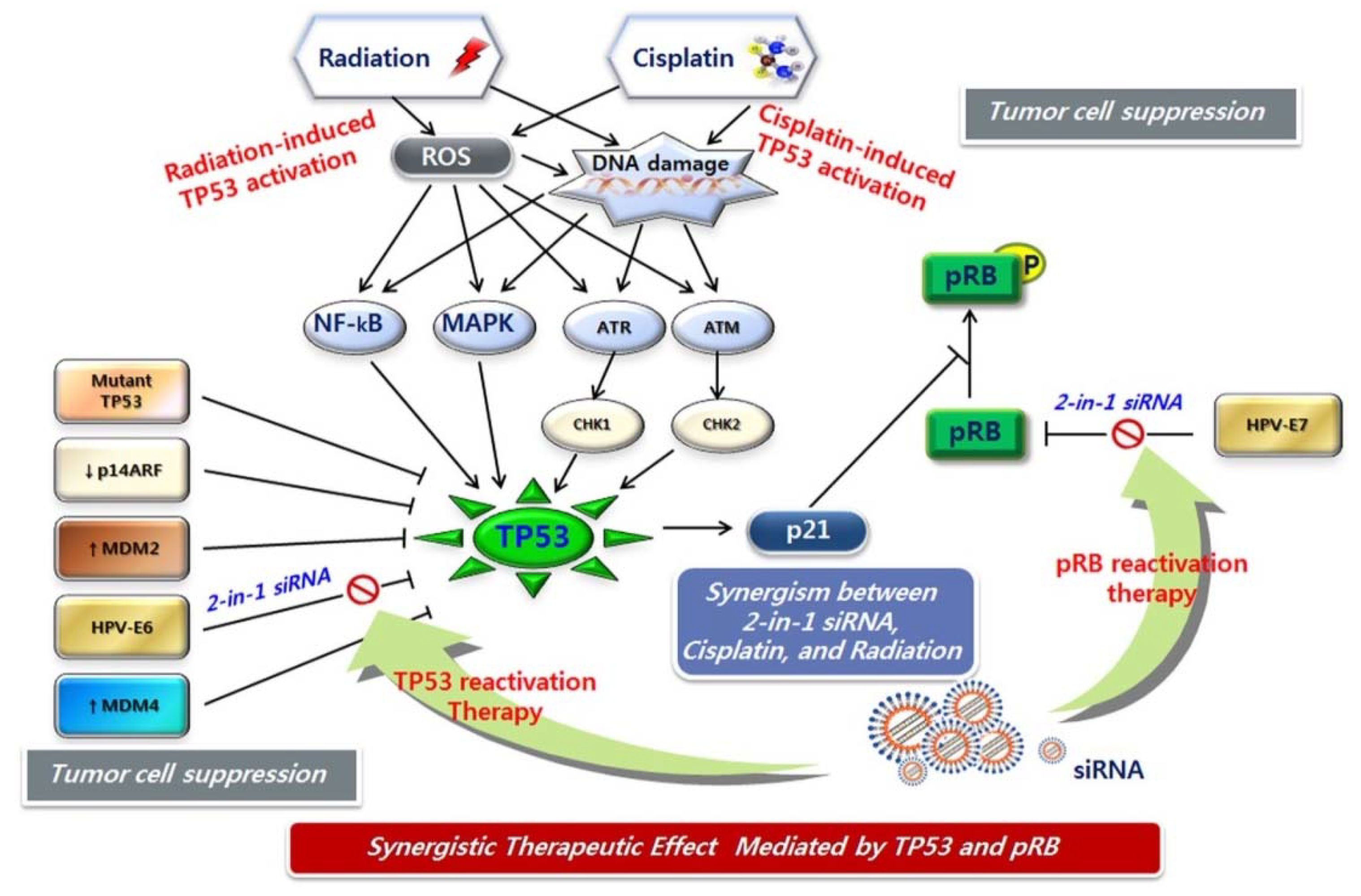
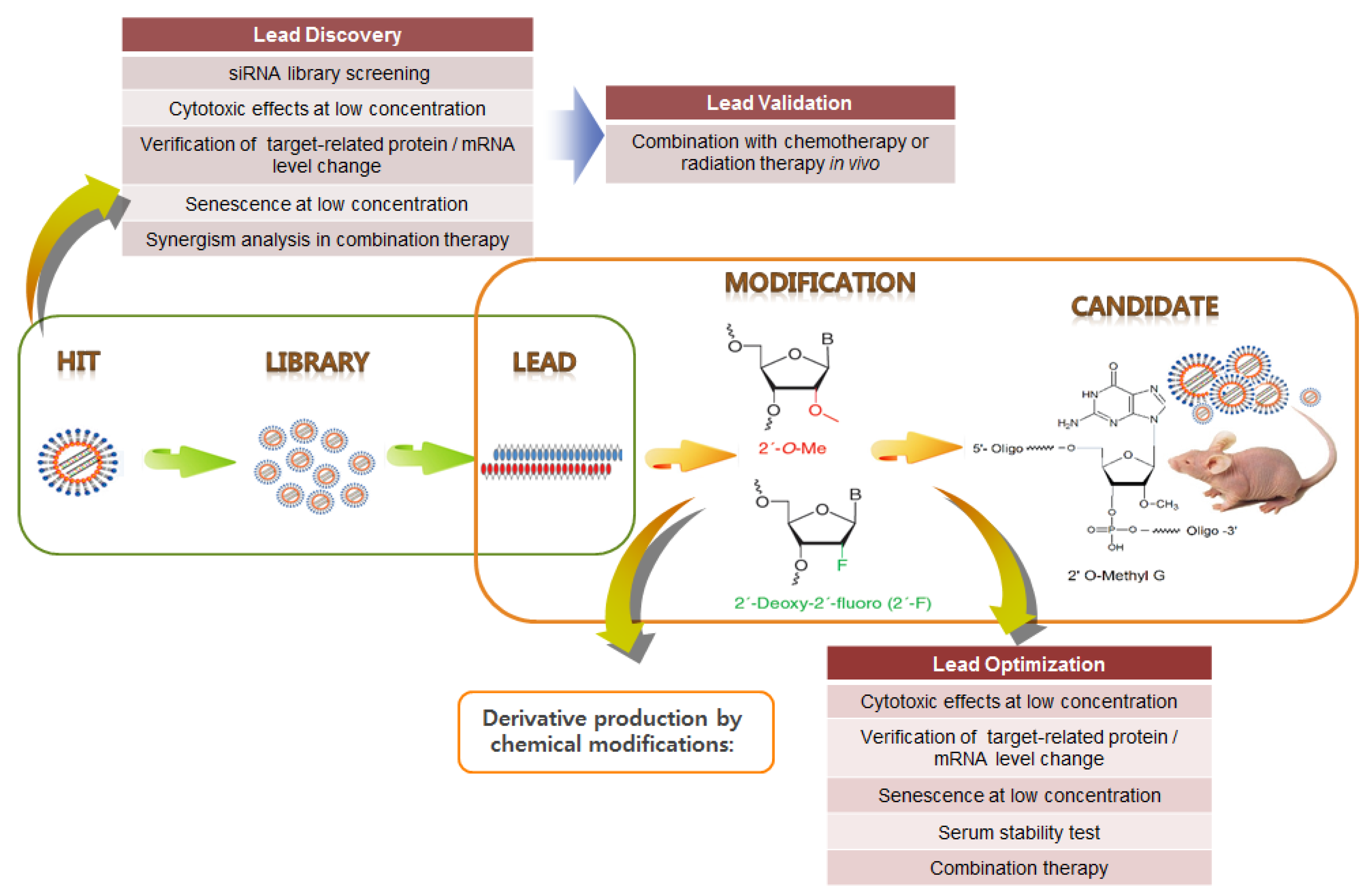
8. siRNA Pooling Technology, a Promising RNAi Therapy
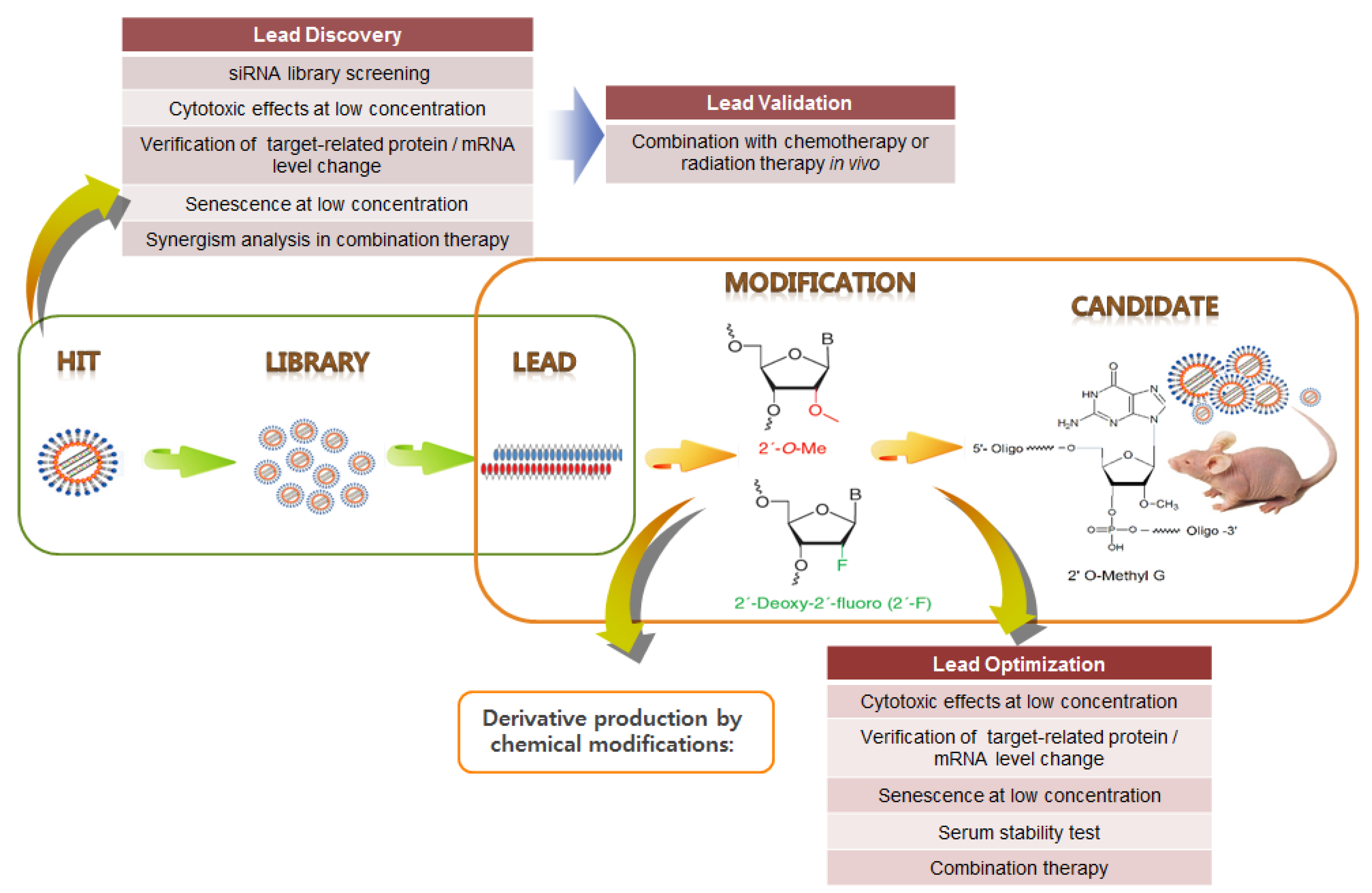
HPV siRNA Nanoparticles for Targeted Anticancer Therapy
| Candidate | Target | Focus of the study | Phase | Delivery | Sponsor |
|---|---|---|---|---|---|
| siRNA-EphA2-DOPC | EphA2 | Advanced solid tumors | Phase 1 not yet recruited | Intravenous | M.D. Anderson Cancer Center |
| TD101 | Mutant keratin | Pachyonychia congenita | Phase 1 completed | Intradermal | TransDerm |
| AGN 211745 | VEGF receptor | CNV, AMD | Phase 2 terminated | Intravitreal | Allergan |
| Bevasiranib | VEGF | DME | Phase 2 completed | Intravitreal | Opko Health, Inc. |
| AMD | Phase 3 withdrawn | ||||
| SV40 siRNA vectors | BCR-ABL | CML | Observational study completed | Hadassah Medical Organization | |
| CALAA-01 | M2 subunit of ribonucleotide reductase | Solid tumor | Phase 1 terminated | Intravenous, cyclodextrin | Calando Pharmaceuticals |
| siRNA IL-10 | IL-10 | Preeclampsia | Observational study terminated | National Taiwan University Hospital | |
| SYL1001 | Receptor TrpV1 | Ocular pain dry eye | Phase 1 completed | Eye drop | Sylentis, S.A. |
| EZN-2968 Antisense oligonucleotide | HIF | Liver cancer or lymphoma | Phase 1 completed | Intravenous | Enzon Pharmaceuticals |
| ALN-VSP02 | VEGF/ Kinesin spindle protein | Solid tumor | Phase 1 completed | Intravenous, SNALP liposome | Alnylam Pharmaceuticals |
| ALN-RSV01 | RSV | Lung transplant patients/RSV infection | Phase 2 completed | Intranasal | |
| ALN-TTR02 | Transthyretin | Amyloidosis | Phase 2 completed | Intravenous, SNALP liposome | |
| ALN-PCS02 | PCSK9 | Hypercholesterolemia | Phase 1 completed | Intravenous, SNALP liposome | |
| Miravirsen | miR-122 | Hepatitis C virus | Phase 2 recruiting | Subcutaneous | SantarisPharma A/S |
| TKM-080301 | Polo-like kinase-1 | Advanced solid tumors | Phase 1 completed | Intravenous, SNALP liposome | Tekmira Pharmaceuticals |
| TKM-EBOLA | Viral RNA | Ebola infection (biodefense) | Phase 1 terminated | ||
| PRO-040201 | Apolipoprotein B | Hypercholesterolemia | Phase 1 terminated | ||
| Atu027 | Protein kinase N3 | Advanced solid tumors | Phase 1 completed | Intravenous, AtuPLEXlipoplex | Silence Therapeutics |
| siG12D LODER | Mutated KRAS oncogene | Pancreatic cancer | Phase 2 / Phase 1 recruiting | Intratumoral | Silenseed, Ltd. |
| I5NP (QPI-1002) | TP53 | ARF | Phase 1 completed | Intravenous naked siRNA | Quark Pharmaceuticals |
| Kidney transplantation | Phase 1/2 completed | ||||
| PF-04523655 | RTP801 | DME | Phase 2 completed | Intravitreal | |
| QPI-1007 | Caspase-2 | Optic nerve atrophy NAION | Phase 1 completed | Intravitreal | Quark Pharmaceuticals |
| SYL040012 | β-2 adrenergic receptor | Glaucoma, ocular hypertension | Phase 1/2 completed | Eye drop | Sylentis, S.A. |
| RXI-109 | Connective tissue growth factor | Dermal scarring | Phase 2 Recruiting | Intradermal | RXi Pharmaceuticals |
| EZN-2968 Antisense Oligonucleotide | HIF-1 | Advanced solid tumors with liver metastases | Phase 1 completed | Intravenous | National Cancer Institute |
| rHIV7-shI-TAR-CCR5RZ | Viral RNA and host factor | AIDS lymphoma | Phase 1 recruiting | Lentiviral | City of Hope Medical Center |
| Excellair | Syk kinase | Asthma | Phase 2 | Inhalation | ZaBeCor |
| FANG vaccine | Furin | Advanced cancer, ovarian cancer, melanoma | Phase 1/2 recruiting | Plasmid | Gradalis, Inc. |
| CEQ508 | β-catenin | Familial adenomatous polyposis (FAP) | Phase 1 | Bacterial | Cequent Pharmaceuticals |
9. Conclusion and Future Perspectives
Acknowledgements
Grant Support
Author Contributions
Conflicts of Interest
References
- Cogliano, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F. Carcinogenicity of human papillomaviruses. Lancet Oncol. 2005, 6, 204. [Google Scholar]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [PubMed]
- zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [PubMed]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [PubMed]
- Syrjanen, S. The role of human papillomavirus infection in head and neck cancers. Ann. Oncol. 2010, 21, vii243–vii 245. [Google Scholar] [CrossRef] [PubMed]
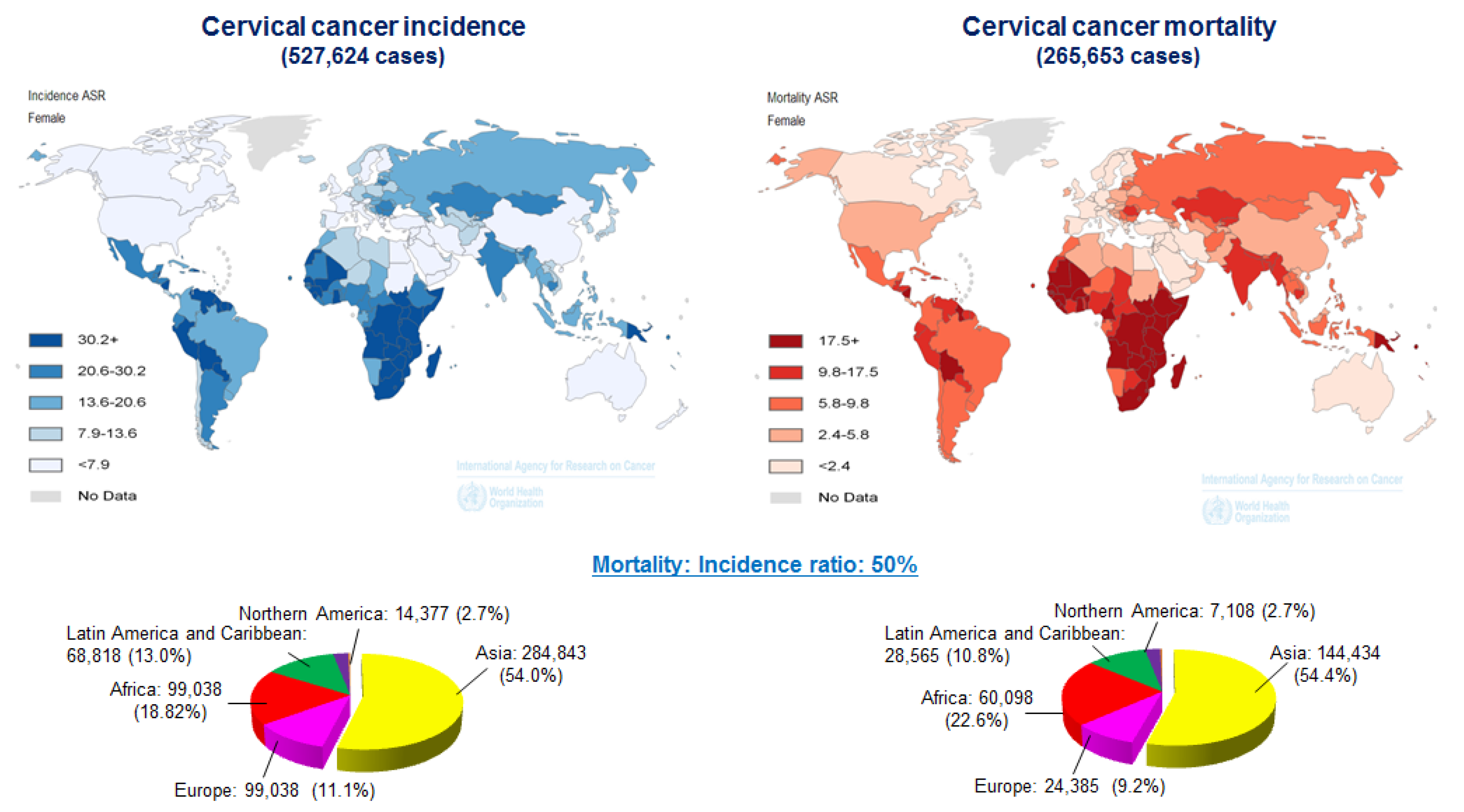
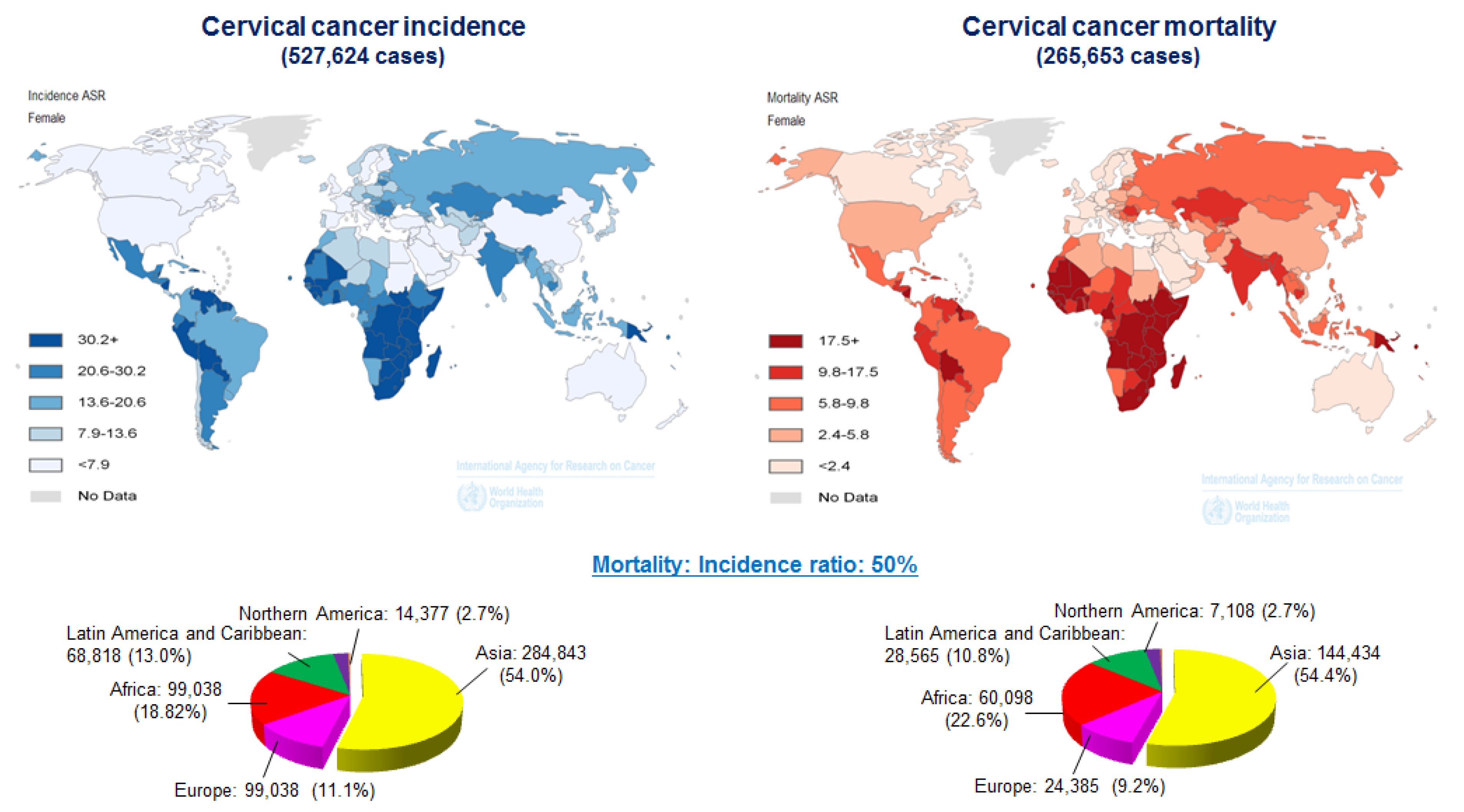
- Arbyn, M.; Castellsague, X.; de Sanjose, S.; Bruni, L.; Saraiya, M.; Bray, F.; Ferlay, J. Worldwide burden of cervical cancer in 2008. Ann. Oncol. 2011, 22, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Derkay, C.S.; Wiatrak, B. Recurrent respiratory papillomatosis: A review. The Laryngoscope 2008, 118, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Munger, K. Mechanisms of genomic instability in human cancer: Insights from studies with human papillomavirus oncoproteins. Int. J. Cancer 2004, 109, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.S.; Lowy, D.R.; Schiller, J.T. Papillomavirus polypeptides E6 and E7 are zinc-binding proteins. J. Virol. 1989, 63, 1404–1407. [Google Scholar] [PubMed]
- Sherman, L.; Schlegel, R. Serum- and calcium-induced differentiation of human keratinocytes is inhibited by the E6 oncoprotein of human papillomavirus type 16. J. Virol. 1996, 70, 3269–3279. [Google Scholar] [PubMed]
- Kanda, T.; Watanabe, S.; Zanma, S.; Sato, H.; Furuno, A.; Yoshiike, K. Human papillomavirus type 16 E6 proteins with glycine substitution for cysteine in the metal-binding motif. Virology 1991, 185, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Nomine, Y.; Masson, M.; Charbonnier, S.; Zanier, K.; Ristriani, T.; Deryckere, F.; Sibler, A.P.; Desplancq, D.; Atkinson, R.A.; Weiss, E.; et al. Structural and functional analysis of E6 oncoprotein: Insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol. Cell 2006, 21, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Dowhanick, J.J.; McBride, A.A.; Howley, P.M. Suppression of cellular proliferation by the papillomavirus E2 protein. J. Virol. 1995, 69, 7791–7799. [Google Scholar] [PubMed]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, M.T.; Smits, H.L.; Briet, M.A.; van den Tweel, J.G.; Struyk, A.P.; van der Noordaa, J.; ter Schegget, J. Uniformity of the splicing pattern of the E6/E7 transcripts in human papillomavirus type 16-transformed human fibroblasts, human cervical premalignant lesions and carcinomas. J. Gen. Virol. 1990, 71, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, H.; Jin, M.H.; Shimizu, K.; Akutsu, N.; Shino, Y.; Simizu, B. Transcription-modulatory activity of full-length E6 and E6*I proteins of human papillomavirus type 16. Virology 1994, 203, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Tao, M.; McCoy, J.P., Jr.; Zheng, Z.M. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J. Virol. 2006, 80, 4249–4263. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, M.; Zheng, Z.M. Oncogenes and RNA splicing of human tumor viruses. Emerg. Microbes Infec. 2014, 3. [Google Scholar] [CrossRef]
- Ajiro, M.; Jia, R.; Zhang, L.; Liu, X.; Zheng, Z.M. Intron definition and a branch site adenosine at nt 385 control RNA splicing of HPV16 E6*I and E7 expression. PLoS ONE 2012, 7, e46412. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Tomaic, V.; Banks, L. The human papillomavirus (HPV) E6* proteins from high-risk, mucosal HPVs can direct degradation of cellular proteins in the absence of full-length E6 protein. J. Virol. 2009, 83, 9863–9874. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.M.; Wang, X. Regulation of cellular miRNA expression by human papillomaviruses. Biochim. Biophys. Acta 2011, 1809, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.K.; Li, Y.; Hafner, M.; Banerjee, N.S.; Tang, S.; Briskin, D.; Meyers, C.; Chow, L.T.; Xie, X.; et al. MicroRNAs are biomarkers of oncogenic human papillomavirus infections. Proc. Natl. Acad. Sci. USA 2014, 111, 4262–4267. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.S.; Vass, W.C.; Lowy, D.R.; Schiller, J.T. In vitro biological activities of the E6 and E7 genes vary among human papillomaviruses of different oncogenic potential. J. Virol. 1991, 65, 292–298. [Google Scholar] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Beaudenon, S.; Howley, P.M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA 1995, 92, 2563–2567. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Clifford, G.; Buonaguro, F.M. Classification of weakly carcinogenic human papillomavirus types: Addressing the limits of epidemiology at the borderline. Infect. Agents Cancer 2009, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Banks, L. Inhibition of e6 induced degradation of p53 is not sufficient for stabilization of p53 protein in cervical tumour derived cell lines. Oncogene 1999, 18, 3309–3315. [Google Scholar] [CrossRef] [PubMed]
- Tungteakkhun, S.S.; Duerksen-Hughes, P.J. Cellular binding partners of the human papillomavirus E6 protein. Arch. Virol. 2008, 153, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.K.; McCoy, J.P.; Banerjee, N.S.; Rader, J.S.; Broker, T.R.; Meyers, C.; Chow, L.T.; Zheng, Z.M. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA 2009, 15, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, S.M.; Syrjanen, K.J. New concepts on the role of human papillomavirus in cell cycle regulation. Ann. Med. 1999, 31, 175–187. [Google Scholar] [CrossRef] [PubMed]
- McMurray, H.R.; Nguyen, D.; Westbrook, T.F.; McAnce, D.J. Biology of human papillomaviruses. Int. J. Exp. Pathol. 2001, 82, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Demers, G.W.; Galloway, D.A. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J. Virol. 1991, 65, 473–478. [Google Scholar] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sampath, A.; Raychaudhuri, P.; Bagchi, S. Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene 2001, 20, 4740–4749. [Google Scholar] [CrossRef] [PubMed]
- Heck, D.V.; Yee, C.L.; Howley, P.M.; Munger, K. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proc. Natl. Acad. Sci. USA 1992, 89, 4442–4446. [Google Scholar] [CrossRef] [PubMed]
- Charette, S.T.; McCance, D.J. The E7 protein from human papillomavirus type 16 enhances keratinocyte migration in an AKT-dependent manner. Oncogene 2007, 26, 7386–7390. [Google Scholar] [CrossRef] [PubMed]
- DiPaolo, J.A.; Woodworth, C.D.; Popescu, N.C.; Notario, V.; Doniger, J. Induction of human cervical squamous cell carcinoma by sequential transfection with human papillomavirus 16 DNA and viral Harvey ras. Oncogene 1989, 4, 395–399. [Google Scholar] [PubMed]
- Durst, M.; Gallahan, D.; Jay, G.; Rhim, J.S. Glucocorticoid-enhanced neoplastic transformation of human keratinocytes by human papillomavirus type 16 and an activated ras oncogene. Virology 1989, 173, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.; Cannon, R.E.; Karrison, T.; Beck-Engeser, G.; Huo, D.; Tennant, R.W.; Jensen, H.; Kast, W.M.; Krausz, T.; Meredith, S.C.; et al. Strong synergy between mutant ras and HPV16 E6/E7 in the development of primary tumors. Oncogene 2004, 23, 3972–3979. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Paredes, A.; De la Cruz-Hernandez, E.; Martinez-Ramirez, I.; Duenas-Gonzalez, A.; Lizano, M. E6 variants of human papillomavirus 18 differentially modulate the protein kinase B/phosphatidylinositol 3-kinase (AKT/PI3K) signaling pathway. Virology 2009, 383, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Pim, D.; Banks, L. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. J. Gen. Virol. 1997, 78, 2607–2613. [Google Scholar] [PubMed]
- Brehm, A.; Nielsen, S.J.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999, 18, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, N.; Torchia, J.; Mymryk, J.S. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene 2003, 22, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.G.; Lee, D.; Kim, J.; Seo, T.; Choe, J. Human papillomavirus type 16 E7 binds to E2F1 and activates E2F1-driven transcription in a retinoblastoma protein-independent manner. J. Biol. Chem. 2002, 277, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.C.; Hollstein, M. Clinical implications of the p53 tumor-suppressor gene. N. Engl. J. Med. 1993, 329, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Hengstermann, A.; Linares, L.K.; Ciechanover, A.; Whitaker, N.J.; Scheffner, M. Complete switch from Mdm2 to human papillomavirus E6-mediated degradation of p53 in cervical cancer cells. Proc. Natl. Acad. Sci. USA 2001, 98, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Hernandez, T.; Robinson, A.; Rodriguez-Tome, P.; Flores, T.; Hollstein, M.; Harris, C.C.; Montesano, R. Iarc database of p53 gene mutations in human tumors and cell lines: Updated compilation, revised formats and new visualisation tools. Nucl. Acids Res. 1998, 26, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Munger, K.; Byrne, J.C.; Howley, P.M. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 1991, 88, 5523–5527. [Google Scholar] [CrossRef] [PubMed]
- Lowy, D.R.; Schiller, J.T. Prophylactic human papillomavirus vaccines. J. Clin. Invest. 2006, 116, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, A.R.; Palefsky, J.M.; Goldstone, S.; Moreira, E.D., Jr.; Penny, M.E.; Aranda, C.; Vardas, E.; Moi, H.; Jessen, H.; Hillman, R.; et al. Efficacy of quadrivalent HPV vaccine against HPV infection and disease in males. N. Engl. J. Med. 2011, 364, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Beerheide, W.; Bernard, H.U.; Tan, Y.J.; Ganesan, A.; Rice, W.G.; Ting, A.E. Potential drugs against cervical cancer: Zinc-ejecting inhibitors of the human papillomavirus type 16 E6 oncoprotein. J. Natl. Cancer Inst. 1999, 91, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Beerheide, W.; Sim, M.M.; Tan, Y.J.; Bernard, H.U.; Ting, A.E. Inactivation of the human papillomavirus-16 E6 oncoprotein by organic disulfides. Bioorganic Med. Chem. 2000, 8, 2549–2560. [Google Scholar] [CrossRef]
- Butz, K.; Denk, C.; Ullmann, A.; Scheffner, M.; Hoppe-Seyler, F. Induction of apoptosis in human papillomaviruspositive cancer cells by peptide aptamers targeting the viral E6 oncoprotein. Proc. Natl. Acad. Sci. USA 2000, 97, 6693–6697. [Google Scholar] [CrossRef] [PubMed]
- Butz, K.; Ristriani, T.; Hengstermann, A.; Denk, C.; Scheffner, M.; Hoppe-Seyler, F. SiRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene 2003, 22, 5938–5945. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Milner, J. Selective silencing of viral gene expression in HPV-positive human cervical carcinoma cells treated with siRNA, a primer of RNA interference. Oncogene 2002, 21, 6041–6048. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Z.; Androphy, E.; Chen, J.; Baleja, J.D. Design and characterization of helical peptides that inhibit the E6 protein of papillomavirus. Biochemistry 2004, 43, 7421–7431. [Google Scholar] [CrossRef] [PubMed]
- Wesierska-Gadek, J.; Schloffer, D.; Kotala, V.; Horky, M. Escape of p53 protein from E6-mediated degradation in Hela cells after cisplatin therapy. Int. J. Cancer 2002, 101, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Yoshinouchi, M.; Yamada, T.; Kizaki, M.; Fen, J.; Koseki, T.; Ikeda, Y.; Nishihara, T.; Yamato, K. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol. Ther. 2003, 8, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.F.; Rao, Z.G.; Zhang, J.R. Effects of anti-HPV16 E6-ribozyme on the proliferation and apoptosis of human cervical cancer cell line CaSki. Acad. J. First Med. Coll. PLA 2002, 22, 496–498. [Google Scholar]
- Ajiro, M.; Zheng, Z.M. E6^E7, a novel splice isoform protein of human papillomavirus 16, stabilizes viral E6 and E7 oncoproteins via HSP90 and GRP78. MBio 2015, 6, e02068–e02014. [Google Scholar] [CrossRef]
- Guo, C.P.; Liu, K.W.; Luo, H.B.; Chen, H.B.; Zheng, Y.; Sun, S.N.; Zhang, Q.; Huang, L. Potent anti-tumor effect generated by a novel human papillomavirus (HPV) antagonist peptide reactivating the pRb/E2F pathway. PLoS ONE 2011, 6, e17734. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, G.H.; Shibata, R.; Wang, J.; Ray, A.S.; Wu, S.; Doerrfler, E.; Reiser, H.; Lee, W.A.; Birkus, G.; Christensen, N.D.; et al. Gs-9191 is a novel topical prodrug of the nucleotide analog 9-(2-phosphonylmethoxyethyl)guanine with antiproliferative activity and possible utility in the treatment of human papillomavirus lesions. Antimicrob. Agents Chemother. 2009, 53, 2777–2784. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.H.; Han, H.D.; Noh, K.H.; Kim, T.W.; Son, S.W. Chitosan hydrogel containing GMCSF and a cancer drug exerts synergistic anti-tumor effects via the induction of CD8+ T cell-mediated anti-tumor immunity. Clin. Exp. Metastasis 2009, 26, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, R.; Morales-Peza, N.; Castelan-Sanchez, I.; Garcia-Villa, E.; Tapia, R.; Cid-Arregui, A.; Garcia-Carranca, A.; Lopez-Bayghen, E.; Gariglio, P. Heparin (GAG-hed) inhibits LCR activity of human papillomavirus type 18 by decreasing AP1 binding. BMC Cancer 2006, 6. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Monie, A.; Pang, X.; Hung, C.F.; Wu, T.C. Vascular disrupting agent DMXAA enhances the antitumor effects generated by therapeutic HPV DNA vaccines. J. Biomed. Sci. 2011, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Hoory, T.; Monie, A.; Wu, A.; Wang, M.C.; Hung, C.F. Treatment with demethylating agent, 5-aza-2′-deoxycytidine enhances therapeutic hpv DNA vaccine potency. Vaccine 2009, 27, 4363–4369. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Bharti, A.C.; Hussain, S.; Mahata, S.; Hedau, S.; Kailash, U.; Kashyap, V.; Bhambhani, S.; Roy, M.; Batra, S.; et al. Elimination of high-risk human papillomavirus type HPV16 infection by ‘Praneem’ polyherbal tablet in women with early cervical intraepithelial lesions. J. Cancer Res. Clin. Oncol. 2009, 135, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Milrot, E.; Jackman, A.; Kniazhanski, T.; Gonen, P.; Flescher, E.; Sherman, L. Methyl jasmonate reduces the survival of cervical cancer cells and downregulates HPV E6 and E7, and survivin. Cancer Lett. 2012, 319, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.X.; Huang, G.Q.; Zhang, S.H. Soyasaponins inhibit the proliferation of hela cells by inducing apoptosis. Exp. Toxicol. Pathol. 2007, 59, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Oh, P.S.; Lim, K.T. Hela cells treated with phytoglycoprotein (150 kDa) were killed by activation of caspase 3 via inhibitory activities of NK-kappaB and AP-1. J. Biomed. Sci. 2007, 14, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Maher, D.M.; Bell, M.C.; O’Donnell, E.A.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Curcumin suppresses human papillomavirus oncoproteins, restores p53, Rb, and PTPN13 proteins and inhibits benzo[a]pyrene-induced upregulation of HPV E7. Mol. Carcinog. 2011, 50, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wu, L.J.; Tashiro, S.; Onodera, S.; Li, L.H.; Ikejima, T. Silymarin augments human cervical cancer Hela cell apoptosis via P38/JNK MAPK pathways in serum-free medium. J. Asian Nat. Prod. Res. 2005, 7, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Munagala, R.; Kausar, H.; Munjal, C.; Gupta, R.C. Withaferin a induces p53-dependent apoptosis by repression of HPV oncogenes and upregulation of tumor suppressor proteins in human cervical cancer cells. Carcinogenesis 2011, 32, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Ahn, W.S.; Huh, S.W.; Bae, S.M.; Lee, I.P.; Lee, J.M.; Namkoong, S.E.; Kim, C.K.; Sin, J.I. A major constituent of green tea, EGCG, inhibits the growth of a human cervical cancer cell line, CaSki cells, through apoptosis, G(1) arrest, and regulation of gene expression. DNA Cell Biol. 2003, 22, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Yu, K.A.; Oh, W.K.; Baeg, T.W.; Oh, H.C.; Ahn, J.S.; Jang, W.C.; Kim, J.W.; Lim, J.S.; Choe, Y.K.; et al. Inhibitory effect of jaceosidin isolated from artemisiaargyi on the function of E6 and E7 oncoproteins of HPV 16. J. Ethnopharmacol. 2005, 98, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Craigo, J.; Callahan, M.; Huang, R.C.; DeLucia, A.L. Inhibition of human papillomavirus type 16 gene expression by nordihydroguaiaretic acid plant lignan derivatives. Antivir. Res. 2000, 47, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Thompson, C.D.; Roberts, J.N.; Muller, M.; Lowy, D.R.; Schiller, J.T. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2006, 2, e69. [Google Scholar] [CrossRef] [PubMed]
- Haasnoot, J.; Westerhout, E.M.; Berkhout, B. RNA interference against viruses: Strike and counterstrike. Nat. Biotechnol. 2007, 25, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Sui, G.; Soohoo, C.; Affar el, B.; Gay, F.; Shi, Y.; Forrester, W.C. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 5515–5520. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Y.; DeRuiter, S.L.; Turner, D.L. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 6047–6052. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; McManus, M.T.; Nguyen, T.; Shen, C.H.; Sharp, P.A.; Eisen, H.N.; Chen, J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc. Natl. Acad. Sci. USA 2003, 100, 2718–2723. [Google Scholar] [CrossRef] [PubMed]
- Kitabwalla, M.; Ruprecht, R.M. RNA interference—A new weapon against HIV and beyond. N. Engl. J. Med. 2002, 347, 1364–1367. [Google Scholar] [CrossRef] [PubMed]
- Milner, J. RNA interference for treating cancers caused by viral infection. Exp. Opin. Biol. Ther. 2003, 3, 459–467. [Google Scholar] [CrossRef]
- Sima, N.; Wang, W.; Kong, D.; Deng, D.; Xu, Q.; Zhou, J.; Xu, G.; Meng, L.; Lu, Y.; Wang, S.; et al. RNA interference against HPV16 E7 oncogene leads to viral E6 and E7 suppression in cervical cancer cells and apoptosis via upregulation of Rb and p53. Apoptosis 2008, 13, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Yamato, K.; Yamada, T.; Kizaki, M.; Ui-Tei, K.; Natori, Y.; Fujino, M.; Nishihara, T.; Ikeda, Y.; Nasu, Y.; Saigo, K.; et al. New highly potent and specific E6 and E7 siRNAs for treatment of HPV16 positive cervical cancer. Cancer Gene Ther. 2008, 15, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Putral, L.; Hengst, K.; Minto, K.; Saunders, N.A.; Leggatt, G.; McMillan, N.A. Inhibition of cervical cancer cell growth in vitro and in vivo with lentiviral-vector delivered short hairpin RNA targeting human papillomavirus E6 and E7 oncogenes. Cancer Gene Ther. 2006, 13, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.H.; Alexander, K.A. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in Hela cells. J. Virol. 2003, 77, 6066–6069. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Tao, M.; McCoy, J.P., Jr.; Zheng, Z.M. Short-term induction and long-term suppression of HPV16 oncogene silencing by RNA interference in cervical cancer cells. Oncogene 2006, 25, 2094–2104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Peng, C.; Li, B.; Wang, F.; Zhou, C.; Hong, D.; Ye, F.; Cheng, X.; Lu, W.; Xie, X. Transcriptional gene silencing of HPV16 E6/E7 induces growth inhibition via apoptosis in vitro and in vivo. Gynecol. Oncol. 2012, 124, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Erkin, O.C.; Kwon, M.J.; Kim, S.H.; Jung, J.I.; Oh, Y.K.; Her, S.W.; Ju, W.; Choi, Y.L.; Song, S.Y.; et al. The synergistic therapeutic effect of cisplatin with human papillomavirus E6/E7 short interfering RNA on cervical cancer cell lines in vitro and in vivo. Int. J. Cancer 2012, 130, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Kuo, T.F.; Chen, Y.J.; Chiu, C.C.; Lu, Y.C.; Li, H.F.; Shen, C.R.; Cheng, A.J. Highly potent and specific siRNAs against E6 or E7 genes of HPV16- or HPV18-infected cervical cancers. Cancer Gene Ther. 2010, 17, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Lu, W.; Ye, F.; Hu, Y.; Xie, X. Gene silencing of HPV16 E6/E7 induced by promoter-targeting siRNA in SiHa cells. Br. J. Cancer 2009, 101, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Jonson, A.L.; Rogers, L.M.; Ramakrishnan, S.; Downs, L.S., Jr. Gene silencing with siRNA targeting E6/E7 as a therapeutic intervention in a mouse model of cervical cancer. Gynecol. Oncol. 2008, 111, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Lea, J.S.; Sunaga, N.; Sato, M.; Kalahasti, G.; Miller, D.S.; Minna, J.D.; Muller, C.Y. Silencing of HPV 18 oncoproteins with RNA interference causes growth inhibition of cervical cancer cells. Reprod. Sci. 2007, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Courtete, J.; Sibler, A.P.; Zeder-Lutz, G.; Dalkara, D.; Oulad-Abdelghani, M.; Zuber, G.; Weiss, E. Suppression of cervical carcinoma cell growth by intracytoplasmic codelivery of anti-oncoprotein E6 antibody and small interfering RNA. Mol. Cancer Ther. 2007, 6, 1728–1735. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Saito, M.; Iwasaki, E.; Ochiya, T.; Takei, Y.; Hayashi, S.; Ono, A.; Hirao, N.; Nakamura, M.; Kubushiro, K.; et al. Intratumor injection of small interfering RNA-targeting human papillomavirus 18 E6 and E7 successfully inhibits the growth of cervical cancer. Int. J. Oncol. 2006, 29, 541–548. [Google Scholar] [PubMed]
- Koivusalo, R.; Krausz, E.; Helenius, H.; Hietanen, S. Chemotherapy compounds in cervical cancer cells primed by reconstitution of p53 function after short interfering RNA-mediated degradation of human papillomavirus 18 E6 mRNA: Opposite effect of siRNA in combination with different drugs. Mol. Pharmacol. 2005, 68, 372–382. [Google Scholar] [PubMed]
- Putral, L.N.; Bywater, M.J.; Gu, W.; Saunders, N.A.; Gabrielli, B.G.; Leggatt, G.R.; McMillan, N.A. RNA interference against human papillomavirus oncogenes in cervical cancer cells results in increased sensitivity to cisplatin. Mol. Pharmacol. 2005, 68, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Huang, S.Y.; Chen, T.T.; Chen, J.C.; Chiou, C.L.; Huang, T.M. Cisplatin restores p53 function and enhances the radiosensitivity in HPV16 E6 containing SiHa cells. J. Cell. Biochem. 2004, 91, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Feldser, D.M.; Kostova, K.K.; Winslow, M.M.; Taylor, S.E.; Cashman, C.; Whittaker, C.A.; Sanchez-Rivera, F.J.; Resnick, R.; Bronson, R.; Hemann, M.T.; et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010, 468, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Karnezis, A.N.; Garcia, D.; Madriles, F.; Kortlever, R.M.; Rostker, F.; Brown Swigart, L.; Pham, D.M.; Seo, Y.; Evan, G.I.; et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010, 468, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microrna processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Loewer, A.; Mock, C.; Lahav, G. Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol. 2011, 7, 488. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Murray, D. Ionizing radiation-induced responses in human cells with differing TP53 status. Int. J. Mol. Sci. 2013, 14, 22409–22435. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Rivlin, N.; Shoshana, O.Y.; Ezra, O.; Madar, S.; Goldfinger, N.; Rotter, V. Chemotherapeutic agents induce the expression and activity of their clearing enzyme CYP3A4 by activating p53. Carcinogenesis 2013, 34, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Loewer, A.; Batchelor, E.; Gaglia, G.; Lahav, G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell 2010, 142, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. P53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Shi, J.; Jung, S.H.; Chen, X.; Cho, K.H. Attractor landscape analysis reveals feedback loops in the p53 network that control the cellular response to DNA damage. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Pan, D.; Pan, L.Z.; Hill, R.; Marcato, P.; Shmulevitz, M.; Vassilev, L.T.; Lee, P.W. Stabilisation of p53 enhances reovirus-induced apoptosis and virus spread through p53-dependent NF-kappaB activation. Br. J. Cancer 2011, 105, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Bartz, S.R.; Zhang, Z.; Burchard, J.; Imakura, M.; Martin, M.; Palmieri, A.; Needham, R.; Guo, J.; Gordon, M.; Chung, N.; et al. Small interfering RNA screens reveal enhanced cisplatin cytotoxicity in tumor cells having both BRCA network and TP53 disruptions. Mol. Cell Biol. 2006, 26, 9377–9386. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.A.; Ye, F.; Marshall, C.B.; Lehmann, B.D.; Pendleton, C.S.; Shyr, Y.; Arteaga, C.L.; Pietenpol, J.A. RNA interference (RNAi) screening approach identifies agents that enhance paclitaxel activity in breast cancer cells. Breast Cancer Res. 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Honma, K.; Iwao-Koizumi, K.; Takeshita, F.; Yamamoto, Y.; Yoshida, T.; Nishio, K.; Nagahara, S.; Kato, K.; Ochiya, T. RPN2 gene confers docetaxel resistance in breast cancer. Nat. Med. 2008, 14, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.; Deeds, S.L.; Weinstein, E.J. A screen of shRNAs targeting tumor suppressor genes to identify factors involved in A549 paclitaxel sensitivity. Oncol. Rep. 2007, 18, 1499–1505. [Google Scholar] [PubMed]
- Swanton, C.; Nicke, B.; Marani, M.; Kelly, G.; Downward, J. Initiation of high frequency multi-drug resistance following kinase targeting by siRNAs. Cell Cycle 2007, 6, 2001–2004. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Cochrane, M.; Leggatt, G.R.; Payne, E.; Choyce, A.; Zhou, F.; Tindle, R.; McMillan, N.A. Both treated and untreated tumors are eliminated by short hairpin RNA-based induction of target-specific immune responses. Proc. Natl. Acad. Sci. USA 2009, 106, 8314–8319. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.L.; Green, N.; Seymour, L.W.; Stevenson, M. Paclitaxel combined with siRNA targeting HPV16 oncogenes improves cytotoxicity for cervical carcinoma. Cancer Gene Ther. 2009, 16, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Eifel, P.J.; Lu, J.; Grigsby, P.W.; Levenback, C.; Stevens, R.E.; Rotman, M.; Gershenson, D.M.; Mutch, D.G. Pelvic radiation with concurrent chemotherapy compared with pelvic and para-aortic radiation for high-risk cervical cancer. N. Engl. J. Med. 1999, 340, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Keys, H.M.; Bundy, B.N.; Stehman, F.B.; Muderspach, L.I.; Chafe, W.E.; Suggs, C.L., 3rd; Walker, J.L.; Gersell, D. Cisplatin, radiation, and adjuvant hysterectomy compared with radiation and adjuvant hysterectomy for bulky stage Ib cervical carcinoma. N. Engl. J. Med. 1999, 340, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Peters, W.A., 3rd; Liu, P.Y.; Barrett, R.J., 2nd; Stock, R.J.; Monk, B.J.; Berek, J.S.; Souhami, L.; Grigsby, P.; Gordon, W., Jr.; Alberts, D.S. Concurrent chemotherapy and pelvic radiation therapy compared with pelvic radiation therapy alone as adjuvant therapy after radical surgery in high-risk early-stage cancer of the cervix. J. Clin. Oncol. 2000, 18, 1606–1613. [Google Scholar] [PubMed]
- Monk, B.J.; Sill, M.W.; McMeekin, D.S.; Cohn, D.E.; Ramondetta, L.M.; Boardman, C.H.; Benda, J.; Cella, D. Phase III trial of four cisplatin-containing doublet combinations in stage IVb, recurrent, or persistent cervical carcinoma: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2009, 27, 4649–4655. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.H.; Blessing, J.A.; McQuellon, R.P.; Thaler, H.T.; Cella, D.; Benda, J.; Miller, D.S.; Olt, G.; King, S.; Boggess, J.F.; et al. Phase III study of cisplatin with or without paclitaxel in stage IVb, recurrent, or persistent squamous cell carcinoma of the cervix: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2004, 22, 3113–3119. [Google Scholar] [CrossRef] [PubMed]
- Long, H.J., 3rd; Bundy, B.N.; Grendys, E.C., Jr.; Benda, J.A.; McMeekin, D.S.; Sorosky, J.; Miller, D.S.; Eaton, L.A.; Fiorica, J.V.; Gynecologic Oncology Group Study. Randomized phase III trial of cisplatin with or without topotecan in carcinoma of the uterine cervix: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2005, 23, 4626–4633. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Yakushiji, M.; Noda, K.; Ikeda, M.; Kudoh, R.; Yajima, A.; Tomoda, Y.; Terashima, Y.; Takeuchi, S.; Hiura, M.; et al. Phase II study of irinotecan and cisplatin as first-line chemotherapy in advanced or recurrent cervical cancer. Oncology 2000, 58, 31–37. [Google Scholar] [CrossRef]
- Pectasides, D.; Fountzilas, G.; Papaxoinis, G.; Pectasides, E.; Xiros, N.; Sykiotis, C.; Koumarianou, A.; Psyrri, A.; Panayiotides, J.; Economopoulos, T. Carboplatin and paclitaxel in metastatic or recurrent cervical cancer. Int. J. Gynecol. Cancer 2009, 19, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.A.; Blessing, J.A.; Nagourney, R.A.; McMeekin, D.S.; Lele, S.; Zweizig, S.L. Cisplatin plus gemcitabine in previously treated squamous cell carcinoma of the cervix: A phase II study of the Gynecologic Oncology Group. Gynecol. Oncol. 2006, 100, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Sill, M.W.; Burger, R.A.; Gray, H.J.; Buekers, T.E.; Roman, L.D. Phase II trial of bevacizumab in the treatment of persistent or recurrent squamous cell carcinoma of the cervix: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2009, 27, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.A.; Blessing, J.A.; Vaccarello, L.; Roman, L.D. Phase II clinical trial of docetaxel in refractory squamous cell carcinoma of the cervix: A Gynecologic Oncology Group Study. Am. J. Clin. Oncol. 2007, 30, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Pow, E.H.; Kwong, D.L.; McMillan, A.S.; Wong, M.C.; Sham, J.S.; Leung, L.H.; Leung, W.K. Xerostomia and quality of life after intensity-modulated radiotherapy vs. conventional radiotherapy for early-stage nasopharyngeal carcinoma: Initial report on a randomized controlled clinical trial. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Pignol, J.P.; Olivotto, I.; Rakovitch, E.; Gardner, S.; Sixel, K.; Beckham, W.; Vu, T.T.; Truong, P.; Ackerman, I.; Paszat, L. A multicenter randomized trial of breast intensity-modulated radiation therapy to reduce acute radiation dermatitis. J. Clin. Oncol. 2008, 26, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Lim, D.S. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol. 2000, 1, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Steel, G.G.; Peckham, M.J. Exploitable mechanisms in combined radiotherapy-chemotherapy: The concept of additivity. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Lahav, G.; Rosenfeld, N.; Sigal, A.; Geva-Zatorsky, N.; Levine, A.J.; Elowitz, M.B.; Alon, U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 2004, 36, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Yamato, K.; Yamada, T.; Kizaki, M.; Ui-Tei, K.; Natori, Y.; Fujino, M.; Nishihara, T.; Ikeda, Y.; Nasu, Y.; Saigo, K.; et al. New highly potent and specific E6 and E7 siRNAs for treatment of hpv16 positive cervical cancer. Cancer Gene Ther. 2008, 15, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Yamato, K.; Egawa, N.; Endo, S.; Ui-Tei, K.; Yamada, T.; Saigo, K.; Hyodo, I.; Kiyono, T.; Nakagawa, I. Enhanced specificity of HPV16 E6E7 siRNA by RNA-DNA chimera modification. Cancer Gene Ther. 2011, 18, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Egusquiaguirre, S.P.; Igartua, M.; Hernandez, R.M.; Pedraz, J.L. Nanoparticle delivery systems for cancer therapy: Advances in clinical and preclinical research. Clin. Transl. Oncol. 2012, 14, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Truong, N.P.; Gu, W.; Prasadam, I.; Jia, Z.; Crawford, R.; Xiao, Y.; Monteiro, M.J. An influenza virus-inspired polymer system for the timed release of siRNA. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Cun, B.; Song, X.; Jia, R.; Wang, H.; Zhao, X.; Liu, B.; Ge, S.; Fan, X. Cell growth inhibition in HPV 18 positive uveal melanoma cells by E6/E7 siRNA. Tumour Biol. 2013, 34, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Hu, J.; Wang, B.; Zhao, L.; Ji, K.; Guo, B.; Yin, D.; Du, Y.; Kopecko, D.J.; et al. Plasmid-based E6-specific siRNA and co-expression of wild-type p53 suppresses the growth of cervical cancer in vitro and in vivo. Cancer Lett. 2013, 335, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.S.; Rajasekaran, N.; Ju, W.; Shin, Y.K. Human Papillomavirus: Current and Future RNAi Therapeutic Strategies for Cervical Cancer. J. Clin. Med. 2015, 4, 1126-1155. https://doi.org/10.3390/jcm4051126
Jung HS, Rajasekaran N, Ju W, Shin YK. Human Papillomavirus: Current and Future RNAi Therapeutic Strategies for Cervical Cancer. Journal of Clinical Medicine. 2015; 4(5):1126-1155. https://doi.org/10.3390/jcm4051126
Chicago/Turabian StyleJung, Hun Soon, Nirmal Rajasekaran, Woong Ju, and Young Kee Shin. 2015. "Human Papillomavirus: Current and Future RNAi Therapeutic Strategies for Cervical Cancer" Journal of Clinical Medicine 4, no. 5: 1126-1155. https://doi.org/10.3390/jcm4051126
APA StyleJung, H. S., Rajasekaran, N., Ju, W., & Shin, Y. K. (2015). Human Papillomavirus: Current and Future RNAi Therapeutic Strategies for Cervical Cancer. Journal of Clinical Medicine, 4(5), 1126-1155. https://doi.org/10.3390/jcm4051126