Abstract
Human papillomaviruses (HPVs) are small DNA viruses; some oncogenic ones can cause different types of cancer, in particular cervical cancer. HPV-associated carcinogenesis provides a classical model system for RNA interference (RNAi) based cancer therapies, because the viral oncogenes E6 and E7 that cause cervical cancer are expressed only in cancerous cells. Previous studies on the development of therapeutic RNAi facilitated the advancement of therapeutic siRNAs and demonstrated its versatility by siRNA-mediated depletion of single or multiple cellular/viral targets. Sequence-specific gene silencing using RNAi shows promise as a novel therapeutic approach for the treatment of a variety of diseases that currently lack effective treatments. However, siRNA-based targeting requires further validation of its efficacy in vitro and in vivo, for its potential off-target effects, and of the design of conventional therapies to be used in combination with siRNAs and their drug delivery vehicles. In this review we discuss what is currently known about HPV-associated carcinogenesis and the potential for combining siRNA with other treatment strategies for the development of future therapies. Finally, we present our assessment of the most promising path to the development of RNAi therapeutic strategies for clinical settings.
1. Introduction
Human papillomaviruses (HPVs) are small DNA viruses with a genome size ~8 kb long. They infect cutaneous or mucosal epithelial cells, genital tissues, and the upper respiratory tract. To date, over 200 genetically distinct subtypes of HPV have been identified, and approximately 90 genotypes have been fully characterized. Among these types, the high-risk HPVs (HR-HPVs), including HPV-16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73, and 82, are associated with more than 90% of cervical cancers, and to a lesser extent with other anogenital cancers and head and neck cancers [1,2,3,4]. Moreover, HPV-16 is associated with a small number of head and neck neoplasias, particularly tonsillar and oro-pharyngeal cancers [5]. Low-risk HPVs (LR-HPVs), including HPV-6, 11, 40, 42, 43, 44, 54, 61, 70, 72, and 81, have been linked to benign epithelial lesions [6]. In contrast to HR-HPV, infections with LR-HPV types 6 and 11 are associated with genital warts, essentially all laryngeal papillomas, and recurrent respiratory papillomatosis [7]. Over 50% of HPV-positive cervical cancers are associated with HPV-16, followed by HPV-18 (12%), HPV-45 (8%), and HPV-31 (5%) [8,9]. The HPV genome is composed of an early region (E) that encodes open reading frames involved in the regulation of viral replication and the viral life cycle, and a late region (L) that encodes two ORFs (L1 and L2) that form the viral capsid. During the course of HPV-mediated cancer development, the viral DNAs are frequently integrated into host cell chromosomes, and the proteins encoded by the viral genes play a critical role in carcinogenesis.
2. HPV and Cervical Cancer
Cervical cancer is one of the most common types of gynecological malignancies worldwide. According to the World Health Organization, the global prevalence of HPV infections in 2012 was approximately 630 million cases. Of these, 190 million were clinical infections leading to 528,000 new diagnoses of cervical carcinoma and ~266,000 deaths (Figure 1) (http://globocan.iarc.fr) [10]. Advances in research continue to improve the precautionary methods available in developed countries. Clinical and molecular epidemiological studies have clearly demonstrated that the major cause of cervical cancer is infection with HR-HPVs, such as types 16 and 18. The proteins encoded by HPV E6 and E7 are the primary oncogenes that play critical roles in HPV-positive cervical carcinomas. Inactivation of tumor suppressor protein p53 (TP53) and retinoblastoma (RB) is one of the main mechanism by which E6 and E7 induce carcinogenesis. Moreover, expression of the HR-HPV E6/E7 genes directly contributes to malignant progression by subverting genomic stability, which is also necessary for the induction of premalignant alterations [11].
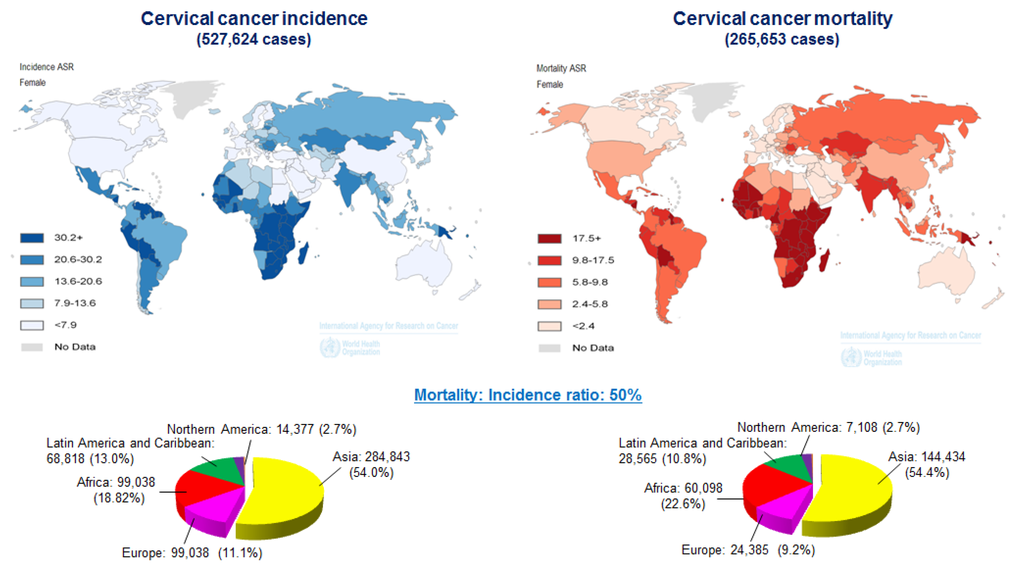
Figure 1.
Worldwide cervical cancer incidence and mortality in 2012. Geographical distribution of cervical cancer incidence (left) and mortality (right) worldwide. Approximately 85% of cases occur in developing countries. Mortality: incidence rate ratios for cervical cancer were obtained from GLOBCAN (http://globocan.iarc.fr/).
Figure 1.
Worldwide cervical cancer incidence and mortality in 2012. Geographical distribution of cervical cancer incidence (left) and mortality (right) worldwide. Approximately 85% of cases occur in developing countries. Mortality: incidence rate ratios for cervical cancer were obtained from GLOBCAN (http://globocan.iarc.fr/).
The Role of E6 and E7 in Carcinogenesis
The HPV E6 protein is a small basic polypeptide of approximately 150 amino acids that contains two zinc-finger motifs [12,13], each consisting of a CXXC-X29-CXXC sequence and the PDZ-binding epitope at C-terminal [14,15]. Overexpression of the E2 protein in HPV18-positive cervical carcinoma cells can negatively affect E6/E7 expression and inhibit cell proliferation [16,17]. In some types, further regulation is achieved through splice donor sites that give rise to truncated forms of E6, denoted as E6*I~IV, dependent upon the position of the downstream splice acceptors [18,19]. The most abundant splice RNA, E6*I, actually function as an E7 mRNA for efficient E7 translation [20,21,22]. The HR-HPV E6 protein and the truncated E6*I peptide destabilize several host proteins involved in cell growth and differentiation [19,23]. Oncogenic HPV infection also deregulates the expression of oncogenic and tumor suppressive miRNAs via E6-TP53 and E7-pRb pathways [24,25].
After viral integration, E6 is expressed, and it facilitates several cellular changes that prolong the cellular lifespan by blocking apoptosis and increasing telomerase activity. The transcriptional activator role of E6 may be coupled to its ability to immortalize and transform cells [26]. E6 binds to cellular proteins, particularly to the HECT domain of ubiquitin ligase E3A (UBE3A), alternatively known as E6-associated protein (E6AP), and to E6BP (reticulocalbin 2, an EF-hand calcium-binding domain). E6BP is a 320 amino acid protein with a high affinity for calcium. E6 can bind to E6BP in the absence of TP53. UBE3A interacts with HPV E6 at a conserved LXXLL motif and forms ternary complexes with TP53, which are then degraded through ubiquitin-dependent mechanisms [27,28,29]. Moreover, E6 can block the translocation of TP53 into the nucleus [30] and thereby inhibit the gene expression regulatory functions of TP53. The consequence of TP53 degradation and blocking of TP53 transport into the nucleus disrupts TP53-mediated cell cycle control, allowing continued cell division despite DNA damage. HR-HPV E6 can also impair apoptosis by accelerated degradation of Bak, c-Myc, FADD, and procaspase-8. Furthermore, E6 binds to E6TP1, hADA3, tuberin, CBP/p300, and Gps2, interfering with the function of these proteins to negatively regulate cell proliferation. HPV also suppresses the innate immune system through binding of E6 to IRF-3, as well as E6-mediated downregulation of TLR9 expression [31]. Also oncogenic HPV E6 is capable of regulating the expression of many cellular miRNAs like miR-34a [32].
E7 ORF encodes an acidic phosphoprotein with zinc-binding motifs consisting of CXXC-X29-CXXC [33], which are essential for proper protein folding and stability. The E7 protein is primarily localized to the nucleus and has been shown to induce cellular proliferation, immortalization, and transformation [34]. HR-HPV E7 confers transforming activities and can immortalize human keratinocytes via interactions with factors involved in the regulation of cell growth [35]. Most E7 proteins contain a strictly conserved LXCXE-binding motif that associates with members of the RB family of tumor suppressors, resulting in ubiquitin-mediated targeted degradation of the associated RB family members [36]. Binding of E7 to hyper-phosphorylated RB (pRB) results in the release of the E2F transcription factor, activating gene transcription [37]. In addition, HR-HPV E7 mediates the degradation of pRB [38]. Both HR and LR-HPVs possess the ability for E7 to bind to pRB, although the interaction between LR-HPVs E7 and pRB is much weaker [39]. Furthermore, HR-HPV E7 not only interacts with cyclins A and E, but also increases the levels of these proteins. In addition, E7 expression results in inactivation of the cyclin-dependent kinase (CDK) inhibitors CDKN1A (p21CIP1) and CDKN1B (p27KIP1) [37]. HPV-16 E7 protein can modulate the cytoplasmic localization of CDKN1B (p27KIP1) and may, in turn, regulate tumor metastasis/aggressiveness through the PI3K/AKT pathway [40]. E7 proteins act by binding with histone deacetylases, which are normally recruited to repress transcription via promoters containing E2F-binding sites.
In addition, a synergistic effect between Ras and E6/E7 genes in cellular transformation has been reported [41,42,43]. E6/E7 expression can activate PI3K and MAPKs, particularly ERK1 and ERK2 [44]. It appears that several transcription factors contribute to HPV-mediated carcinogenesis, including the TATA box-binding protein (TBP), a component of the NURD histone deacetylase complex, Mi2β [45,46], the acetyl transferases p300/CBP and p300/CBP-associated factors (P/CAF) [47], and E2F [48]. Thus, the E6 and E7 oncoproteins are thought to immortalize cells, primarily through interference with the TP53 and pRB tumor suppressor proteins. In addition, cervical cancer displays notably increased or decreased expression of a large number of cellular oncogenic or tumor suppressive miRNAs. The elevated expression of miR-16, miR-25, miR-92a, and miR-378 and the decreased expression of miR-22, miR-27a, miR-29a, and miR-100 were attributed to viral oncoprotein E6 or E7 [24,25]. These observations are not surprising because both E6 and E7 modulate numbers of major transcription factors such as MYC, TP53, and E2F, which are upstream molecules for a large number of these miRNA genes.
3. TP53 Mutations in Human Cancer
TP53 is the most commonly mutated gene in human cancer [49]. Genetic alterations of TP53 in human tumors include allelic losses, missense, frame-shift mutations and intragenic deletions, and epigenetic changes are also observed. Our analysis of 17,584 tumor samples with a variety of human cancer types in The Cancer Genome Atlas (TCGA) database showed that the TP53 gene was frequently mutated in ovarian (90.66%), uterine carcinosarcoma (89.5%), esophageal (71.9%), head and neck (70.18%), lung (60%), colorectal (54.14%), and other cancers. In the case of cervical cancer, mutations in the TP53 gene are rarely reported, with an occurrence of only 5.1%. Tumor suppression by TP53 is primarily regulated through Mdm2-mediated ubiquitination of TP53. However, in HPV-positive cervical cancer cells, the degradation of TP53 is completely converted from Mdm2 to E6-mediated ubiquitination [50]. Thus, the level of TP53 protein in cervical carcinomas remains remarkably low, despite TP53 signaling activation, including oncogenic addiction and DNA damage by reactive oxygen species (ROS). Therefore, most HPV-associated cervical carcinomas, unlike many other cancers, usually carry the wild-type (WT) TP53 gene [51,52]. Here, we also focus on the role of TP53 and the implications of TP53-based anticancer therapies for cervical cancer.
4. Therapeutics against HPVs
In 2006, the U.S. Food and Drug Administration approved Gardasil, the first quadrivalent cancer vaccine, for use in women 9–26 years of age for the prevention of cervical cancer, precancerous genital lesions, and genital warts caused by HPV6, HPV11, HPV16, and HPV18 [53,54]. In 2009, FDA approved Cervarix, another vaccine to prevent cervical cancer and precancerous lesions caused by human papillomavirus (HPV) types 16 and 18. The vaccine is approved for use in girls and women ages 10 years through 25 years. However, current vaccine efficacy is not evident for the therapy of cervical cancer patients.
For cervical cancer therapy, E6 or the E6/UBE3A complex deserves special attention as a specific target. In Table 1, we discussed several strategies that target E6 or the E6/E6-AP complex that have been developed, including various therapies that employ cytotoxic drugs, a zinc-ejecting inhibitor of the viral E6 oncoprotein, an E6-AP mimetic epitope peptide (mimotope), an anti-E6 ribozyme, peptide aptamers that target the viral E6 oncoprotein, siRNAs that target the viral E6 oncogene, and combinations of all these therapies [55,56,57,58,59,60,61,62,63]. Based on recent reports a possible new strategy is to induce viral E6 and E7 instability by using HSP90 and GRP78 inhibitors for the treatment of cervical cancer [64]. An E7 antagonist peptide has been studied to determine its therapeutic efficacy. The E7 antagonist showed antitumor effects through pRB reactivation both in vitro and in vivo [65]. GS-9191, a nucleotide analog prodrug, showed an antiproliferative effect in vitro, and its topical application reduced the size of papillomas in the cottontail rabbit papillomavirus model [66]. Chitosan hydrogel containing granulocyte-macrophage colony-stimulating factor (GM-CSF) in combination with anticancer drugs, cyclophosphamide in particular, results in antitumor effects through CD8+ T cell immunity [67]. Heparin-like glycosaminoglycans have been shown to inhibit tumor growth by the downregulation of HPV18 long control region activity in transgenic mice [68]. Finally, 5-aza-2′-deoxycytidine, a demethylating agent, and 5,6-dimethyl xanthenone-4-acetic acid, a vascular disrupting agent, have each been combined with therapeutic HPV DNA vaccines [69,70], showing significant antitumor therapeutic effects in vivo.

Table 1.
HPV-targeting therapeutics.
| Reagent | References | Note | |
|---|---|---|---|
| HPV E7 antagonist | Peptide | [65] | |
| E6-binding aptamer | Peptide | [58] | |
| E6-AP mimetic epitope | Helical peptides | [60] | |
| Organic disulfides containing dithiobisamine moiety | Organic compound | [55] | |
| Anti-E6 ribozyme | RNA molecule | [63] | |
| GS-9191 | Nucleotide analog prodrug | [66] | |
| 5-aza-2′-deoxycytidine (DAC) | Nucleotide analog | [69] | Combination with therapeutic HPV DNA vaccine |
| 5,6-dimethylxanthenone-4-acetic acid (DMXAA) | Small molecule | [70] | |
| Cisplatin | Platinum compound | [61] | Alone or combination treatment |
| 4,4′-dithiodimorpholine | Zinc-ejecting inhibitor | [55] | |
| Glycosaminoglycans (GAGs) | Heparin-like | [68] | |
| Chitosan hydrogel | Natural biopolymer | [67] | Combination treatment |
| Methyl jasmonate | Plant hormone | [72] | Plant-originated products |
| Epigallocatechingallate (EGCG) | Plant-derived natural compound | [78] | |
| Nordihydroguaiaretic acid (NDGA) | Plant lignan derivative | [80] | |
| Silymarin | Plant flavonoid | [76] | |
| Jaceosidin (4′,5,7-trihydroxy-3′,6-dimethoxyflavone) | Plant-derived natural compound | [79] | |
| Withaferin A | Plant-derived natural compound | [77] | |
| Praneem tablet | Plant extract | [71] | |
| Curcumin | Plant extract | [75] | |
| Phytoglycoprotein | Plant-originated glycoprotein | [74] | |
| Soyasaponins | Plant-derived natural compound | [73] | |
| Carrageenan | Compound from red algae | [81] | |
| 17-N-allylyamino-17-demethoxygeldanamycin (17-AAG) | A derivative of the antibiotic geldanamycin | [64] | Combination treatment with GRP78 inhibitor |
Additionally, several plant-derived compounds have been investigated to determine their therapeutic potential in cervical cancer (Table 1). In clinical trials, intravaginally administered Praneem, a polyherbal formulation, was shown to eliminate HPV16 infection in early cervical intraepithelial lesions [71]. Methyl jasmonate, soyasaponin, phytoglycoprotein, curcumin, silymarin, withaferin A, and epigallocatechingallate (EGCG) showed therapeutic effects via repression of viral oncogenes, upregulation of tumor suppressor genes, or induction of apoptosis in vitro [72,73,74,75,76,77,78]. Withaferin A treatment reduced tumor volume by approximately 70% in a xenograft model. Interestingly, a few of these natural compounds specifically target HPV. For example, jaceosidin inhibits the functions of the E6 and E7 oncoproteins in HPV16-positive cervical cancer cells [79], and nordihydroguaiaretic acid also inhibits HPV16 gene expression [80]. Carrageenan, a compound produced by red algae, inhibits HPV infection through direct binding to the viral capsid, and through a heparan sulfate proteoglycan-independent inhibitory effect [81].
4.1. RNAi-based Therapeutics against HPVs
Recently, novel antiviral RNAi therapies have been developed and tested in clinical trials with short interfering RNAs (siRNAs) [82]. siRNAs have been demonstrated to be capable of selective silencing of endogenous genes in mammalian cells [83,84], and of selectively silencing viral genes in virus-induced diseases [85,86,87]. Remarkably, it has been reported that RNAi targeting of E7 or E6/E7 promotes the accumulation of TP53 and/or pRB, eventually leading to the induction of apoptosis and/or senescence in HPV16-positive cervical cancer cell lines [59,62,88,89], as well as in HPV18-positive human cervical cancer cells [90,91]. It has been reported earlier that, silencing of both E6/E7 produces greater anticancer activity than E6 alone [92,93,94]. Several studies of RNAi targeting of the HPV E6 or E7 oncogenes are summarized in Table 2.
Table 2.
List of RNAi studies targeting HPV E6 or E7.
| References | Cell line | Target Transcripts | Note | ||
|---|---|---|---|---|---|
| Cun et al., 2013 | [149] | OCM1, OM431, VUP and SP6.5 | Synthesized siRNA | E6 & E7 mRNA | |
| Li et al., 2013 | [150] | SiHa | Plasmid-based E6-specific siRNA | E6 mRNA | Dual coexpressed-E6-specific siRNA and wild type TP53 |
| Zhou et al., 2012 | [93] | SiHa | Synthesized siRNA | E6/E7 mRNA | BALB/C nude mice, intratumoral injections every other dayduring a 12-day period |
| Jung et al., 2012 | [94] | HeLa, CaSki, SiHa | Synthesized siRNA | E6/E7 mRNA | BALB/C nude mice, intravenous injections in combination with Cisplatin |
| Chang et al., 2010 | [95] | CaSki, HeLa | siRNA Plasmid | E6 only or E6/E7 mRNA | BALB/C nude mice, intratumoral injections twice a week for two weeks |
| Hong et al., 2009 | [96] | SiHa | Synthesized siRNA | E6/E7 mRNA | |
| Jonson et al., 2008 | [97] | CaSki | Synthesized siRNA | E6/E7 mRNA | nu/nu mice, intratumoral injections every three days for 35 days |
| Sima et al., 2008 | [88] | SiHa, CaSki | shRNA | E6/E7 mRNA | |
| Lea et al., 2007 | [98] | HeLa | Synthesized siRNA | E6 only or E6/E7 mRNA | |
| Courtete et al., 2007 | [99] | CaSki, SiHa, HeLa | Synthesized siRNA | E6 mRNA | |
| Fujii et al., 2006 | [100] | SKG-IIIa, SKG-II, HeLa | Synthesized siRNA | E6 & E7 mRNA | Nude mice, Intratumoral infections for 10 days |
| Tang et al., 2006 | [20] | CaSki and SiHa, HeLa | Synthesized siRNA | E6 or E6*I mRNA and E7 mRNA | |
| Yamato et al., 2008 | [89] | CaSki and SiHa, | Synthesized siRNA | E6 mRNA | |
| Koivusalo et al.,2005 | [101] | HeLa | Synthesized siRNA | E6 mRNA | siRNA combination with different drugs |
| Putral et al., 2005 | [102] | SiHa, CaSki | Synthesized siRNA | E6* mRNA and full length E6 mRNA | |
| Yoshinouchi et al., 2003 | [62] | SiHa | Synthesized siRNA | E6* mRNA and full length E6 mRNA | |
| Butz et al., 2003 | [58] | HeLa | Vector and Synthesized siRNA | full length E6 mRNA | |
| Hall and Alexander, 2003 | [91] | HeLa | Synthesized siRNA | E7 mRNA | |
| Jiang and Milner, 2002 | [59] | CaSki, SiHa | Synthesized siRNA | E6 and E7 mRNA |
Zhou et al. [93] recently reported that two siRNAs targeting the E6/E7 promoter and E7 transcripts produced E6 and E7 mRNA knockdown, increased TP53 protein levels, decreased CDKN2A (p16INK4A) protein levels, and exhibited SiHa cell growth inhibition via apoptosis. In xenografted mice, siRNA transfection of cells in vitro or intratumoral siRNA injection also inhibited tumor growth and induced apoptosis. Another study reveals that HPV16 E6/E7 silencing by this promoter-targeting siRNA was related to histone modification associated with histone H3-Lys9 methylation [96]. Chang et al. [95] designed nine siRNAs that specifically targeted E6 or E6/E7 mRNA of HPV16 or HPV18. Intratumoral administration of potent siRNA resulted in the inhibition of tumor growth and induction of apoptosis in vivo, suggesting that siRNA treatment shows potential as an adjuvant therapy for cervical cancer. The therapeutic mechanism of siRNA-mediated silencing of both E6 and E7 mainly depends on the reactivation of TP53 and pRB, inducing apoptosis and strong cellular senescence. In our recent study, anti-angiogenesis and inhibition of lung metastasis were determined to constitute additional mechanisms operating in combination therapy with E6/E7-specific siRNA and cisplatin [94]. Hall et al. [91] examined an siRNA sequence that targets the type I transcript of E6/E7 mRNA and found reduced levels of full-length E6 and E7 mRNA in cells. They confirmed that this siRNA sequence transfected cells show reduced E6 mRNA levels, leading to increase of the expression level of TP53 protein. They also demonstrated that transfection with E6/E7-targeting siRNA inhibited DNA replication and induced cellular senescence in HPV 18-positive human cervical cancer cells. Hall et al. [91] also suggested that siRNA inhibition of E6 and E7 demonstrates potential as a therapeutic intervention for the treatment of cervical cancer. In support of this proposition, Jonson et al. [97] confirmed the therapeutic effect of direct intratumoral injection of HPV16 E6/E7 siRNA using the CaSki xenograft model.
4.2. Anticancer Therapeutic Strategies Targeting Activation of the TP53 Pathway
In cervical cancer, the TP53 pathway is often disrupted. Therefore, functional restoration of WT-TP53 may induce the regression of cervical carcinomas, which can be achieved by abrogating the expression of the E6 or E6/E7 oncogenes, or through cisplatin (cis-diaminedichloroplatinum II; CDDP) or radiation treatment. It has been demonstrated that cisplatin therapy allows TP53 to escape from E6-mediated degradation, thereby facilitating TP53 accumulation in the nucleoli of HeLa cells [61]. Concurrent treatment with cisplatin and radiotherapy also restores TP53 function and enhances the radiosensitivity of HPV16-positive SiHa cells [103]. Additional reports have arrived at the same remarkable conclusion [104,105,106]. The restoration of TP53 function in established tumors (including lymphomas, sarcomas, and hepatocellular carcinomas) causes regression of tumors in vivo and could represent an effective new approach to treating cancer. Evans et al. [104] used the Eμ-myc mouse model to validate the effects of modulation of the TP53 pathway on various aspects of tumor biology. They developed a TP53 knock-in model, in which the WT-TP53 gene is replaced by a chimeric TP53 protein containing an estrogen receptor (ER) domain linked to the WT-TP53 sequence. TP53 restoration markedly increased tumor cell death and significantly prolonged survival of the mice. Jacks et al. utilized a mouse model, in which TP53 was restored by removal of the STOP cassette following activation of a tamoxifen-inducible Cre recombinase. Interestingly, in lymphomas, TP53 restoration induces apoptotic cell death; whereas, in sarcomas, it induces cell-cycle arrest with signs of cellular senescence [105].
Lowe et al. developed a mouse model of hepatocellular carcinoma [106]. In this study, purified embryonic liver cells were transformed by introduction of an activated H-ras oncogene, and TP53 expression was downregulated using a doxycycline-repressible short hairpin RNA directed against TP53. TP53 induction in this hepatocarcinoma model led to growth arrest with senescence rather than apoptotic cell death. In addition, the efficiency of TP53-mediated tumor clearance is stage-specific and dependent on the overall heterogeneity and aggressiveness of tumor cell populations [107,108]. Silencing or reactivation of TP53 may also regulate the expression level of host microRNAs regulations in cervical cancer [32,109]. Numerous other recent approaches investigating TP53 protein dynamics have provided new insights into the topology of the signaling network. The majority of studies revealed clear differences in gene expression when TP53 is pulsing and when TP53 expression is sustained at high levels for several hours. In response to ultraviolet light, TP53 shows a single graded pulse; whereas, in response to double-strand breaks caused by γ irradiation, TP53 exhibits a series of repeated pulses [109,110]. TP53 was also activated following the administration of various anticancer chemotherapeutic agents [111]. However, the ability of a TP53 pulse to arrest the cell cycle and activate its target genes, such as CDKN1A, depends on specific posttranslational modifications, which occur only in response to severe extrinsic cell damage [112]. Depending upon its expression level, TP53 can promote either DNA damage repair or survival of damaged cells. In recent years, another treatment method has emerged, in which the TP53 pulses are sustained by Nutlin-3, an MDM2 inhibitor that can be employed alone or in combination with chemo/radiotherapy; this appears to be a valid approach for the treatment of tumors [113,114]. Sustained induction of TP53 triggers multiple cellular programs ranging from transient responses, such as DNA repair and cell cycle arrest, to terminal fates, such as apoptotic cell death and senescence. As an alternative strategy, stabilization of TP53 by Nutlin-3a significantly enhanced reovirus-induced apoptosis [115]. Overall, combinations of conventional therapies can induce frequent TP53 pulses and sustain their expression level, leading to irreparable damage and apoptosis of cancer cells.
Our unpublished live cell imaging results on reporter activity also revealed that combination of a HPV E6/E7 siRNA pool with a chemotherapeutic agent sustained TP53 dynamics over a long period, and led to immediate cell death. Analyzing the TP53 dynamics can be an important part of a signal in cancer, directly influencing different functional cellular outcomes, such as growth, survival, and death. Overall, stabilization and maximal activation of the TP53 signaling network can facilitate determination of the optimal therapeutic strategy for cervical cancer.
5. RNAi-based Combination Therapeutics against HPV
Several RNAi studies have been performed in human tumor cell lines using synthetic siRNAs or short hairpin RNA (shRNA) libraries to identify modulators of drug sensitivity [116,117,118,119,120,121,122]. These studies address whether combination therapy with siRNA may significantly enhance the sensitivity of cancer cells and may help to prevent chemo/radio resistance resulting from low-dose chemo/radio therapy. From a clinical perspective, if the combined effect is equal to the sum of the effects of the individual modalities, then the effect of the multiple modalities is additive. If the combined effect is greater than the predicted effect of the multiple individual modalities, however, then the interaction is synergistic. The multiple modalities may also interact in an antagonistic fashion. Statistical analysis using the Chou-Talalay method must be applied in order to determine the synergistic, additive, or antagonistic effects of RNAi-based combination therapies.
In our study, we validated the role of 2-in-1 antiviral siRNAs targeting the HPV E6 and E7 oncogenes as a potential sensitizer to CDDP [94] and radiation therapy (unpublished) for the treatment of cervical carcinoma. Thus, the combination of siRNAs targeting E6 or E6/E7 may have synergistic effects on the restoration of TP53 and/or pRB function, and may be a more effective therapeutic modality for the treatment of cervical cancer. Putral et al. [102] investigated CDDP co-therapy, and found that shRNAs to E6 increased CDDP sensitivity in HeLa cells. Tang et al. [92] also reveals the silencing of HPV16 E6 and E7 expression by the shRNA in cervical cancer cells; at late passages the cells develop a resistant program in the presence of E7 siRNAs. Another recent study revealed that, intratumoral injection of both exonic (E6/E7-Exon) and intronic (E6/E7-Intron) siRNAs co-administration with intravenous injection of paclitaxel treatment restored the tumor-suppressive effect more effectively than co-administration of paclitaxel treatment with (E6/E7-Exon) siRNA. Also, both E6/E7 siRNA co-administrations with paclitaxel treatment resulted in similar tumor-suppressive effect compared with paclitaxel alone, and showed some extended survival [123]. Further studies are required to overcome all the key points mentioned above and to develop clinically applicable combination therapies based on RNAi. Most importantly, to establish proof-of-concept for RNAi-based combination therapeutics, the mechanism underlying the synergy between the treatments should be elucidated. The results of in vivo experiments have demonstrated that combination therapy is significantly superior to either modality alone. Moreover, assessments to verify the absence of off-target effects and minimal induction of interferon are a prerequisite for the clinical application of RNAi-based combination therapeutics.
6. Unmet Medical Needs of Cervical Cancer
In the 1990s, the Gynecologic Oncology Group (GOG) conducted a randomized trial for radiotherapy in combination with three concurrent chemotherapy regimens—CDDP alone (group 1); CDDP, fluorouracil, and hydroxyurea (group 2); and hydroxyurea alone (group 3)—in patients with locally advanced cervical cancer (stage IIB, III, or IVA without involvement of the para-aortic lymph nodes) [124]. The groups receiving CDDP (groups 1 and 2) showed a higher rate of progression-free survival (PFS) than the group receiving hydroxyurea alone (group 3). The overall survival (OS) rate was also significantly higher in groups 1 and 2 than in group 3. Moreover, the highest combined frequency of grade 3 (moderate) and grade 4 (severe) adverse effects was observed in group 2, which means that radiation in combination with CDDP produced fewer side-effects than with the other triple agent combination. As a result, CDDP has been proposed for use as a standard radiotherapy and chemotherapy drug for locally advanced cervical cancer based on that trial.
Morris et al. [125] compared the effect of administering radiotherapy to a pelvic and para-aortic field with that of administering pelvic radiation in combination with chemotherapy (fluorouracil and CDDP) in women with advanced cervical cancer (stages IIB through IVA or stage IB or IIA with a tumor diameter of at least 5 cm or involvement of the pelvic lymph nodes). Kaplan-Meier analysis revealed that disease-free survival (DFS) and OS rates were significantly higher in patients treated with radiotherapy and chemotherapy than in those treated with radiotherapy alone. They found that the addition of chemotherapy to radiation treatment significantly improved survival among women with locally advanced cervical cancer. In the combined therapy group, the reported rate of local relapse was 19%, and the rate of distance relapse was 14%. Keys et al. [126] determined that weekly infusions of CDDP during radiotherapy improved PFS and OS among patients with bulky stage IB cervical cancer. They also found that chemoradiotherapy followed by radical hysterectomy significantly reduced the risk of disease recurrence and death in women with bulky stage IB cervical cancers. However, the chemoradiotherapy group exhibited higher frequencies of transient grade 3/4 adverse hematologic effects and adverse gastrointestinal effects. Furthermore, although residual tumors were not more frequently detected in the combined-therapy group than in the radiation alone group, residual masses in the combined-therapy group were reported as often as 48%.
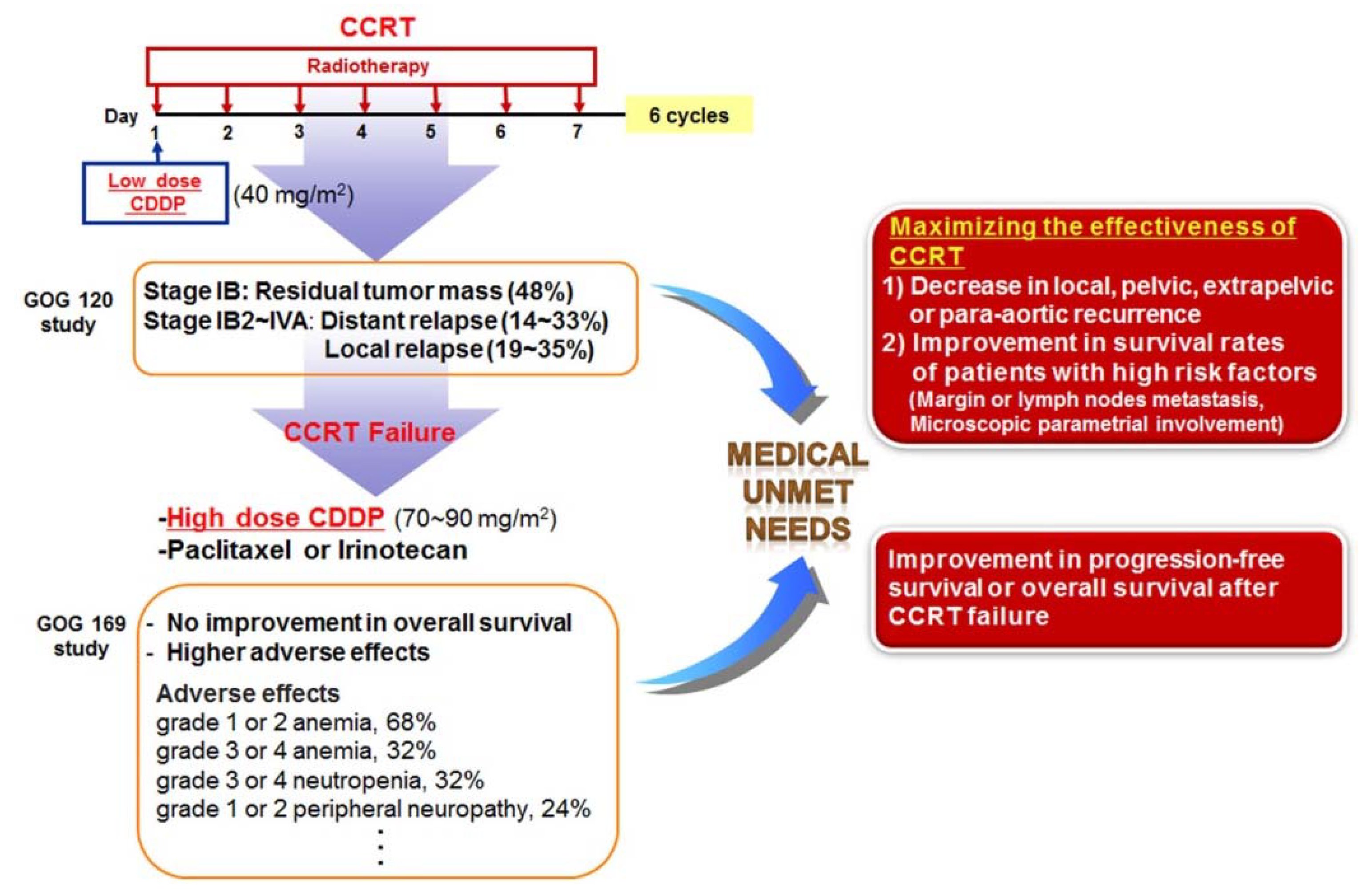
Collectively, these randomized trials for cervical cancer all demonstrated that concomitant treatment with CDDP and radiotherapy produced more favorable outcomes than radiotherapy alone. Therefore, CDDP chemotherapy with concurrent radiation (CCRT) is recommended as the first-line therapy for patients with disease at stage IIB or greater, and as an adjuvant therapy following radical hysterectomy for patients with locally advanced cervical cancer (Figure 2) [125,126,127]. Although this practice guideline for cervical cancer has improved the survival rates for patients undergoing CCRT, CCRT failure still occurs, including residual cancers, adverse effects, local recurrences, or distance recurrences, as mentioned above [124,125,126]. In addition, when primary therapy with CCRT fails to treat cervical cancer, most patients may no longer be sensitive to radiation therapy alone or CDDP chemotherapy alone, and single-agent therapy cannot be considered as a secondary therapy following failure of CCRT. Thus, patients with recurrent or metastatic squamous cell carcinoma of the cervix receive high-dose CDDP plus paclitaxel combination therapy as a palliative chemotherapy [128,129] (Figure 3).
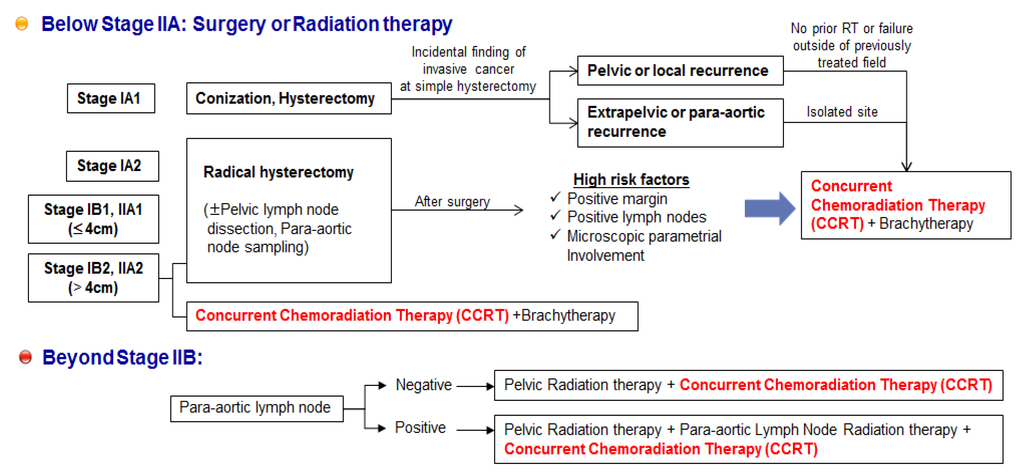
Figure 2.
Practice guideline summary for cervical cancer. Concurrent chemoradiation therapy (CCRT) is recommended as the first-line therapy for patients with disease at stage IIB or greater, and as an adjuvant therapy following radical hysterectomy for patients with locally advanced cervical cancer.
Figure 2.
Practice guideline summary for cervical cancer. Concurrent chemoradiation therapy (CCRT) is recommended as the first-line therapy for patients with disease at stage IIB or greater, and as an adjuvant therapy following radical hysterectomy for patients with locally advanced cervical cancer.
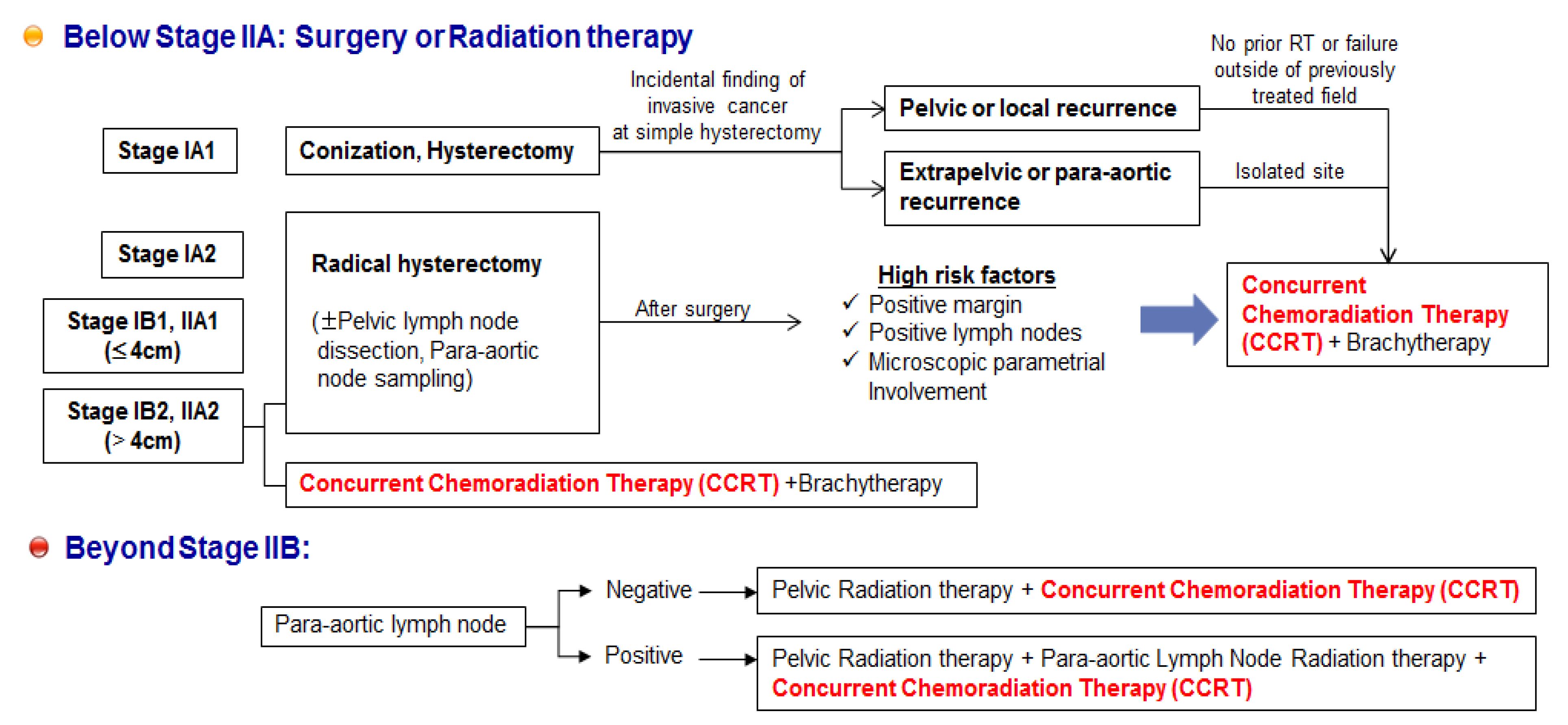
A phase III GOG study was conducted to determine whether CDDP plus paclitaxel improved response rate, PFS, or survival compared with CDDP alone in patients with stage IVB, recurrent, or persistent squamous cell carcinoma of the cervix [129]. The combination of CDDP and paclitaxel appears to be superior to CDDP alone with respect to the objective response rate, PFS, and sustained quality of life, but with increased adverse effects and no improvement in OS in patients with stage IVB, recurrent, or persistent squamous cell carcinoma of the cervix. Another GOG study reported that the combination of topotecan and CDDP produced superior response and OS compared to CDDP alone [130]. Therefore, unmet medical needs remain regarding two points for the treatment of cervical cancer (Figure 3). First, in cases of early lesions with high risk factors (margin or lymph node metastasis, or microscopic parametrial involvement) after surgery, and in advanced lesions beyond stage IIB, it is important to maximize the effectiveness of CDDP-based concurrent CCRT, in order to decrease recurrence and improve the survival rate.
Second, we must consider patients who experienced recurrence and metastasis following failure of CCRT. Clinical studies have been conducted for methods to improve PFS, OS, and to reduce severe adverse events. Although at present, CDDP is considered to be the most active cytotoxic agent available, various combinations of chemotherapy with paclitaxel, carboplatin, topotecan, irinotecan, gemcitabine, bevacizumab, or docetaxel have been employed in clinical studies of metastatic or recurrent cervical cancer to alleviate poor prognosis of recurrent and metastatic cervical cancer [128,129,130,131,132,133,134,135]. In summary, the evidence discussed here suggests that novel anticancer therapeutics, in combination with CCRT, show promise as therapies to control cervical cancer.
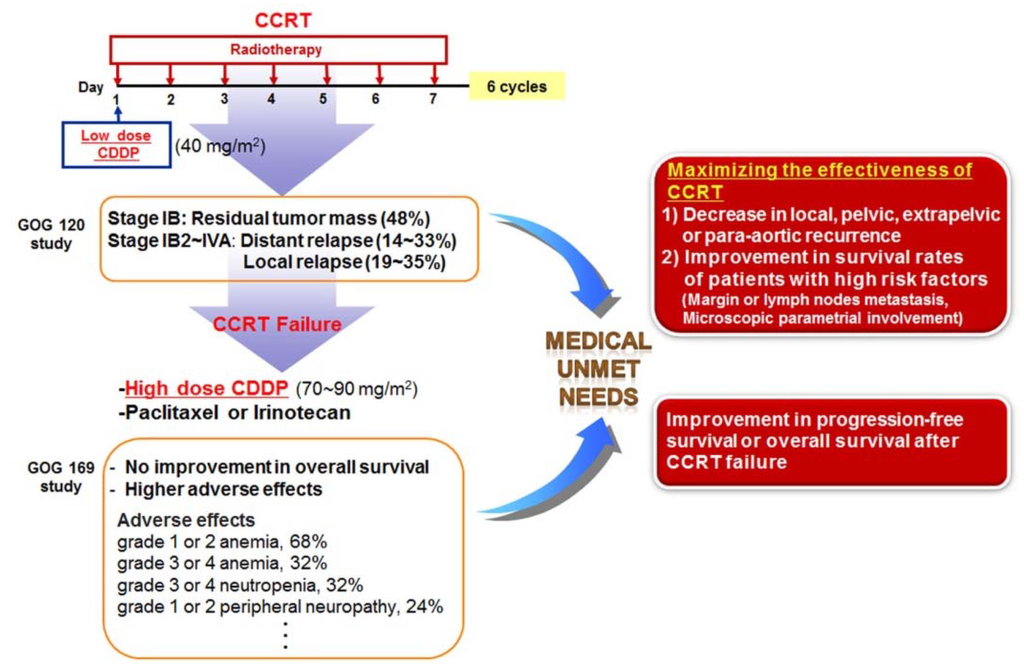
Figure 3.
Unmet medical needs of cervical cancer. It is important to maximize the effectiveness of CCRT in order to decrease recurrence and improve the survival rate. Following failure of CCRT, the clinical endpoint is to improve progression-free survival, overall survival, and the reduction of severe adverse events.
Figure 3.
Unmet medical needs of cervical cancer. It is important to maximize the effectiveness of CCRT in order to decrease recurrence and improve the survival rate. Following failure of CCRT, the clinical endpoint is to improve progression-free survival, overall survival, and the reduction of severe adverse events.
7. Potential Combination Therapies for Cervical Cancer
Radiation therapy constitutes the use of various forms of radiation to safely and effectively treat cancer. The ability of radiation therapy to provide both curative and palliative treatment has underscored its importance in cancer therapy. In the clinical setting, radiation can be delivered in a targeted manner to the area of interest, allowing for its specific delivery to tumor tissues. In the last few decades, technological advancements in the delivery of radiation and in treatment planning have resulted in improved patient outcomes, particularly in the reduction of normal tissue toxicity [136,137]. The generation of hydroxyl radicals is thought to be the initiating event that leads to the majority of the biological damage created by ionizing radiation.
DNA damage can take the form of either single-strand breaks (SSBs) or double-strand breaks (DSBs). SSBs are more easily repaired by the cell, and hence are less likely to be mutagenic or lethal. DSBs are the most lethal DNA damage induced by radiotherapy. The ATM kinase initiates a signaling pathway that is primarily induced by DSBs and that acts by phosphorylating hundreds of proteins [138]. CHK2 is an important effector molecule targeted by ATM. The ATR kinase activates a pathway principally induced by UV damage that involves CHK1 kinase [139,140,141]. Among the targets of ATM and ATR is TP53, which plays a key role in controlling DNA-damage-induced G1/S and G2/M checkpoints. TP53 can activate CDKN1A, which facilitates cell cycle arrest and DNA repair, or induced apoptosis if repair is impossible. In addition, JNK and p38, members of the MAPK family, are highly activated in response to ionizing radiation. TP53 also interacts with several crucial components of the DNA repair machinery and is situated at a crossroads among radiation-induced DNA repair, apoptosis, and senescence (Figure 4).
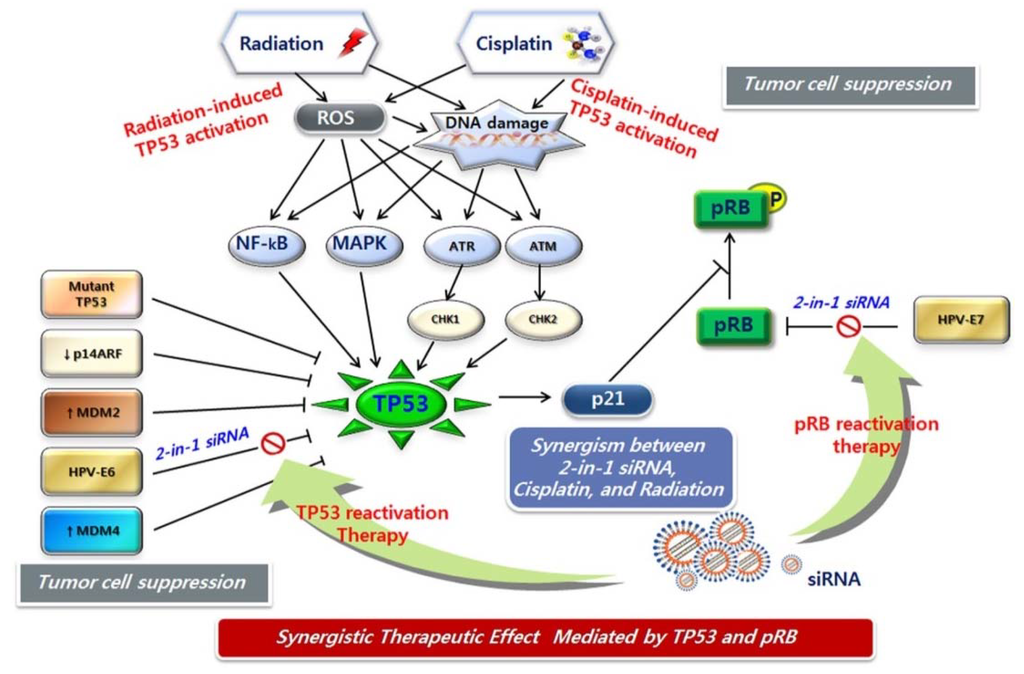
Figure 4.
Dual activation of TP53 and pRB in combination therapy with CCRT and HPV E6/E7 siRNA. Cisplatin and radiation therapies induce TP53 activation by DNA damage signal transduction. HPV E6/E7 siRNA treatment results in TP53 and pRb reactivation. Combination therapy with CCRT and HPV E6/E7 siRNA may fully activate functional TP53 and pRb through different transduction mechanisms, leading to strong cellular response.
Figure 4.
Dual activation of TP53 and pRB in combination therapy with CCRT and HPV E6/E7 siRNA. Cisplatin and radiation therapies induce TP53 activation by DNA damage signal transduction. HPV E6/E7 siRNA treatment results in TP53 and pRb reactivation. Combination therapy with CCRT and HPV E6/E7 siRNA may fully activate functional TP53 and pRb through different transduction mechanisms, leading to strong cellular response.
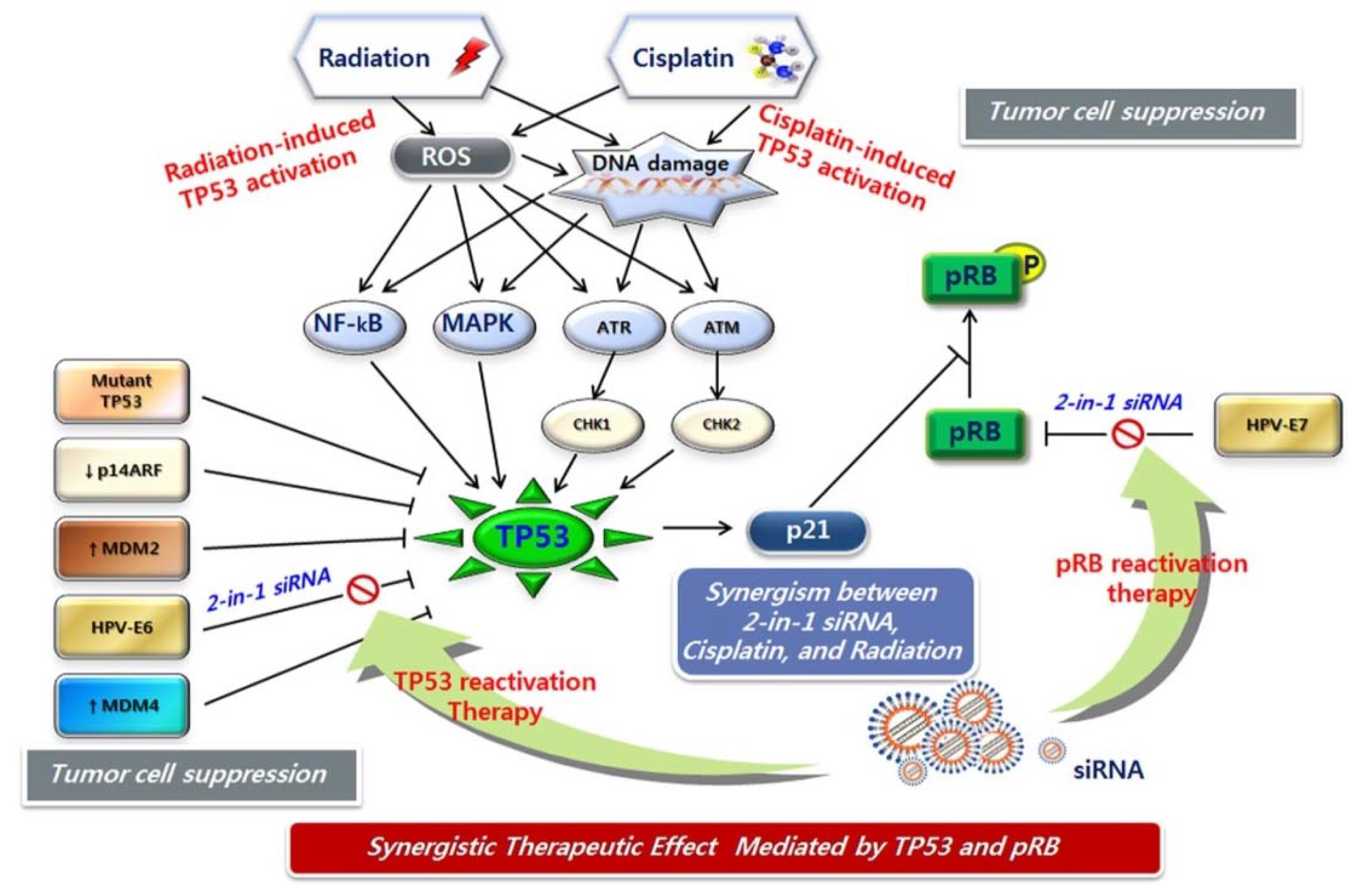
CDDP is the most active and well-known anticancer agent available for clinical use. The mechanism of action of platinum-based drugs was recently reviewed; however, the cellular processing of CDDP, including the regulation of drug uptake and efflux, and the signaling of DNA damage, cell-cycle arrest, DNA repair, and cell death, is still not fully understood [142]. CDDP-induced DNA damage signals through ATM/ATR activated cell cycle checkpoints (CHK1, CHK2) and leads to DNA repair, cell cycle arrest, and apoptosis, which are mediated by the TP53 pathway (Figure 4). CDDP also triggers the activation of the MAPK pathways in tumor cells. Following DNA damage, the ERK, JNK, and p38 MAPK cascades are activated; these subsequently activate downstream target genes, including TP53.
While both radiation therapy and chemotherapy have been used individually as treatment modalities in cancer patients for some time, the advantages of combined chemo-radiotherapy have only recently been observed clinically. The theoretical framework for the interaction between these two modalities was introduced in 1979 by Steel and Peckham [143]. Radiation therapy affects locoregional control of the tumor mass; whereas, chemotherapy acts on distant metastasis, with no interaction between the two modalities. In addition, chemotherapy co-operates with radiation therapy in the radiation field, leading to increased killing of cancer cells. As mentioned above, CDDP in combination with concurrent radiation is commonly recommended for patients with disease at stage IIB or greater, and for those with locally advanced cervical cancer. Recently, a breakthrough study demonstrated that TP53 was expressed in a wavelike or “pulsed” manner following exposure to ionizing radiation alone [144]. Sustained TP53 expression or an increased number of TP53 pulses increases the probability that TP53 will activate downstream targets involved in the induction of apoptosis and senescence. In addition, activation of the tumor suppressor pRB is part of the mode of action of HPV E6/E7 siRNA (Figure 4). Thus, full reactivation of TP53 is possible, and its dynamics are better sustained by combination therapy with CCRT and HPV E6/E7 siRNA.
To fulfill the medical unmet needs in CCRT failure, further studies are required to optimize the alternative combined therapeutic strategies like HPV E6/E7 siRNA in combination with combined chemotherapy via intravenous delivery system for apoptosis and senescence assay validation, analysis of dynamic behavior of TP53, Chou-Talalay analysis, and other validations with better therapeutic outcome. In theory, combination with HPV E6/E7 siRNA therapy can benefit from synergistic effects and reduced toxicity because of the lowered dose of chemotherapy and/or radiotherapy required.
8. siRNA Pooling Technology, a Promising RNAi Therapy
Long double-stranded RNA (dsRNA) initiates RNAi, which can generate as transcripts from an invading virus, a transposon, or other inappropriately transcribed endogenous sequence. In mammalian cells, Dicer digests dsRNA into a pool of duplex siRNAs that bind with the RNA-induced silencing complex (RISC) to induce specific gene silencing. Naturally, the siRNAs generated by Dicer represent a pool of siRNAs that function together to silence the expression of the targeted gene. Thus, to replicate the process, pools of synthetic siRNAs are utilized to silence individual genes in a specific manner. A reduction in the silencing efficiency of siRNA pools was attributed to the saturation of RISC with non-functional siRNAs that combine with the more functional duplexes for access to the RNA silencing machinery. However, highly potent siRNAs, which are rationally designed, screened, validated for efficacy, and chemically modified for stability, can mimic the pools generated in nature. Because each siRNA has a strong therapeutic effect, this siRNA mixture (pooling technology) reduces the effective concentration of each siRNA and reduces the off-target effects. Therefore, siRNA pools also show potential for therapeutic benefit in HPV E6/E7 targeting-siRNA drug development.
Yamato et al. [145] screened an HPV16 siRNA library and identified several potent E6/E7 siRNAs. Subsequently, these potent siRNAs were modified as RNA–DNA chimeric siRNAs to enhance their specificity [146]. Previously, we identified and validated HPV E6/E7 siRNA hits and then established proof-of-concept of combination therapy with CDDP [94]. Following the RNAi therapeutics pipeline, HPV16- and 18-type siRNA libraries were screened to select highly potent siRNA leads, which were then validated through in vitro and in vivo experiments. Then, siRNA derivatives were designed to increase their stability and specificity via chemical modification (Figure 5). These derivatives were optimized through various analyses, and five siRNA candidates (HPV18 E6/E7 2 type, HPV16 E6/E7 3 type) were ultimately selected (not yet published). These candidates were tested for their in vivo therapeutic efficacy, characterized by siRNA pharmacokinetic/pharmacodynamic (PK/PD) analysis, and applied to siRNA pooling technology. These siRNA pools are likely to be highly effective in various combinations with conventional chemotherapies (CCRT) in alleviating tumors resistant to conventional treatments. In summary, future studies on HPV E6/E7-associated carcinogenesis and its cellular targets have significant implications for the development of potent therapeutic siRNA pools in combination with CCRT for cervical cancer, as well as HPV-associated cancers.
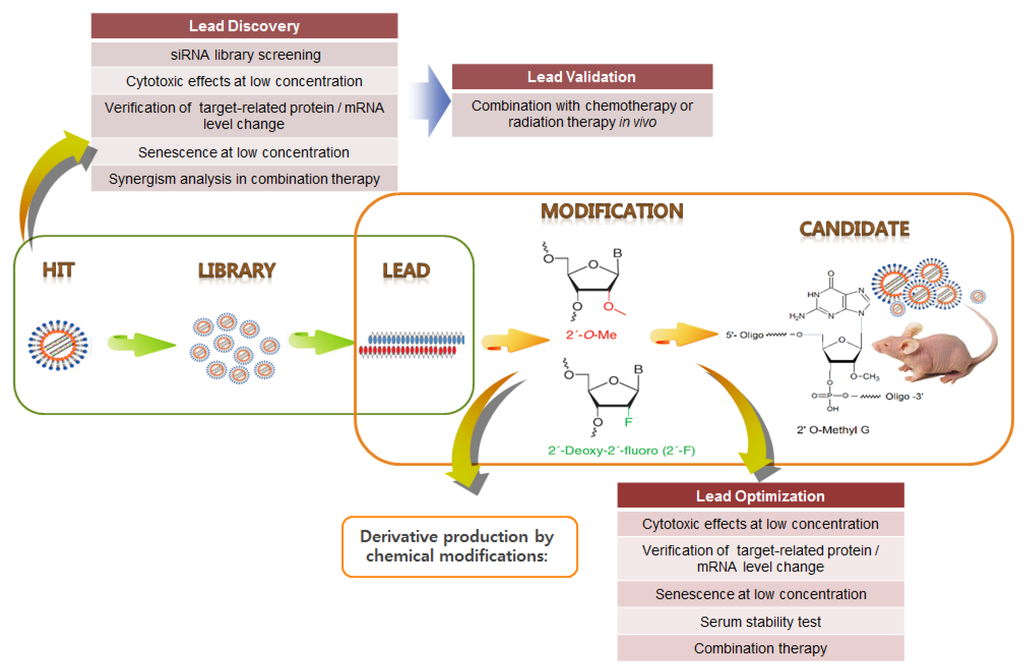
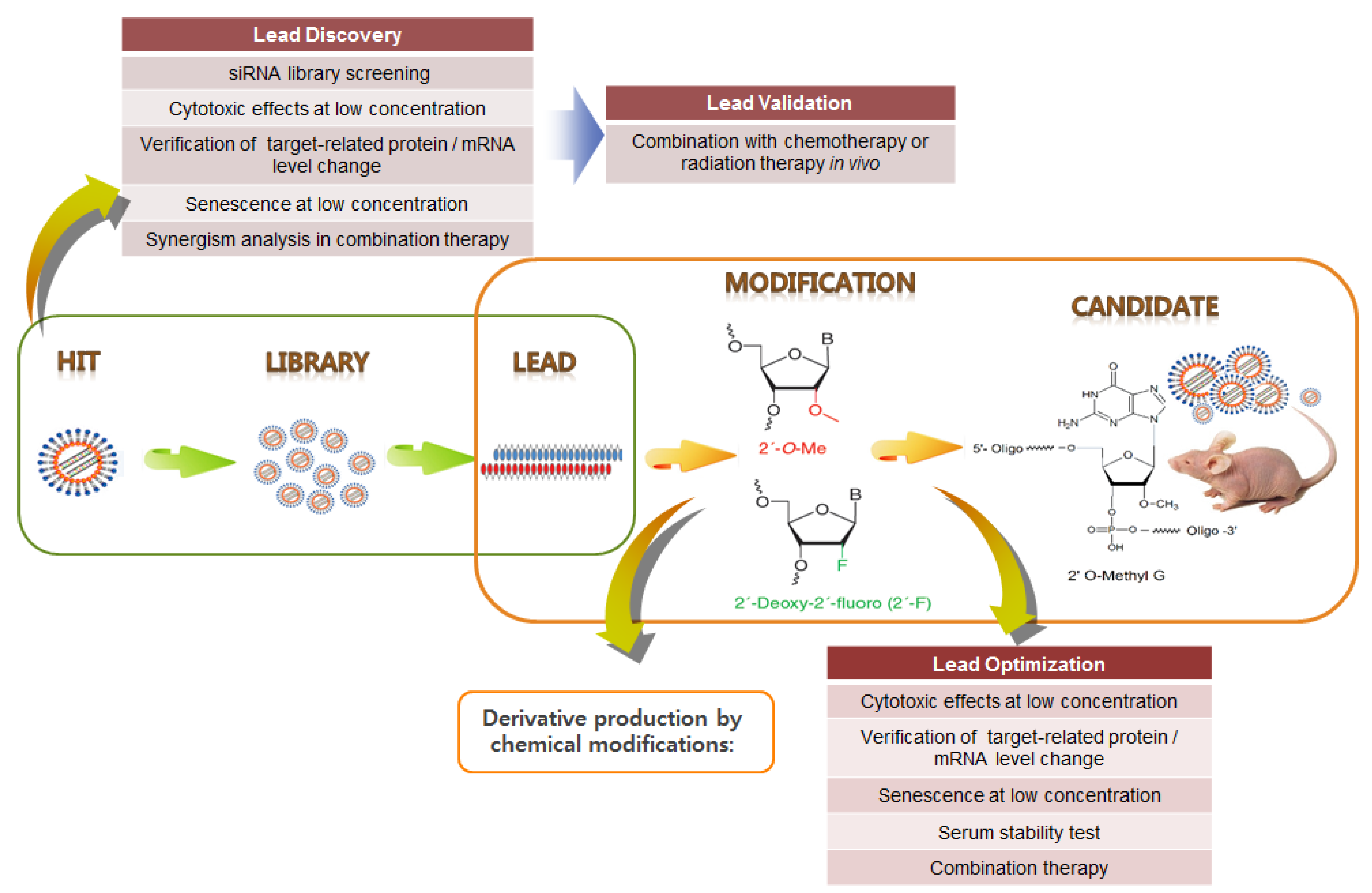
Figure 5.
Example of HPV E6/E7 siRNA drug development pipeline. Following the RNAi therapeutics pipeline, highly potent siRNA leads are chemically modified to increase their stability and specificity. Following optimization, siRNA candidates can be selected.
Figure 5.
Example of HPV E6/E7 siRNA drug development pipeline. Following the RNAi therapeutics pipeline, highly potent siRNA leads are chemically modified to increase their stability and specificity. Following optimization, siRNA candidates can be selected.
HPV siRNA Nanoparticles for Targeted Anticancer Therapy
Over the last few decades, the applications of nanotechnology in medicine have been extensively explored in many areas, particularly in targeted drug delivery systems (DDS). The use of colloidal nanocarriers, including polymeric nanoparticles, polymeric micelles, and liposomes, is an emerging field of interest in nanomedicine. Compared with others, polymeric nanoparticles show great promise and are being widely investigated for application as synthetic vaccines, and delivery of siRNAs and oligonucleotides, antibiotics, and small molecule drugs [147]. The use of flexible polymeric chains in polymeric nanoparticles, such as PEG, are able to confer “stealth” properties to nanoparticles, because PEGylation of the nanoparticle surface prevents opsonin binding by reducing protein interaction. In addition, PEG-coated spherical nanoparticles with a neutral charge exhibit an increased half-life in the blood. Modulation of carrier properties promises more effective siRNA delivery to tumor sites, and could lead to enhanced drug safety and efficacy. Successful escape of siRNA nanoparticles from endosomes and the release of the contents into the cytoplasm are necessary to improve the efficiency of gene silencing [148]. Due to the acidic nature of endosomal/lysosomal vesicles, pH-buffering agents are widely exploited to promote cargo release. Under acidic conditions, various macromolecules with low-pKa amine groups have been shown to exhibit a “proton sponge effect.” When the complexes are internalized into the cell, these nanoparticle complexes are capable of buffering the endosomal vesicles, leading to swelling and lysis, thus releasing the nucleic acids into the cytoplasm. An alternate modification at the distal end of PEGylated nanoparticles with ligands against target cell surface receptors allows uptake via receptor-mediated endocytosis, leading to efficient intracellular siRNA nanoparticle release. To date, few siRNA delivery systems designed for anticancer treatment have reached the clinical trial stage, and the majorities are nanocarriers. The three leading RNAi candidates for cancer therapy were CALAA-01 (Calando Pharmaceuticals), ALN-VSP02 (Alnylam), and Atu027 (Silence Therapeutics) with enrollment starting in 2008–2009. These candidates are being used to clinically validate the three distinct systemic delivery platforms on which they were based: the cyclodextrin-containing polycation RONDEL technology, the AtuPLEX lipoplex technology, and SNALP liposomes. A small number of other DDS development companies, such as Bioneer (SAMi: Self-Assembled-Micelle-inhibitory-RNA), NanoCarrier Co., Ltd. (siRNA micelles), Celsion. EGEN, Inc. (siRNA-lipopolyamine nanoparticles), and Dicerna Pharmaceuticals, Inc. (EnCore) are involved in the process of investigating clinical DDS development. A detailed summary of clinical trials of siRNA-based therapeutics and their sponsor companies are listed in Table 3 [20,98,99,100,101,149,150]. Recently, Alnylam Pharmaceuticals in partnership with Tekmira developed a dual targeted RNAi drug in lipid nanoparticle carrier system, encapsulating two siRNAs to target mRNA of vascular endothelial growth factor (VEGF) and kinesin spindle protein (KSP) mRNA [151]. A better understanding of the intracellular fate of siRNA-nanoparticles will provide more rational guidelines for designing an ideal siRNA-nanoparticle delivery system for targeted cancer therapeutics. Currently, we are also in the process of developing candidate pooled siRNA nanoparticles, and conducting pre-clinical studies and clinical trials for the prevention and therapy of HPV-associated cervical carcinomas.
Table 3.
RNAi-based therapies: Early stage clinical trials.
| Candidate | Target | Focus of the study | Phase | Delivery | Sponsor |
|---|---|---|---|---|---|
| siRNA-EphA2-DOPC | EphA2 | Advanced solid tumors | Phase 1 not yet recruited | Intravenous | M.D. Anderson Cancer Center |
| TD101 | Mutant keratin | Pachyonychia congenita | Phase 1 completed | Intradermal | TransDerm |
| AGN 211745 | VEGF receptor | CNV, AMD | Phase 2 terminated | Intravitreal | Allergan |
| Bevasiranib | VEGF | DME | Phase 2 completed | Intravitreal | Opko Health, Inc. |
| AMD | Phase 3 withdrawn | ||||
| SV40 siRNA vectors | BCR-ABL | CML | Observational study completed | Hadassah Medical Organization | |
| CALAA-01 | M2 subunit of ribonucleotide reductase | Solid tumor | Phase 1 terminated | Intravenous, cyclodextrin | Calando Pharmaceuticals |
| siRNA IL-10 | IL-10 | Preeclampsia | Observational study terminated | National Taiwan University Hospital | |
| SYL1001 | Receptor TrpV1 | Ocular pain dry eye | Phase 1 completed | Eye drop | Sylentis, S.A. |
| EZN-2968 Antisense oligonucleotide | HIF | Liver cancer or lymphoma | Phase 1 completed | Intravenous | Enzon Pharmaceuticals |
| ALN-VSP02 | VEGF/ Kinesin spindle protein | Solid tumor | Phase 1 completed | Intravenous, SNALP liposome | Alnylam Pharmaceuticals |
| ALN-RSV01 | RSV | Lung transplant patients/RSV infection | Phase 2 completed | Intranasal | |
| ALN-TTR02 | Transthyretin | Amyloidosis | Phase 2 completed | Intravenous, SNALP liposome | |
| ALN-PCS02 | PCSK9 | Hypercholesterolemia | Phase 1 completed | Intravenous, SNALP liposome | |
| Miravirsen | miR-122 | Hepatitis C virus | Phase 2 recruiting | Subcutaneous | SantarisPharma A/S |
| TKM-080301 | Polo-like kinase-1 | Advanced solid tumors | Phase 1 completed | Intravenous, SNALP liposome | Tekmira Pharmaceuticals |
| TKM-EBOLA | Viral RNA | Ebola infection (biodefense) | Phase 1 terminated | ||
| PRO-040201 | Apolipoprotein B | Hypercholesterolemia | Phase 1 terminated | ||
| Atu027 | Protein kinase N3 | Advanced solid tumors | Phase 1 completed | Intravenous, AtuPLEXlipoplex | Silence Therapeutics |
| siG12D LODER | Mutated KRAS oncogene | Pancreatic cancer | Phase 2 / Phase 1 recruiting | Intratumoral | Silenseed, Ltd. |
| I5NP (QPI-1002) | TP53 | ARF | Phase 1 completed | Intravenous naked siRNA | Quark Pharmaceuticals |
| Kidney transplantation | Phase 1/2 completed | ||||
| PF-04523655 | RTP801 | DME | Phase 2 completed | Intravitreal | |
| QPI-1007 | Caspase-2 | Optic nerve atrophy NAION | Phase 1 completed | Intravitreal | Quark Pharmaceuticals |
| SYL040012 | β-2 adrenergic receptor | Glaucoma, ocular hypertension | Phase 1/2 completed | Eye drop | Sylentis, S.A. |
| RXI-109 | Connective tissue growth factor | Dermal scarring | Phase 2 Recruiting | Intradermal | RXi Pharmaceuticals |
| EZN-2968 Antisense Oligonucleotide | HIF-1 | Advanced solid tumors with liver metastases | Phase 1 completed | Intravenous | National Cancer Institute |
| rHIV7-shI-TAR-CCR5RZ | Viral RNA and host factor | AIDS lymphoma | Phase 1 recruiting | Lentiviral | City of Hope Medical Center |
| Excellair | Syk kinase | Asthma | Phase 2 | Inhalation | ZaBeCor |
| FANG vaccine | Furin | Advanced cancer, ovarian cancer, melanoma | Phase 1/2 recruiting | Plasmid | Gradalis, Inc. |
| CEQ508 | β-catenin | Familial adenomatous polyposis (FAP) | Phase 1 | Bacterial | Cequent Pharmaceuticals |
VEGF: Vascular endothelial growth factor; DME: Diabetic macular edema; RSV: Respiratory syncytial virus; AMD: Age-related macular degeneration; CNV: Choroidal neovascularization; NAION: Non-arteritic anterior ischemic optic neuropathy; CML: Chronic myeloid leukemia; ARF: Acute renal failure; DME: Diabetic macular edema; PCSK9: Proprotein convertase subtilisin/kexintype 9; HIF: Hypoxia-inducible factor; SNALP: Stable nucleic acid lipid particle.
9. Conclusion and Future Perspectives
In spite of recent progress and various treatment modalities that have proved beneficial to some extent, no effective treatment is currently available for HPV-associated carcinogenesis. Even though the precise molecular targets have been characterized and several approaches for their inhibition have been demonstrated, not currently possessing an effective treatment approach constitutes a problem as important as finding novel methods for known targets and mechanisms. Nevertheless, the use of combinatorial treatment approaches with RNAi appears to be best suited for clinical protocols.
Since its discovery, RNAi has enabled interrogation of the role of individual genes in complex cellular processes. Advancements in RNAi-based screening technologies have fuelled anticipation of new discoveries. The siRNA pooling technique may prove advantageous, as there is an increased probability of at least one highly effective siRNA being present within the population. Moreover, the decreased concentration of each siRNA may reduce the potential for sequence-specific off-target effects. Another advantage over conventional therapies is that silencing of viral oncogenes and genes associated with carcinogenesis selectively enhance the chemosensitivity of tumor cells, which would help make attractive drug targets. However, siRNA-based therapeutics has encountered many barriers in clinical trials, including the stability of the RNA molecule itself, minimization of non-target effects, and limited choice of delivery systems for treatment. Although preliminary results from our study and several preclinical trials are just now becoming available, the results available to date support the safety of this innovative cancer therapy. All of these obstacles must be overcome if siRNA-based treatments for cancer are to be successful. In addition, associations and synergies between siRNA and other chemo/radio therapeutic agents may open new avenues for treatment of cervical cancer and improve the clinical outcome of patients with cervical cancer.
Acknowledgements
We are very grateful to Sung Youl Hong, Ryong Nam Kim, and Young Deug Kim for their critical review and support.
Grant Support
This research was supported by the R&D Program for the Society of the National Research Foundation (NRF) funded by the Ministry of Science, ICT and Future Planning (Grant number: NRF-2013M3C8A1075908) and by the Global Core Research Center (GCRC) grant (Grant number: NRF-2011-0030001) from the National Research Foundation (NRF), Ministry of Education, Science and Technology (MEST), Republic of Korea.
Author Contributions
All authors contributed significantly to the drafting and critical revision of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cogliano, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F. Carcinogenicity of human papillomaviruses. Lancet Oncol. 2005, 6, 204. [Google Scholar]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [PubMed]
- zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [PubMed]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [PubMed]
- Syrjanen, S. The role of human papillomavirus infection in head and neck cancers. Ann. Oncol. 2010, 21, vii243–vii 245. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Castellsague, X.; de Sanjose, S.; Bruni, L.; Saraiya, M.; Bray, F.; Ferlay, J. Worldwide burden of cervical cancer in 2008. Ann. Oncol. 2011, 22, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Derkay, C.S.; Wiatrak, B. Recurrent respiratory papillomatosis: A review. The Laryngoscope 2008, 118, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Munger, K. Mechanisms of genomic instability in human cancer: Insights from studies with human papillomavirus oncoproteins. Int. J. Cancer 2004, 109, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.S.; Lowy, D.R.; Schiller, J.T. Papillomavirus polypeptides E6 and E7 are zinc-binding proteins. J. Virol. 1989, 63, 1404–1407. [Google Scholar] [PubMed]
- Sherman, L.; Schlegel, R. Serum- and calcium-induced differentiation of human keratinocytes is inhibited by the E6 oncoprotein of human papillomavirus type 16. J. Virol. 1996, 70, 3269–3279. [Google Scholar] [PubMed]
- Kanda, T.; Watanabe, S.; Zanma, S.; Sato, H.; Furuno, A.; Yoshiike, K. Human papillomavirus type 16 E6 proteins with glycine substitution for cysteine in the metal-binding motif. Virology 1991, 185, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Nomine, Y.; Masson, M.; Charbonnier, S.; Zanier, K.; Ristriani, T.; Deryckere, F.; Sibler, A.P.; Desplancq, D.; Atkinson, R.A.; Weiss, E.; et al. Structural and functional analysis of E6 oncoprotein: Insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol. Cell 2006, 21, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Dowhanick, J.J.; McBride, A.A.; Howley, P.M. Suppression of cellular proliferation by the papillomavirus E2 protein. J. Virol. 1995, 69, 7791–7799. [Google Scholar] [PubMed]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, M.T.; Smits, H.L.; Briet, M.A.; van den Tweel, J.G.; Struyk, A.P.; van der Noordaa, J.; ter Schegget, J. Uniformity of the splicing pattern of the E6/E7 transcripts in human papillomavirus type 16-transformed human fibroblasts, human cervical premalignant lesions and carcinomas. J. Gen. Virol. 1990, 71, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, H.; Jin, M.H.; Shimizu, K.; Akutsu, N.; Shino, Y.; Simizu, B. Transcription-modulatory activity of full-length E6 and E6*I proteins of human papillomavirus type 16. Virology 1994, 203, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Tao, M.; McCoy, J.P., Jr.; Zheng, Z.M. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J. Virol. 2006, 80, 4249–4263. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, M.; Zheng, Z.M. Oncogenes and RNA splicing of human tumor viruses. Emerg. Microbes Infec. 2014, 3. [Google Scholar] [CrossRef]
- Ajiro, M.; Jia, R.; Zhang, L.; Liu, X.; Zheng, Z.M. Intron definition and a branch site adenosine at nt 385 control RNA splicing of HPV16 E6*I and E7 expression. PLoS ONE 2012, 7, e46412. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Tomaic, V.; Banks, L. The human papillomavirus (HPV) E6* proteins from high-risk, mucosal HPVs can direct degradation of cellular proteins in the absence of full-length E6 protein. J. Virol. 2009, 83, 9863–9874. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.M.; Wang, X. Regulation of cellular miRNA expression by human papillomaviruses. Biochim. Biophys. Acta 2011, 1809, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.K.; Li, Y.; Hafner, M.; Banerjee, N.S.; Tang, S.; Briskin, D.; Meyers, C.; Chow, L.T.; Xie, X.; et al. MicroRNAs are biomarkers of oncogenic human papillomavirus infections. Proc. Natl. Acad. Sci. USA 2014, 111, 4262–4267. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.S.; Vass, W.C.; Lowy, D.R.; Schiller, J.T. In vitro biological activities of the E6 and E7 genes vary among human papillomaviruses of different oncogenic potential. J. Virol. 1991, 65, 292–298. [Google Scholar] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Beaudenon, S.; Howley, P.M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA 1995, 92, 2563–2567. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Clifford, G.; Buonaguro, F.M. Classification of weakly carcinogenic human papillomavirus types: Addressing the limits of epidemiology at the borderline. Infect. Agents Cancer 2009, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Banks, L. Inhibition of e6 induced degradation of p53 is not sufficient for stabilization of p53 protein in cervical tumour derived cell lines. Oncogene 1999, 18, 3309–3315. [Google Scholar] [CrossRef] [PubMed]
- Tungteakkhun, S.S.; Duerksen-Hughes, P.J. Cellular binding partners of the human papillomavirus E6 protein. Arch. Virol. 2008, 153, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.K.; McCoy, J.P.; Banerjee, N.S.; Rader, J.S.; Broker, T.R.; Meyers, C.; Chow, L.T.; Zheng, Z.M. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6. RNA 2009, 15, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, S.M.; Syrjanen, K.J. New concepts on the role of human papillomavirus in cell cycle regulation. Ann. Med. 1999, 31, 175–187. [Google Scholar] [CrossRef] [PubMed]
- McMurray, H.R.; Nguyen, D.; Westbrook, T.F.; McAnce, D.J. Biology of human papillomaviruses. Int. J. Exp. Pathol. 2001, 82, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Demers, G.W.; Galloway, D.A. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J. Virol. 1991, 65, 473–478. [Google Scholar] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sampath, A.; Raychaudhuri, P.; Bagchi, S. Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene 2001, 20, 4740–4749. [Google Scholar] [CrossRef] [PubMed]
- Heck, D.V.; Yee, C.L.; Howley, P.M.; Munger, K. Efficiency of binding the retinoblastoma protein correlates with the transforming capacity of the E7 oncoproteins of the human papillomaviruses. Proc. Natl. Acad. Sci. USA 1992, 89, 4442–4446. [Google Scholar] [CrossRef] [PubMed]
- Charette, S.T.; McCance, D.J. The E7 protein from human papillomavirus type 16 enhances keratinocyte migration in an AKT-dependent manner. Oncogene 2007, 26, 7386–7390. [Google Scholar] [CrossRef] [PubMed]
- DiPaolo, J.A.; Woodworth, C.D.; Popescu, N.C.; Notario, V.; Doniger, J. Induction of human cervical squamous cell carcinoma by sequential transfection with human papillomavirus 16 DNA and viral Harvey ras. Oncogene 1989, 4, 395–399. [Google Scholar] [PubMed]
- Durst, M.; Gallahan, D.; Jay, G.; Rhim, J.S. Glucocorticoid-enhanced neoplastic transformation of human keratinocytes by human papillomavirus type 16 and an activated ras oncogene. Virology 1989, 173, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.; Cannon, R.E.; Karrison, T.; Beck-Engeser, G.; Huo, D.; Tennant, R.W.; Jensen, H.; Kast, W.M.; Krausz, T.; Meredith, S.C.; et al. Strong synergy between mutant ras and HPV16 E6/E7 in the development of primary tumors. Oncogene 2004, 23, 3972–3979. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Paredes, A.; De la Cruz-Hernandez, E.; Martinez-Ramirez, I.; Duenas-Gonzalez, A.; Lizano, M. E6 variants of human papillomavirus 18 differentially modulate the protein kinase B/phosphatidylinositol 3-kinase (AKT/PI3K) signaling pathway. Virology 2009, 383, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Pim, D.; Banks, L. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. J. Gen. Virol. 1997, 78, 2607–2613. [Google Scholar] [PubMed]
- Brehm, A.; Nielsen, S.J.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999, 18, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, N.; Torchia, J.; Mymryk, J.S. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene 2003, 22, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.G.; Lee, D.; Kim, J.; Seo, T.; Choe, J. Human papillomavirus type 16 E7 binds to E2F1 and activates E2F1-driven transcription in a retinoblastoma protein-independent manner. J. Biol. Chem. 2002, 277, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.C.; Hollstein, M. Clinical implications of the p53 tumor-suppressor gene. N. Engl. J. Med. 1993, 329, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Hengstermann, A.; Linares, L.K.; Ciechanover, A.; Whitaker, N.J.; Scheffner, M. Complete switch from Mdm2 to human papillomavirus E6-mediated degradation of p53 in cervical cancer cells. Proc. Natl. Acad. Sci. USA 2001, 98, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Hernandez, T.; Robinson, A.; Rodriguez-Tome, P.; Flores, T.; Hollstein, M.; Harris, C.C.; Montesano, R. Iarc database of p53 gene mutations in human tumors and cell lines: Updated compilation, revised formats and new visualisation tools. Nucl. Acids Res. 1998, 26, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Munger, K.; Byrne, J.C.; Howley, P.M. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 1991, 88, 5523–5527. [Google Scholar] [CrossRef] [PubMed]
- Lowy, D.R.; Schiller, J.T. Prophylactic human papillomavirus vaccines. J. Clin. Invest. 2006, 116, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, A.R.; Palefsky, J.M.; Goldstone, S.; Moreira, E.D., Jr.; Penny, M.E.; Aranda, C.; Vardas, E.; Moi, H.; Jessen, H.; Hillman, R.; et al. Efficacy of quadrivalent HPV vaccine against HPV infection and disease in males. N. Engl. J. Med. 2011, 364, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Beerheide, W.; Bernard, H.U.; Tan, Y.J.; Ganesan, A.; Rice, W.G.; Ting, A.E. Potential drugs against cervical cancer: Zinc-ejecting inhibitors of the human papillomavirus type 16 E6 oncoprotein. J. Natl. Cancer Inst. 1999, 91, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Beerheide, W.; Sim, M.M.; Tan, Y.J.; Bernard, H.U.; Ting, A.E. Inactivation of the human papillomavirus-16 E6 oncoprotein by organic disulfides. Bioorganic Med. Chem. 2000, 8, 2549–2560. [Google Scholar] [CrossRef]
- Butz, K.; Denk, C.; Ullmann, A.; Scheffner, M.; Hoppe-Seyler, F. Induction of apoptosis in human papillomaviruspositive cancer cells by peptide aptamers targeting the viral E6 oncoprotein. Proc. Natl. Acad. Sci. USA 2000, 97, 6693–6697. [Google Scholar] [CrossRef] [PubMed]
- Butz, K.; Ristriani, T.; Hengstermann, A.; Denk, C.; Scheffner, M.; Hoppe-Seyler, F. SiRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene 2003, 22, 5938–5945. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Milner, J. Selective silencing of viral gene expression in HPV-positive human cervical carcinoma cells treated with siRNA, a primer of RNA interference. Oncogene 2002, 21, 6041–6048. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Z.; Androphy, E.; Chen, J.; Baleja, J.D. Design and characterization of helical peptides that inhibit the E6 protein of papillomavirus. Biochemistry 2004, 43, 7421–7431. [Google Scholar] [CrossRef] [PubMed]
- Wesierska-Gadek, J.; Schloffer, D.; Kotala, V.; Horky, M. Escape of p53 protein from E6-mediated degradation in Hela cells after cisplatin therapy. Int. J. Cancer 2002, 101, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Yoshinouchi, M.; Yamada, T.; Kizaki, M.; Fen, J.; Koseki, T.; Ikeda, Y.; Nishihara, T.; Yamato, K. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol. Ther. 2003, 8, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.F.; Rao, Z.G.; Zhang, J.R. Effects of anti-HPV16 E6-ribozyme on the proliferation and apoptosis of human cervical cancer cell line CaSki. Acad. J. First Med. Coll. PLA 2002, 22, 496–498. [Google Scholar]
- Ajiro, M.; Zheng, Z.M. E6^E7, a novel splice isoform protein of human papillomavirus 16, stabilizes viral E6 and E7 oncoproteins via HSP90 and GRP78. MBio 2015, 6, e02068–e02014. [Google Scholar] [CrossRef]
- Guo, C.P.; Liu, K.W.; Luo, H.B.; Chen, H.B.; Zheng, Y.; Sun, S.N.; Zhang, Q.; Huang, L. Potent anti-tumor effect generated by a novel human papillomavirus (HPV) antagonist peptide reactivating the pRb/E2F pathway. PLoS ONE 2011, 6, e17734. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, G.H.; Shibata, R.; Wang, J.; Ray, A.S.; Wu, S.; Doerrfler, E.; Reiser, H.; Lee, W.A.; Birkus, G.; Christensen, N.D.; et al. Gs-9191 is a novel topical prodrug of the nucleotide analog 9-(2-phosphonylmethoxyethyl)guanine with antiproliferative activity and possible utility in the treatment of human papillomavirus lesions. Antimicrob. Agents Chemother. 2009, 53, 2777–2784. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.H.; Han, H.D.; Noh, K.H.; Kim, T.W.; Son, S.W. Chitosan hydrogel containing GMCSF and a cancer drug exerts synergistic anti-tumor effects via the induction of CD8+ T cell-mediated anti-tumor immunity. Clin. Exp. Metastasis 2009, 26, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, R.; Morales-Peza, N.; Castelan-Sanchez, I.; Garcia-Villa, E.; Tapia, R.; Cid-Arregui, A.; Garcia-Carranca, A.; Lopez-Bayghen, E.; Gariglio, P. Heparin (GAG-hed) inhibits LCR activity of human papillomavirus type 18 by decreasing AP1 binding. BMC Cancer 2006, 6. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Monie, A.; Pang, X.; Hung, C.F.; Wu, T.C. Vascular disrupting agent DMXAA enhances the antitumor effects generated by therapeutic HPV DNA vaccines. J. Biomed. Sci. 2011, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Hoory, T.; Monie, A.; Wu, A.; Wang, M.C.; Hung, C.F. Treatment with demethylating agent, 5-aza-2′-deoxycytidine enhances therapeutic hpv DNA vaccine potency. Vaccine 2009, 27, 4363–4369. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Bharti, A.C.; Hussain, S.; Mahata, S.; Hedau, S.; Kailash, U.; Kashyap, V.; Bhambhani, S.; Roy, M.; Batra, S.; et al. Elimination of high-risk human papillomavirus type HPV16 infection by ‘Praneem’ polyherbal tablet in women with early cervical intraepithelial lesions. J. Cancer Res. Clin. Oncol. 2009, 135, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Milrot, E.; Jackman, A.; Kniazhanski, T.; Gonen, P.; Flescher, E.; Sherman, L. Methyl jasmonate reduces the survival of cervical cancer cells and downregulates HPV E6 and E7, and survivin. Cancer Lett. 2012, 319, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.X.; Huang, G.Q.; Zhang, S.H. Soyasaponins inhibit the proliferation of hela cells by inducing apoptosis. Exp. Toxicol. Pathol. 2007, 59, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Oh, P.S.; Lim, K.T. Hela cells treated with phytoglycoprotein (150 kDa) were killed by activation of caspase 3 via inhibitory activities of NK-kappaB and AP-1. J. Biomed. Sci. 2007, 14, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Maher, D.M.; Bell, M.C.; O’Donnell, E.A.; Gupta, B.K.; Jaggi, M.; Chauhan, S.C. Curcumin suppresses human papillomavirus oncoproteins, restores p53, Rb, and PTPN13 proteins and inhibits benzo[a]pyrene-induced upregulation of HPV E7. Mol. Carcinog. 2011, 50, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wu, L.J.; Tashiro, S.; Onodera, S.; Li, L.H.; Ikejima, T. Silymarin augments human cervical cancer Hela cell apoptosis via P38/JNK MAPK pathways in serum-free medium. J. Asian Nat. Prod. Res. 2005, 7, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Munagala, R.; Kausar, H.; Munjal, C.; Gupta, R.C. Withaferin a induces p53-dependent apoptosis by repression of HPV oncogenes and upregulation of tumor suppressor proteins in human cervical cancer cells. Carcinogenesis 2011, 32, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Ahn, W.S.; Huh, S.W.; Bae, S.M.; Lee, I.P.; Lee, J.M.; Namkoong, S.E.; Kim, C.K.; Sin, J.I. A major constituent of green tea, EGCG, inhibits the growth of a human cervical cancer cell line, CaSki cells, through apoptosis, G(1) arrest, and regulation of gene expression. DNA Cell Biol. 2003, 22, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Yu, K.A.; Oh, W.K.; Baeg, T.W.; Oh, H.C.; Ahn, J.S.; Jang, W.C.; Kim, J.W.; Lim, J.S.; Choe, Y.K.; et al. Inhibitory effect of jaceosidin isolated from artemisiaargyi on the function of E6 and E7 oncoproteins of HPV 16. J. Ethnopharmacol. 2005, 98, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Craigo, J.; Callahan, M.; Huang, R.C.; DeLucia, A.L. Inhibition of human papillomavirus type 16 gene expression by nordihydroguaiaretic acid plant lignan derivatives. Antivir. Res. 2000, 47, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Thompson, C.D.; Roberts, J.N.; Muller, M.; Lowy, D.R.; Schiller, J.T. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2006, 2, e69. [Google Scholar] [CrossRef] [PubMed]
- Haasnoot, J.; Westerhout, E.M.; Berkhout, B. RNA interference against viruses: Strike and counterstrike. Nat. Biotechnol. 2007, 25, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Sui, G.; Soohoo, C.; Affar el, B.; Gay, F.; Shi, Y.; Forrester, W.C. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 5515–5520. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Y.; DeRuiter, S.L.; Turner, D.L. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 6047–6052. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; McManus, M.T.; Nguyen, T.; Shen, C.H.; Sharp, P.A.; Eisen, H.N.; Chen, J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc. Natl. Acad. Sci. USA 2003, 100, 2718–2723. [Google Scholar] [CrossRef] [PubMed]
- Kitabwalla, M.; Ruprecht, R.M. RNA interference—A new weapon against HIV and beyond. N. Engl. J. Med. 2002, 347, 1364–1367. [Google Scholar] [CrossRef] [PubMed]
- Milner, J. RNA interference for treating cancers caused by viral infection. Exp. Opin. Biol. Ther. 2003, 3, 459–467. [Google Scholar] [CrossRef]
- Sima, N.; Wang, W.; Kong, D.; Deng, D.; Xu, Q.; Zhou, J.; Xu, G.; Meng, L.; Lu, Y.; Wang, S.; et al. RNA interference against HPV16 E7 oncogene leads to viral E6 and E7 suppression in cervical cancer cells and apoptosis via upregulation of Rb and p53. Apoptosis 2008, 13, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Yamato, K.; Yamada, T.; Kizaki, M.; Ui-Tei, K.; Natori, Y.; Fujino, M.; Nishihara, T.; Ikeda, Y.; Nasu, Y.; Saigo, K.; et al. New highly potent and specific E6 and E7 siRNAs for treatment of HPV16 positive cervical cancer. Cancer Gene Ther. 2008, 15, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Putral, L.; Hengst, K.; Minto, K.; Saunders, N.A.; Leggatt, G.; McMillan, N.A. Inhibition of cervical cancer cell growth in vitro and in vivo with lentiviral-vector delivered short hairpin RNA targeting human papillomavirus E6 and E7 oncogenes. Cancer Gene Ther. 2006, 13, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.H.; Alexander, K.A. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in Hela cells. J. Virol. 2003, 77, 6066–6069. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Tao, M.; McCoy, J.P., Jr.; Zheng, Z.M. Short-term induction and long-term suppression of HPV16 oncogene silencing by RNA interference in cervical cancer cells. Oncogene 2006, 25, 2094–2104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Peng, C.; Li, B.; Wang, F.; Zhou, C.; Hong, D.; Ye, F.; Cheng, X.; Lu, W.; Xie, X. Transcriptional gene silencing of HPV16 E6/E7 induces growth inhibition via apoptosis in vitro and in vivo. Gynecol. Oncol. 2012, 124, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Erkin, O.C.; Kwon, M.J.; Kim, S.H.; Jung, J.I.; Oh, Y.K.; Her, S.W.; Ju, W.; Choi, Y.L.; Song, S.Y.; et al. The synergistic therapeutic effect of cisplatin with human papillomavirus E6/E7 short interfering RNA on cervical cancer cell lines in vitro and in vivo. Int. J. Cancer 2012, 130, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Kuo, T.F.; Chen, Y.J.; Chiu, C.C.; Lu, Y.C.; Li, H.F.; Shen, C.R.; Cheng, A.J. Highly potent and specific siRNAs against E6 or E7 genes of HPV16- or HPV18-infected cervical cancers. Cancer Gene Ther. 2010, 17, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Lu, W.; Ye, F.; Hu, Y.; Xie, X. Gene silencing of HPV16 E6/E7 induced by promoter-targeting siRNA in SiHa cells. Br. J. Cancer 2009, 101, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Jonson, A.L.; Rogers, L.M.; Ramakrishnan, S.; Downs, L.S., Jr. Gene silencing with siRNA targeting E6/E7 as a therapeutic intervention in a mouse model of cervical cancer. Gynecol. Oncol. 2008, 111, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Lea, J.S.; Sunaga, N.; Sato, M.; Kalahasti, G.; Miller, D.S.; Minna, J.D.; Muller, C.Y. Silencing of HPV 18 oncoproteins with RNA interference causes growth inhibition of cervical cancer cells. Reprod. Sci. 2007, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Courtete, J.; Sibler, A.P.; Zeder-Lutz, G.; Dalkara, D.; Oulad-Abdelghani, M.; Zuber, G.; Weiss, E. Suppression of cervical carcinoma cell growth by intracytoplasmic codelivery of anti-oncoprotein E6 antibody and small interfering RNA. Mol. Cancer Ther. 2007, 6, 1728–1735. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Saito, M.; Iwasaki, E.; Ochiya, T.; Takei, Y.; Hayashi, S.; Ono, A.; Hirao, N.; Nakamura, M.; Kubushiro, K.; et al. Intratumor injection of small interfering RNA-targeting human papillomavirus 18 E6 and E7 successfully inhibits the growth of cervical cancer. Int. J. Oncol. 2006, 29, 541–548. [Google Scholar] [PubMed]
- Koivusalo, R.; Krausz, E.; Helenius, H.; Hietanen, S. Chemotherapy compounds in cervical cancer cells primed by reconstitution of p53 function after short interfering RNA-mediated degradation of human papillomavirus 18 E6 mRNA: Opposite effect of siRNA in combination with different drugs. Mol. Pharmacol. 2005, 68, 372–382. [Google Scholar] [PubMed]
- Putral, L.N.; Bywater, M.J.; Gu, W.; Saunders, N.A.; Gabrielli, B.G.; Leggatt, G.R.; McMillan, N.A. RNA interference against human papillomavirus oncogenes in cervical cancer cells results in increased sensitivity to cisplatin. Mol. Pharmacol. 2005, 68, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Huang, S.Y.; Chen, T.T.; Chen, J.C.; Chiou, C.L.; Huang, T.M. Cisplatin restores p53 function and enhances the radiosensitivity in HPV16 E6 containing SiHa cells. J. Cell. Biochem. 2004, 91, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Feldser, D.M.; Kostova, K.K.; Winslow, M.M.; Taylor, S.E.; Cashman, C.; Whittaker, C.A.; Sanchez-Rivera, F.J.; Resnick, R.; Bronson, R.; Hemann, M.T.; et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010, 468, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Karnezis, A.N.; Garcia, D.; Madriles, F.; Kortlever, R.M.; Rostker, F.; Brown Swigart, L.; Pham, D.M.; Seo, Y.; Evan, G.I.; et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010, 468, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microrna processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Loewer, A.; Mock, C.; Lahav, G. Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol. 2011, 7, 488. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Murray, D. Ionizing radiation-induced responses in human cells with differing TP53 status. Int. J. Mol. Sci. 2013, 14, 22409–22435. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Rivlin, N.; Shoshana, O.Y.; Ezra, O.; Madar, S.; Goldfinger, N.; Rotter, V. Chemotherapeutic agents induce the expression and activity of their clearing enzyme CYP3A4 by activating p53. Carcinogenesis 2013, 34, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Loewer, A.; Batchelor, E.; Gaglia, G.; Lahav, G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell 2010, 142, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. P53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Shi, J.; Jung, S.H.; Chen, X.; Cho, K.H. Attractor landscape analysis reveals feedback loops in the p53 network that control the cellular response to DNA damage. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Pan, D.; Pan, L.Z.; Hill, R.; Marcato, P.; Shmulevitz, M.; Vassilev, L.T.; Lee, P.W. Stabilisation of p53 enhances reovirus-induced apoptosis and virus spread through p53-dependent NF-kappaB activation. Br. J. Cancer 2011, 105, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Bartz, S.R.; Zhang, Z.; Burchard, J.; Imakura, M.; Martin, M.; Palmieri, A.; Needham, R.; Guo, J.; Gordon, M.; Chung, N.; et al. Small interfering RNA screens reveal enhanced cisplatin cytotoxicity in tumor cells having both BRCA network and TP53 disruptions. Mol. Cell Biol. 2006, 26, 9377–9386. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.A.; Ye, F.; Marshall, C.B.; Lehmann, B.D.; Pendleton, C.S.; Shyr, Y.; Arteaga, C.L.; Pietenpol, J.A. RNA interference (RNAi) screening approach identifies agents that enhance paclitaxel activity in breast cancer cells. Breast Cancer Res. 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Honma, K.; Iwao-Koizumi, K.; Takeshita, F.; Yamamoto, Y.; Yoshida, T.; Nishio, K.; Nagahara, S.; Kato, K.; Ochiya, T. RPN2 gene confers docetaxel resistance in breast cancer. Nat. Med. 2008, 14, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.; Deeds, S.L.; Weinstein, E.J. A screen of shRNAs targeting tumor suppressor genes to identify factors involved in A549 paclitaxel sensitivity. Oncol. Rep. 2007, 18, 1499–1505. [Google Scholar] [PubMed]
- Swanton, C.; Nicke, B.; Marani, M.; Kelly, G.; Downward, J. Initiation of high frequency multi-drug resistance following kinase targeting by siRNAs. Cell Cycle 2007, 6, 2001–2004. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Cochrane, M.; Leggatt, G.R.; Payne, E.; Choyce, A.; Zhou, F.; Tindle, R.; McMillan, N.A. Both treated and untreated tumors are eliminated by short hairpin RNA-based induction of target-specific immune responses. Proc. Natl. Acad. Sci. USA 2009, 106, 8314–8319. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.L.; Green, N.; Seymour, L.W.; Stevenson, M. Paclitaxel combined with siRNA targeting HPV16 oncogenes improves cytotoxicity for cervical carcinoma. Cancer Gene Ther. 2009, 16, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Eifel, P.J.; Lu, J.; Grigsby, P.W.; Levenback, C.; Stevens, R.E.; Rotman, M.; Gershenson, D.M.; Mutch, D.G. Pelvic radiation with concurrent chemotherapy compared with pelvic and para-aortic radiation for high-risk cervical cancer. N. Engl. J. Med. 1999, 340, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Keys, H.M.; Bundy, B.N.; Stehman, F.B.; Muderspach, L.I.; Chafe, W.E.; Suggs, C.L., 3rd; Walker, J.L.; Gersell, D. Cisplatin, radiation, and adjuvant hysterectomy compared with radiation and adjuvant hysterectomy for bulky stage Ib cervical carcinoma. N. Engl. J. Med. 1999, 340, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Peters, W.A., 3rd; Liu, P.Y.; Barrett, R.J., 2nd; Stock, R.J.; Monk, B.J.; Berek, J.S.; Souhami, L.; Grigsby, P.; Gordon, W., Jr.; Alberts, D.S. Concurrent chemotherapy and pelvic radiation therapy compared with pelvic radiation therapy alone as adjuvant therapy after radical surgery in high-risk early-stage cancer of the cervix. J. Clin. Oncol. 2000, 18, 1606–1613. [Google Scholar] [PubMed]
- Monk, B.J.; Sill, M.W.; McMeekin, D.S.; Cohn, D.E.; Ramondetta, L.M.; Boardman, C.H.; Benda, J.; Cella, D. Phase III trial of four cisplatin-containing doublet combinations in stage IVb, recurrent, or persistent cervical carcinoma: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2009, 27, 4649–4655. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.H.; Blessing, J.A.; McQuellon, R.P.; Thaler, H.T.; Cella, D.; Benda, J.; Miller, D.S.; Olt, G.; King, S.; Boggess, J.F.; et al. Phase III study of cisplatin with or without paclitaxel in stage IVb, recurrent, or persistent squamous cell carcinoma of the cervix: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2004, 22, 3113–3119. [Google Scholar] [CrossRef] [PubMed]
- Long, H.J., 3rd; Bundy, B.N.; Grendys, E.C., Jr.; Benda, J.A.; McMeekin, D.S.; Sorosky, J.; Miller, D.S.; Eaton, L.A.; Fiorica, J.V.; Gynecologic Oncology Group Study. Randomized phase III trial of cisplatin with or without topotecan in carcinoma of the uterine cervix: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2005, 23, 4626–4633. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Yakushiji, M.; Noda, K.; Ikeda, M.; Kudoh, R.; Yajima, A.; Tomoda, Y.; Terashima, Y.; Takeuchi, S.; Hiura, M.; et al. Phase II study of irinotecan and cisplatin as first-line chemotherapy in advanced or recurrent cervical cancer. Oncology 2000, 58, 31–37. [Google Scholar] [CrossRef]
- Pectasides, D.; Fountzilas, G.; Papaxoinis, G.; Pectasides, E.; Xiros, N.; Sykiotis, C.; Koumarianou, A.; Psyrri, A.; Panayiotides, J.; Economopoulos, T. Carboplatin and paclitaxel in metastatic or recurrent cervical cancer. Int. J. Gynecol. Cancer 2009, 19, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Brewer, C.A.; Blessing, J.A.; Nagourney, R.A.; McMeekin, D.S.; Lele, S.; Zweizig, S.L. Cisplatin plus gemcitabine in previously treated squamous cell carcinoma of the cervix: A phase II study of the Gynecologic Oncology Group. Gynecol. Oncol. 2006, 100, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Sill, M.W.; Burger, R.A.; Gray, H.J.; Buekers, T.E.; Roman, L.D. Phase II trial of bevacizumab in the treatment of persistent or recurrent squamous cell carcinoma of the cervix: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2009, 27, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.A.; Blessing, J.A.; Vaccarello, L.; Roman, L.D. Phase II clinical trial of docetaxel in refractory squamous cell carcinoma of the cervix: A Gynecologic Oncology Group Study. Am. J. Clin. Oncol. 2007, 30, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Pow, E.H.; Kwong, D.L.; McMillan, A.S.; Wong, M.C.; Sham, J.S.; Leung, L.H.; Leung, W.K. Xerostomia and quality of life after intensity-modulated radiotherapy vs. conventional radiotherapy for early-stage nasopharyngeal carcinoma: Initial report on a randomized controlled clinical trial. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Pignol, J.P.; Olivotto, I.; Rakovitch, E.; Gardner, S.; Sixel, K.; Beckham, W.; Vu, T.T.; Truong, P.; Ackerman, I.; Paszat, L. A multicenter randomized trial of breast intensity-modulated radiation therapy to reduce acute radiation dermatitis. J. Clin. Oncol. 2008, 26, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Lim, D.S. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol. 2000, 1, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Steel, G.G.; Peckham, M.J. Exploitable mechanisms in combined radiotherapy-chemotherapy: The concept of additivity. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Lahav, G.; Rosenfeld, N.; Sigal, A.; Geva-Zatorsky, N.; Levine, A.J.; Elowitz, M.B.; Alon, U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 2004, 36, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Yamato, K.; Yamada, T.; Kizaki, M.; Ui-Tei, K.; Natori, Y.; Fujino, M.; Nishihara, T.; Ikeda, Y.; Nasu, Y.; Saigo, K.; et al. New highly potent and specific E6 and E7 siRNAs for treatment of hpv16 positive cervical cancer. Cancer Gene Ther. 2008, 15, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Yamato, K.; Egawa, N.; Endo, S.; Ui-Tei, K.; Yamada, T.; Saigo, K.; Hyodo, I.; Kiyono, T.; Nakagawa, I. Enhanced specificity of HPV16 E6E7 siRNA by RNA-DNA chimera modification. Cancer Gene Ther. 2011, 18, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Egusquiaguirre, S.P.; Igartua, M.; Hernandez, R.M.; Pedraz, J.L. Nanoparticle delivery systems for cancer therapy: Advances in clinical and preclinical research. Clin. Transl. Oncol. 2012, 14, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Truong, N.P.; Gu, W.; Prasadam, I.; Jia, Z.; Crawford, R.; Xiao, Y.; Monteiro, M.J. An influenza virus-inspired polymer system for the timed release of siRNA. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Cun, B.; Song, X.; Jia, R.; Wang, H.; Zhao, X.; Liu, B.; Ge, S.; Fan, X. Cell growth inhibition in HPV 18 positive uveal melanoma cells by E6/E7 siRNA. Tumour Biol. 2013, 34, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Hu, J.; Wang, B.; Zhao, L.; Ji, K.; Guo, B.; Yin, D.; Du, Y.; Kopecko, D.J.; et al. Plasmid-based E6-specific siRNA and co-expression of wild-type p53 suppresses the growth of cervical cancer in vitro and in vivo. Cancer Lett. 2013, 335, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).