
Role of Factor H and Related Proteins in Regulating Complement Activation in the Macula, and Relevance to Age-Related Macular Degeneration

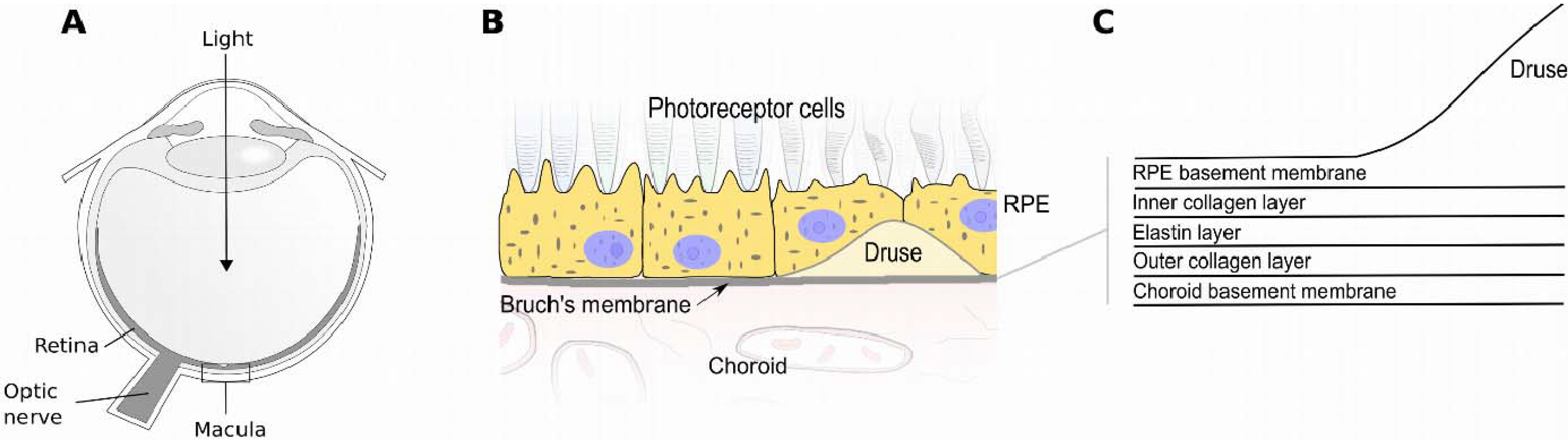{kind=link}
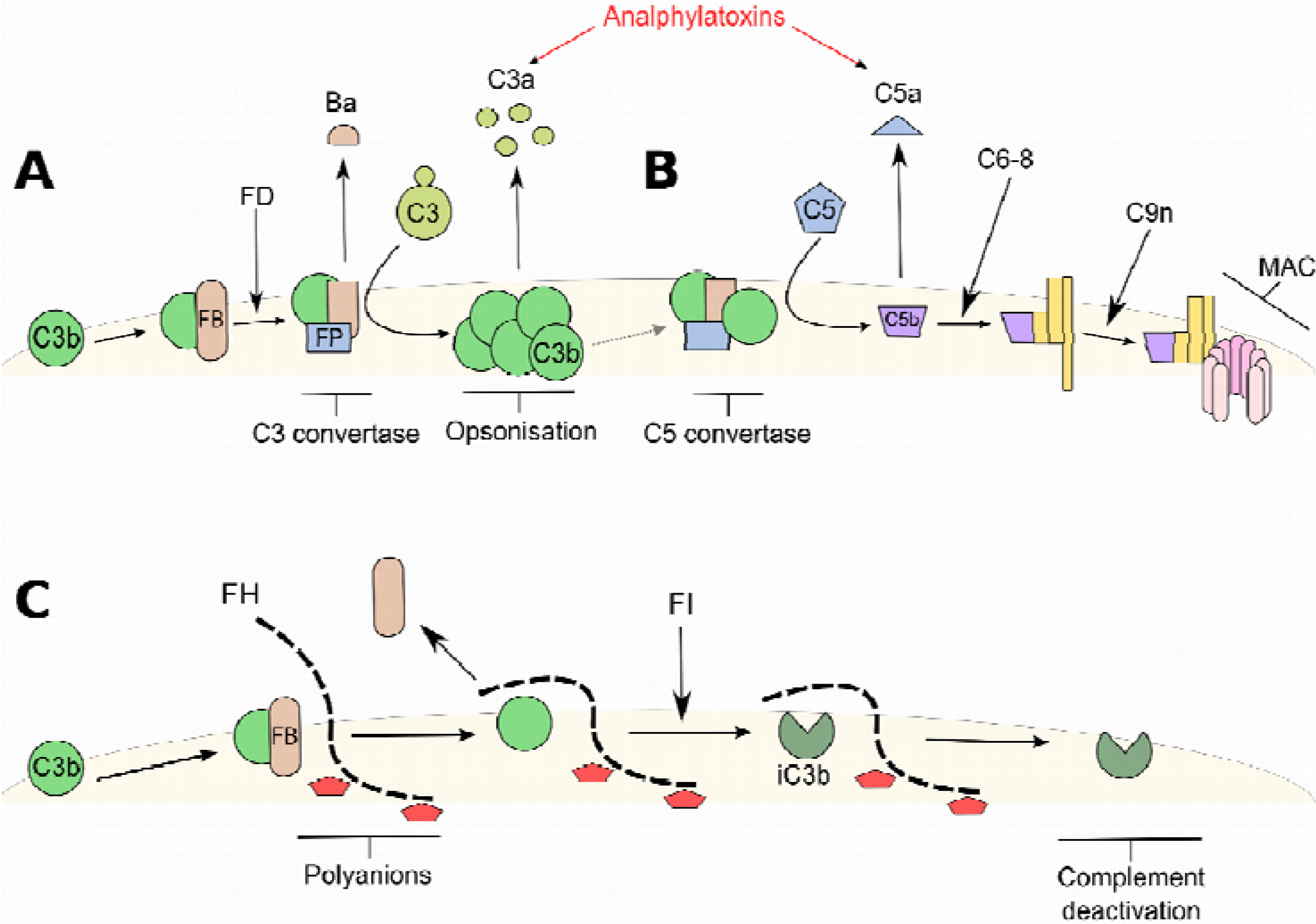{kind=link}
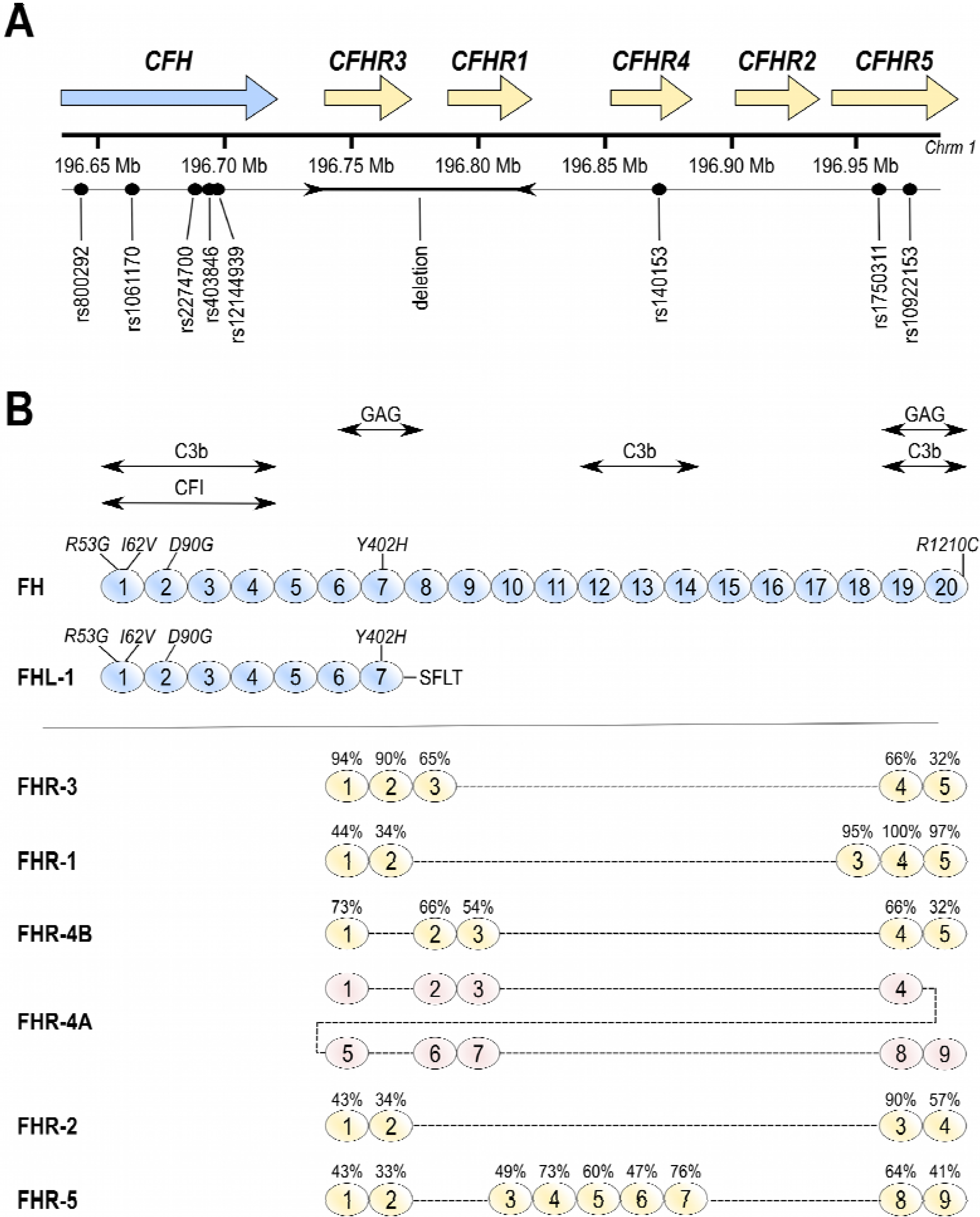{kind=link}
Abstract
:1. Introduction
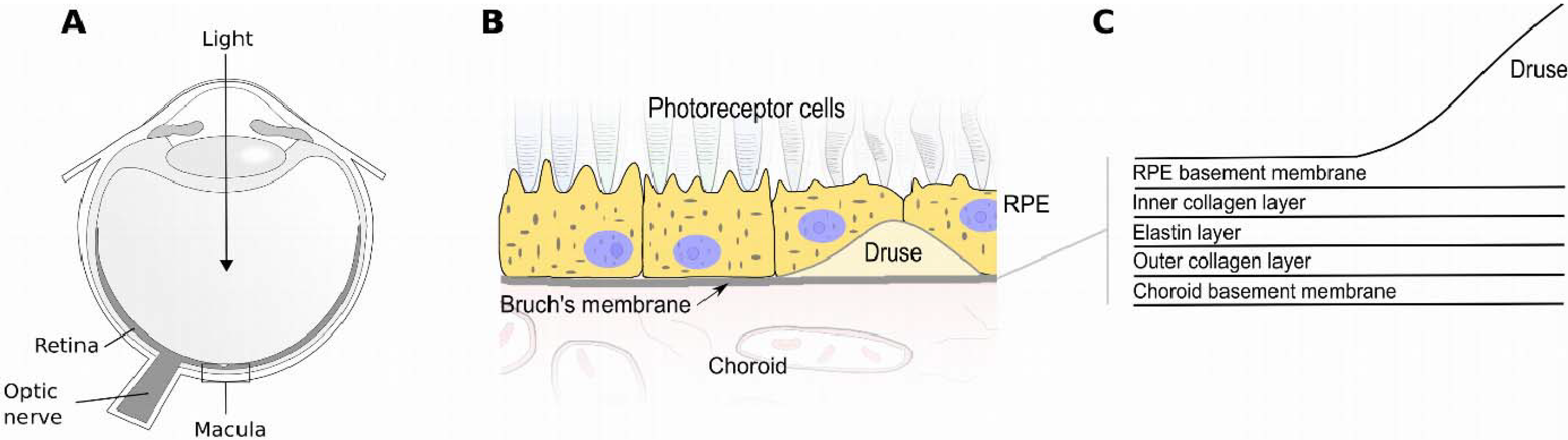
2. Factor H (FH) and the Alternative Complement Pathway
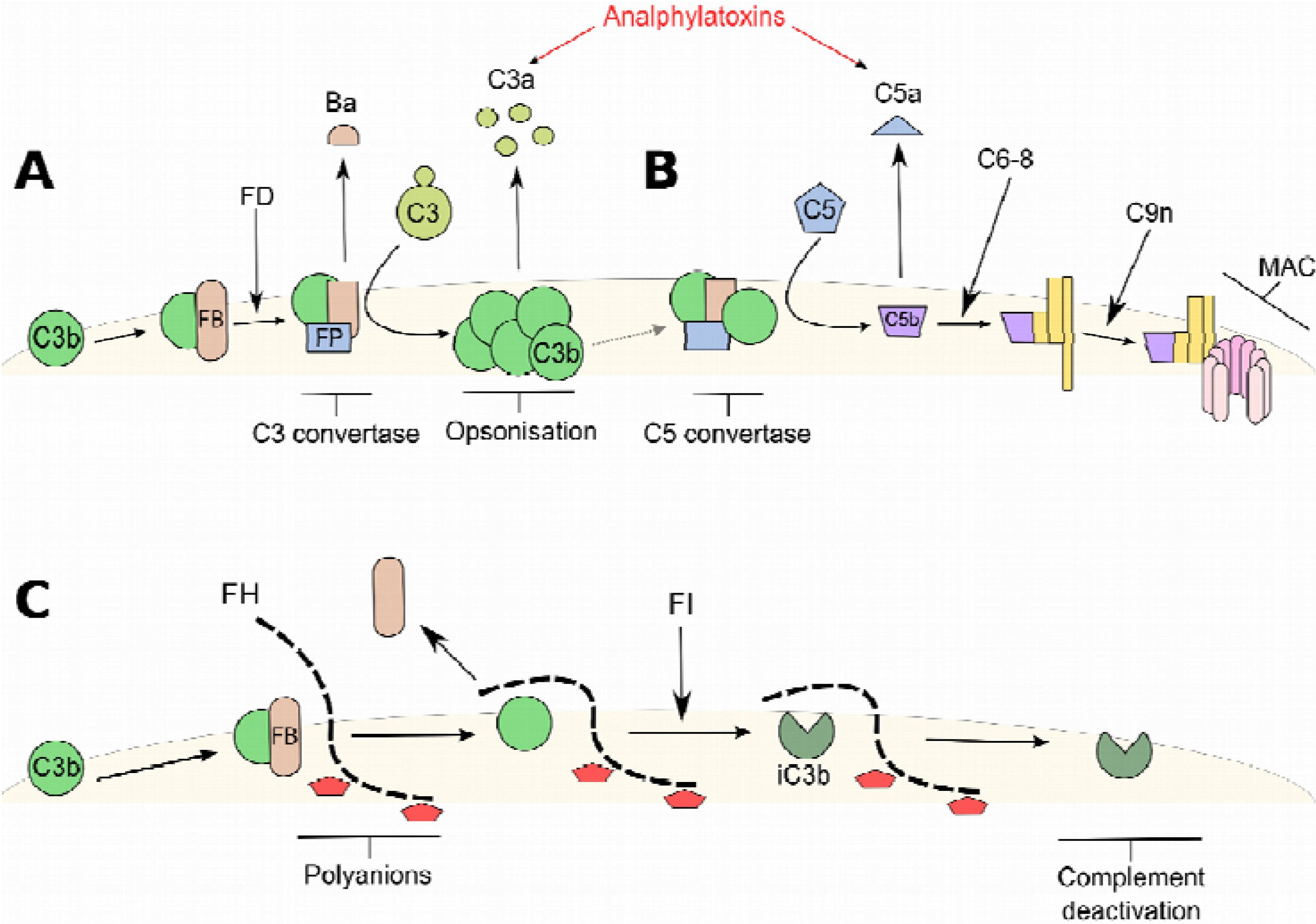
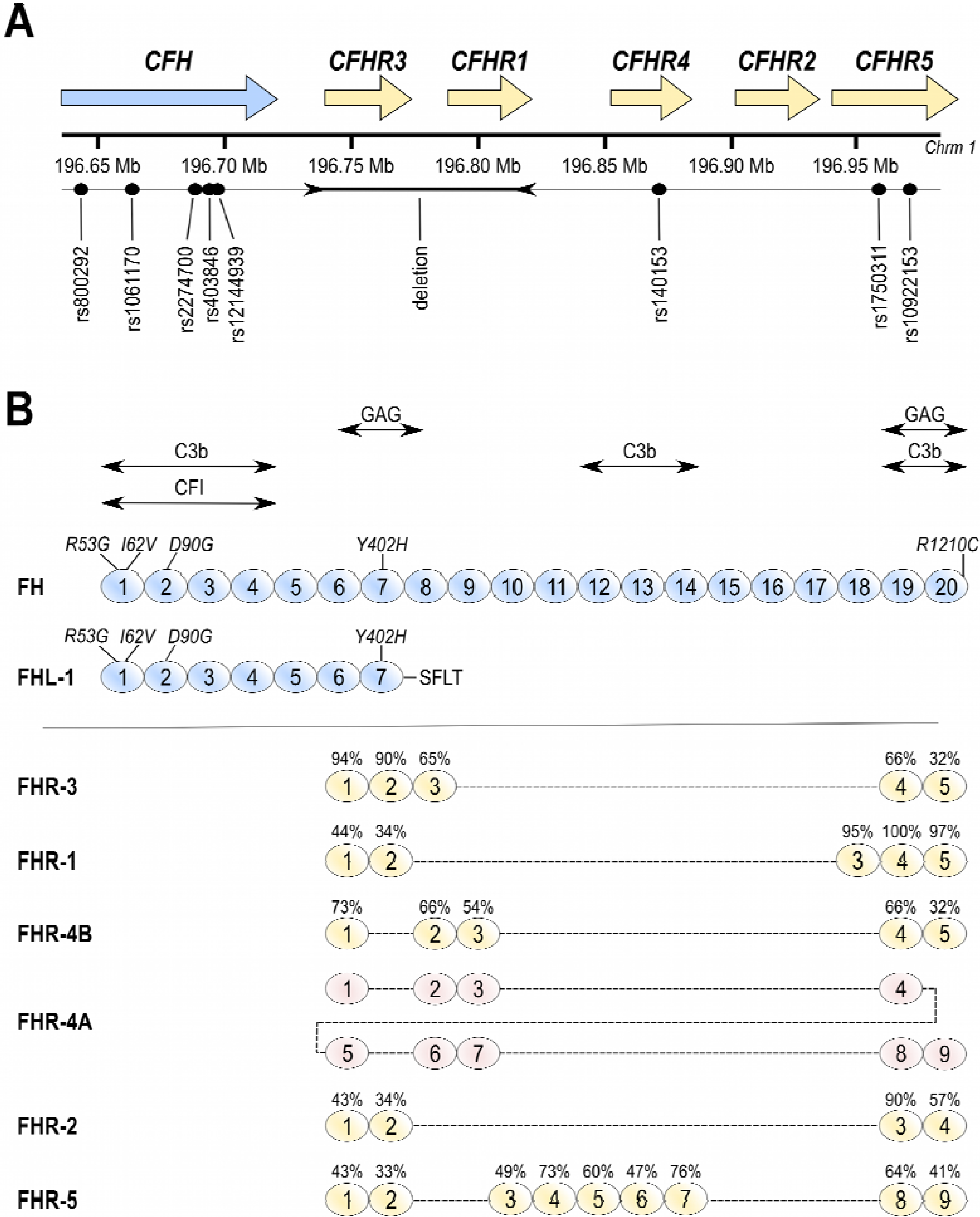
3. Factor H-Related Proteins
4. Genetics of AMD and Functional Implications
5. Tissue Interactions of FH and FHL-1
6. Effects of Aging on Ocular Immunity
7. Complement Based Therapies for AMD
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sarks, S.; Cherepanoff, S.; Killingsworth, M.; Sarks, J. Relationship of Basal Laminar Deposit and Membranous Debris to the Clinical Presentation of Early Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 968–977. [Google Scholar] [CrossRef]
- Hageman, G.S.; Mullins, R.F.; Russell, S.R.; Johnson, L.V.; Anderson, D.H. Vitronectin is a constituent of ocular drusen and the vitronectin gene is expressed in human retinal pigmented epithelial cells. FASEB J. 1999, 13, 477–484. [Google Scholar] [PubMed]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 432–433. [Google Scholar] [CrossRef]
- Schramm, E.C.; Clark, S.J.; Triebwasser, M.P.; Raychaudhuri, S.; Seddon, J.M.; Atkinson, J.P. Genetic variants in the complement system predisposing to age-related macular degeneration: A review. Mol. Immunol. 2014, 61, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; Bishop, P.; Day, A. Complement factor H and age-related macular degeneration: The role of glycosaminoglycan recognition in disease pathology. Biochem. Soc. Trans. 2010, 38, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Gennaro, R.; Simonic, T.; Negri, A.; Mottola, C.; Secchi, C.; Ronchi, S.; Romeo, D. C5a fragment of bovine complement. Purification, bioassays, amino-acid sequence and other structural studies. Eur. J. Biochem. 1986, 155, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.M. Complement factor H and the haemolytic uraemic syndrome. Lancet 2001, 358, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.H.; Harris, C.L.; Pickering, M.C. Dense deposit disease. Mol. Immunol. 2011, 48, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Ritter, M.; Bolz, M.; Haidinger, M.; Deák, G.; Sacu, S.; Säemann, M.; Schmidt-Erfurth, U. Functional and morphological macular abnormalities in membranoproliferative glomerulonephritis type II. Br. J. Ophthalmol. 2010, 94, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Strohmeyer, R.; Ramirez, M.; Cole, G.J.; Mueller, K.; Rogers, J. Association of factor H of the alternative pathway of complement with agrin and complement receptor 3 in the Alzheimer’s disease brain. J. Neuroimmunol. 2002, 131, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, I.B.; Riet, L.; Gevers, T.; Dam, G.B.; Kuppevelt, T.H.; David, G.; Küsters, B.; Waal, R.M.W.; Verbeek, M.M. Sulfation of heparan sulfate associated with amyloid-β plaques in patients with Alzheimer’s disease. Acta Neuropathol. 2009, 119, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Hebecker, M.; Józsi, M. Factor H-related protein 4 activates complement by serving as a platform for the assembly of an alternative pathway C3 convertase via its interaction with C3b. J. Biol. Chem. 2012, 287, 19528–19536. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Lauer, N.; Hartmann, A.; Stippa, S.; Keilhauer, C.N.; Oppermann, M.; Pandey, M.K.; Kohl, J.; Zipfel, P.F.; Weber, B.H.F.; et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum. Mol. Genet. 2010, 19, 4694–4704. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, H.U.; Buhlmann, D.; Hortschansky, P.; Chen, Q.; Böhm, S.; Kemper, M.J.; Wallich, R.; Hartmann, A.; Hallström, T.; Zipfel, P.F.; et al. Human Factor H-Related Protein 2 (CFHR2) Regulates Complement Activation. PLoS One 2013, 8, e78617. [Google Scholar] [CrossRef] [PubMed]
- De Jorge, G.E.; Caesar, J.J.E.; Malik, T.H.; Patel, M.; Colledge, M.; Johnson, S.; Hakobyan, S.; Morgan, B.P.; Harris, C.L.; Pickering, M.C.; et al. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc. Natl. Acad. Sci. 2013, 110, 4685–4690. [Google Scholar] [CrossRef] [PubMed]
- Tortajada, A.; Yébenes, H.; Abarrategui-Garrido, C.; Anter, J.; García-Fernández, J.M.; Martínez-Barricarte, R.; Alba-Domínguez, M.; Malik, T.H.; Bedoya, R.; Cabrera Pérez, R.; et al. C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation. J. Clin. Investig. 2013, 123, 2434–2446. [Google Scholar] [CrossRef] [PubMed]
- Józsi, M.; Richter, H.; Löschmann, I.; Skerka, C.; Buck, F.; Beisiegel, U.; Erdei, A.; Zipfel, P.F. FHR-4A: A new factor H-related protein is encoded by the human FHR-4 gene. Eur. J. Hum. Genet. 2005, 13, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Schmidt, C.Q.; White, A.M.; Hakobyan, S.; Morgan, B.P.; Bishop, P.N. Identification of Factor H-like Protein 1 as the Predominant Complement Regulator in Bruch’s Membrane: Implications for Age-Related Macular Degeneration. J. Immunol. 2014, 193, 4962–4970. [Google Scholar] [CrossRef] [PubMed]
- Montes, T.; Tortajada, A.; Morgan, B.P.; Rodríguez de Córdoba, S.; Harris, C.L. Functional basis of protection against age-related macular degeneration conferred by a common polymorphism in complement factor B. Proc. Natl. Acad. Sci. 2009, 106, 4366–4371. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Yu, Y.; Miller, E.C.; Reynolds, R.; Tan, P.L.; Gowrisankar, S.; Goldstein, J.I.; Triebwasser, M.; Anderson, H.E.; Zerbib, J.; et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 2013, 45, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Iartchouk, O.; Chin, K.; Tan, P.L.; Tai, A.K.; Ripke, S.; Gowrisankar, S.; Vemuri, S.; Montgomery, K.; Yu, Y.; et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011, 43, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; McKeigue, P.M.; Skerka, C.; Hayward, C.; Rudan, I.; Vitart, V.; Polasek, O.; Armbrecht, A.-M.; Yates, J.R.W.; Vatavuk, Z.; et al. Genetic influences on plasma CFH and CFHR1 concentrations and their role in susceptibility to age-related macular degeneration. Hum. Mol. Genet. 2013, 22, 4857–4869. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.O.; Ritter, R.; Abel, K.J.; Manning, A.; Panhuysen, C.; Farrer, L.A. Complement factor H polymorphism and age-related macular degeneration. Science 2005, 308, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.; Spencer, K.L.; Kwan, S.Y.; Noureddine, M.; Gilbert, J.R.; et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.-Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef] [PubMed]
- Day, A.J.; Willis, A.C.; Ripoche, J.; Sim, R.B. Sequence polymorphism of human complement factor H. Immunogenetics 1988, 27, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Sofat, R.; Casas, J.P.; Webster, A.R.; Bird, A.C.; Mann, S.S.; Yates, J.R.W.; Moore, A.T.; Sepp, T.; Cipriani, V.; Bunce, C.; et al. Complement factor H genetic variant and age-related macular degeneration: Effect size, modifiers and relationship to disease subtype. Int. J. Epidemiol. 2012, 41, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Corral, P.; Pérez-Caballero, D.; Huarte, O.; Simckes, A.M.; Goicoechea, E.; López-Trascasa, M.; de Córdoba, S.R. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am. J. Hum. Genet. 2002, 71, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Manuelian, T.; Hellwage, J.; Meri, S.; Caprioli, J.; Noris, M.; Heinen, S.; Jozsi, M.; Neumann, H.P.; Remuzzi, G.; Zipfel, P.F. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J. Clin. Investig. 2003, 111, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Józsi, M.; Heinen, S.; Hartmann, A.; Ostrowicz, C.W.; Hälbich, S.; Richter, H.; Kunert, A.; Licht, C.; Saunders, R.E.; Perkins, S.J.; et al. Factor H and atypical hemolytic uremic syndrome: Mutations in the C-terminus cause structural changes and defective recognition functions. J. Am. Soc. Nephrol. 2006, 17, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Triebwasser, M.P.; Wong, E.K.S.; Schramm, E.C.; Thomas, B.; Reynolds, R.; Mardis, E.R.; Atkinson, J.P.; Daly, M.; Raychaudhuri, S.; et al. Whole-exome sequencing identifies rare, functional CFH variants in families with macular degeneration. Hum. Mol. Genet. 2014, 23, 5283–5293. [Google Scholar] [CrossRef] [PubMed]
- Ripoche, J.; Day, A.J.; Harris, T.J.; Sim, R.B. The complete amino acid sequence of human complement factor H. Biochem. J. 1988, 249, 593–602. [Google Scholar] [PubMed]
- Hughes, A.E.; Orr, N.; Esfandiary, H.; Diaz-Torres, M.; Goodship, T.; Chakravarthy, U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat. Genet. 2006, 38, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.S.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.M.; Anderson, D.H.; Johnson, L.V.; Radeke, M.J.; Kavanagh, D.; Richards, A.; Atkinson, J.; et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: Characterization, ethnic distribution and evolutionary implications. Ann. Med. 2006, 38, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A.P.; Deakin, J.A.; Schmidt, C.Q.; Blaum, B.S.; Egan, C.; Ferreira, V.P.; Pangburn, M.K.; Lyon, M.; Uhrin, D.; Barlow, P.N. Structure Shows That a Glycosaminoglycan and Protein Recognition Site in Factor H Is Perturbed by Age-related Macular Degeneration-linked Single Nucleotide Polymorphism. J. Biol. Chem. 2007, 282, 18960–18968. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.A.; Edwards, A.O.; Ryu, E.; Tosakulwong, N.; Baratz, K.H.; Brown, W.L.; Charbel Issa, P.; Scholl, H.P.; Pollok-Kopp, B.; Schmid-Kubista, K.E.; et al. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum. Mol. Genet. 2010, 19, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Sjöberg, A.P.; Trouw, L.A.; Clark, S.J.; Sjölander, J.; Heinegård, D.; Sim, R.B.; Day, A.J.; Blom, A.M. The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. J. Biol. Chem. 2007, 282, 10894–10900. [Google Scholar] [CrossRef] [PubMed]
- Haapasalo, K.; Jarva, H.; Siljander, T.; Tewodros, W.; Vuopio-Varkila, J.; Jokiranta, T.S. Complement factor H allotype 402H is associated with increased C3b opsonization and phagocytosis of Streptococcus pyogenes. Mol. Microbiol. 2008, 70, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Higman, V.A.; Mulloy, B.; Perkins, S.J.; Lea, S.M.; Sim, R.B.; Day, A.J. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J. Biol. Chem. 2006, 281, 24713–24720. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Bishop, P.N.; Day, A.J. The Proteoglycan Glycomatrix: A Sugar Microenvironment Essential for Complement Regulation. Front. Immunol. 2013, 4, 412. [Google Scholar] [PubMed]
- Clark, S.J.; Perveen, R.; Hakobyan, S.; Morgan, B.P.; Sim, R.B.; Bishop, P.N.; Day, A.J. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch’s membrane in human retina. J. Biol. Chem. 2010, 285, 30192–30202. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Ridge, L.A.; Herbert, A.P.; Hakobyan, S.; Mulloy, B.; Lennon, R.; Würzner, R.; Morgan, B.P.; Uhrín, D.; Bishop, P.N.; et al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J. Immunol. 2013, 190, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, M.; Manuelian, T.; Jozsi, M.; Brandt, E.; Jokiranta, T.S.; Heinen, S.; Meri, S.; Skerka, C.; Gotze, O.; Zipfel, P.F. The C-terminus of complement regulator Factor H mediates target recognition: Evidence for a compact conformation of the native protein. Clin. Exp. Immunol. 2006, 144, 342–352. [Google Scholar] [CrossRef]
- Józsi, M.; Oppermann, M.; Lambris, J.D.; Zipfel, P.F. The C-terminus of complement factor H is essential for host cell protection. Mol. Immunol. 2007, 44, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Ugarte, M.; Osborne, N.N.; Brown, L.A.; Bishop, P.N. Iron, zinc, and copper in retinal physiology and disease. Surv. Ophthalmol. 2013, 58, 585–609. [Google Scholar] [CrossRef] [PubMed]
- Weismann, D.; Hartvigsen, K.; Lauer, N.; Bennett, K.L.; Scholl, H.P.N.; Issa, P.C.; Cano, M.; Brandstätter, H.; Tsimikas, S.; Skerka, C.; et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 2011, 478, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Nelson, K.C.; Wu, M.; Sternberg, P.; Jones, D.P. Oxidative damage and protection of the RPE. Prog. Retin. Eye Res. 2000, 19, 205–221. [Google Scholar] [CrossRef]
- Keenan, T.D.L.; Pickford, C.E.; Holley, R.J.; Clark, S.J.; Lin, W.; Dowsey, A.W.; Merry, C.L.; Day, A.J.; Bishop, P.N. Age-dependent changes in heparan sulfate in human Bruch’s membrane: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5370–5379. [Google Scholar] [CrossRef]
- Clark, S.J.; Keenan, T.D.L.; Fielder, H.L.; Collinson, L.J.; Holley, R.J.; Merry, C.L.R.; van Kuppevelt, T.H.; Day, A.J.; Bishop, P.N. Mapping the differential distribution of glycosaminoglycans in the adult human retina, choroid, and sclera. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6511–6521. [Google Scholar] [CrossRef]
- Keenan, T.D.L.; Clark, S.J.; Unwin, R.D.; Ridge, L.A.; Day, A.J.; Bishop, P.N. Mapping the differential distribution of proteoglycan core proteins in the adult human retina, choroid, and sclera. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7528–7538. [Google Scholar] [CrossRef]
- Feyzi, E.; Saldeen, T.; Larsson, E.; Lindahl, U.; Salmivirta, M. Age-dependent Modulation of Heparan Sulfate Structure and Function. J. Biol. Chem. 1998, 273, 13395–13398. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.R.; Gallo, R.L. Glycosaminoglycans and their proteoglycans: Host-Associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2006, 20, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451. [Google Scholar] [CrossRef]
- Yehoshua, Z.; de Amorim Garcia Filho, C.A.; Nunes, R.P.; Gregori, G.; Penha, F.M.; Moshfeghi, A.A.; Zhang, K.; Sadda, S.; Feuer, W.; Rosenfeld, P.J. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: The COMPLETE study. Ophthalmology 2014, 121, 693–701. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, S.J.; Bishop, P.N. Role of Factor H and Related Proteins in Regulating Complement Activation in the Macula, and Relevance to Age-Related Macular Degeneration. J. Clin. Med. 2015, 4, 18-31. https://doi.org/10.3390/jcm4010018
Clark SJ, Bishop PN. Role of Factor H and Related Proteins in Regulating Complement Activation in the Macula, and Relevance to Age-Related Macular Degeneration. Journal of Clinical Medicine. 2015; 4(1):18-31. https://doi.org/10.3390/jcm4010018
Chicago/Turabian StyleClark, Simon J., and Paul N. Bishop. 2015. "Role of Factor H and Related Proteins in Regulating Complement Activation in the Macula, and Relevance to Age-Related Macular Degeneration" Journal of Clinical Medicine 4, no. 1: 18-31. https://doi.org/10.3390/jcm4010018
APA StyleClark, S. J., & Bishop, P. N. (2015). Role of Factor H and Related Proteins in Regulating Complement Activation in the Macula, and Relevance to Age-Related Macular Degeneration. Journal of Clinical Medicine, 4(1), 18-31. https://doi.org/10.3390/jcm4010018