iPS Cells for Modelling and Treatment of Retinal Diseases

 ,
, 
Abstract
:
1. Introduction
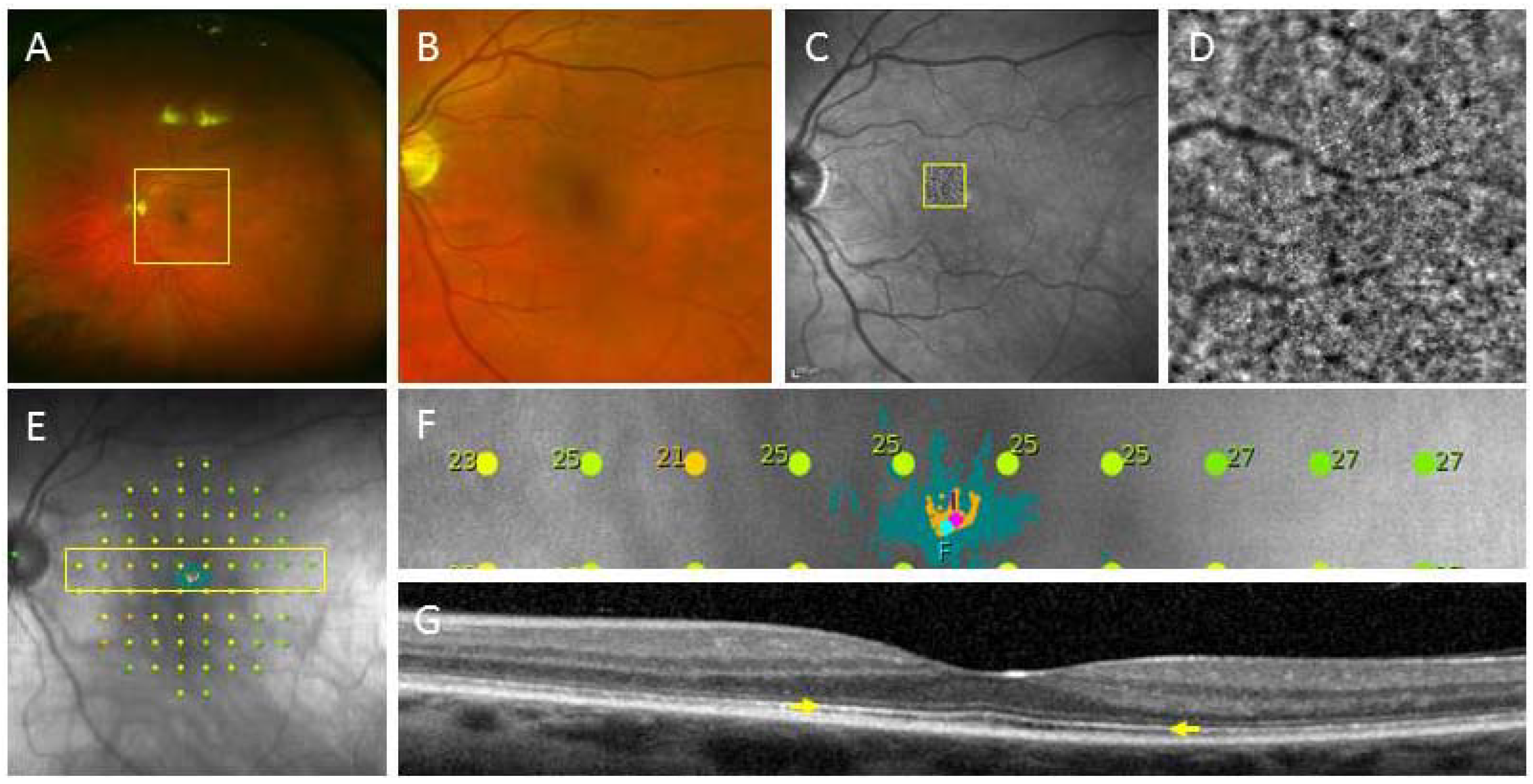
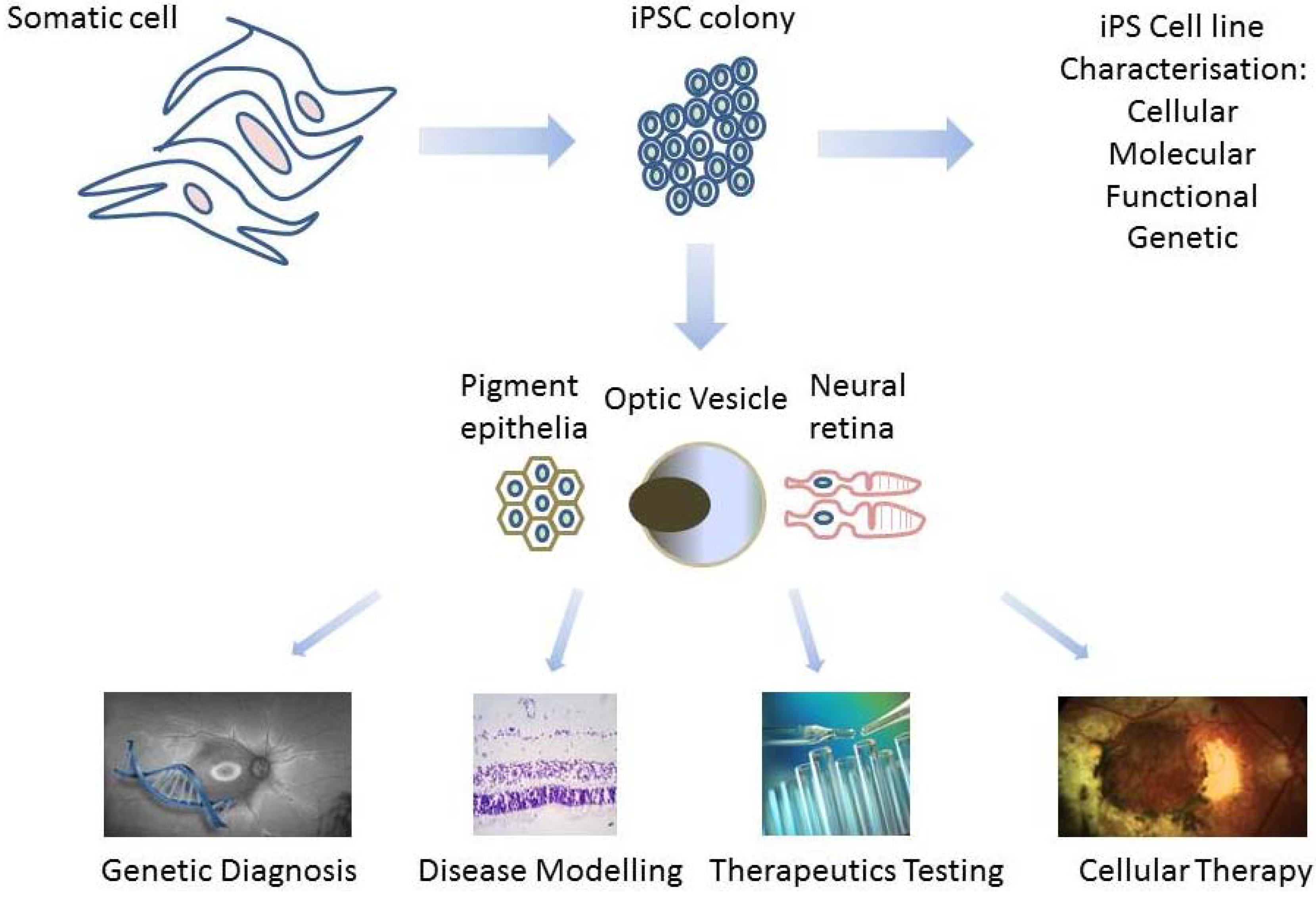
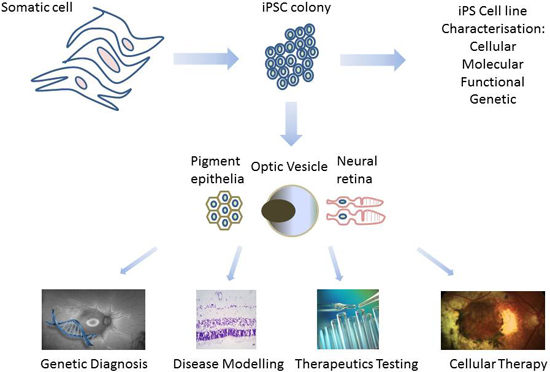
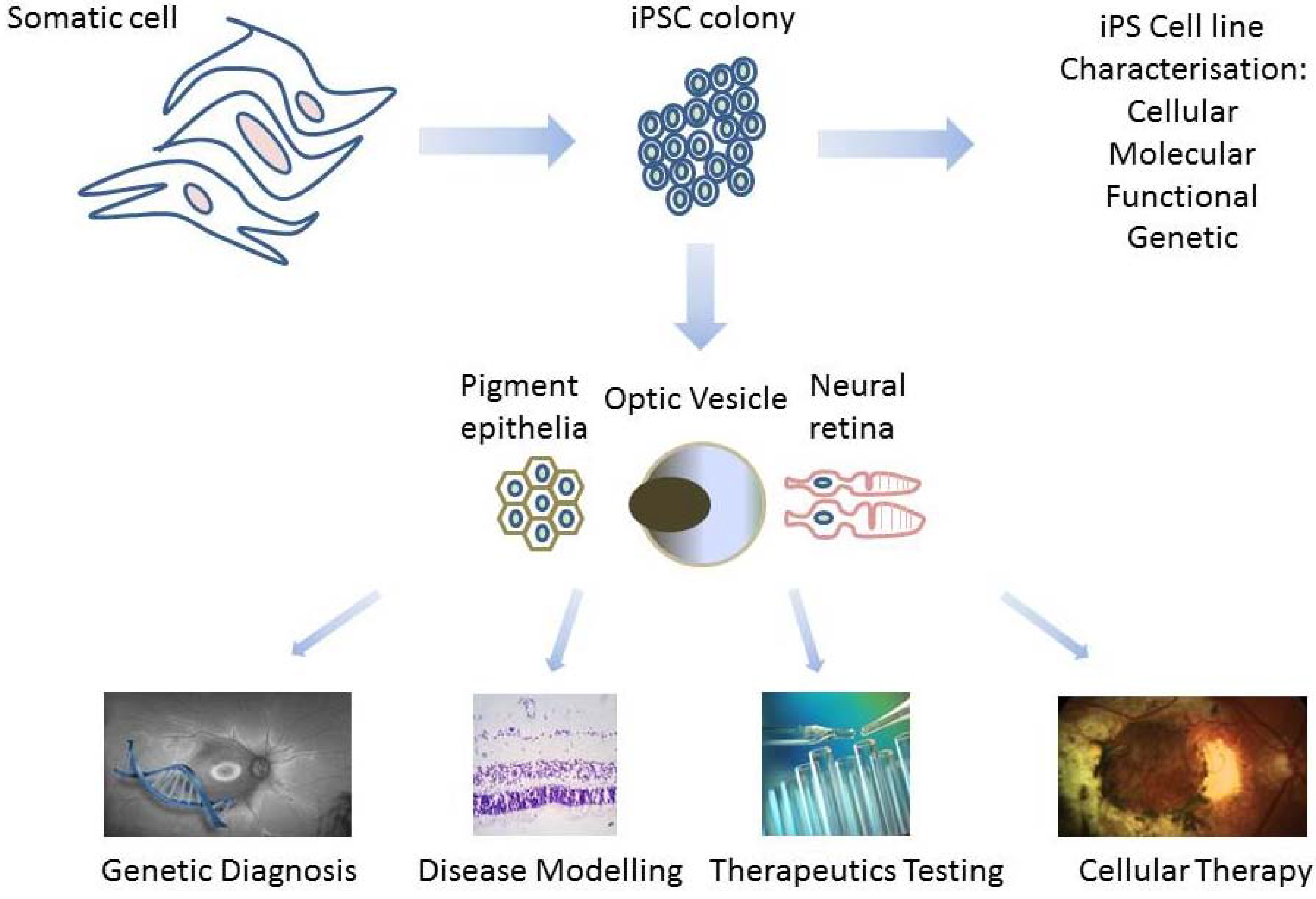
2. Derivation of Patient-Specific Retinal Cells from iPSC for Clinical Use
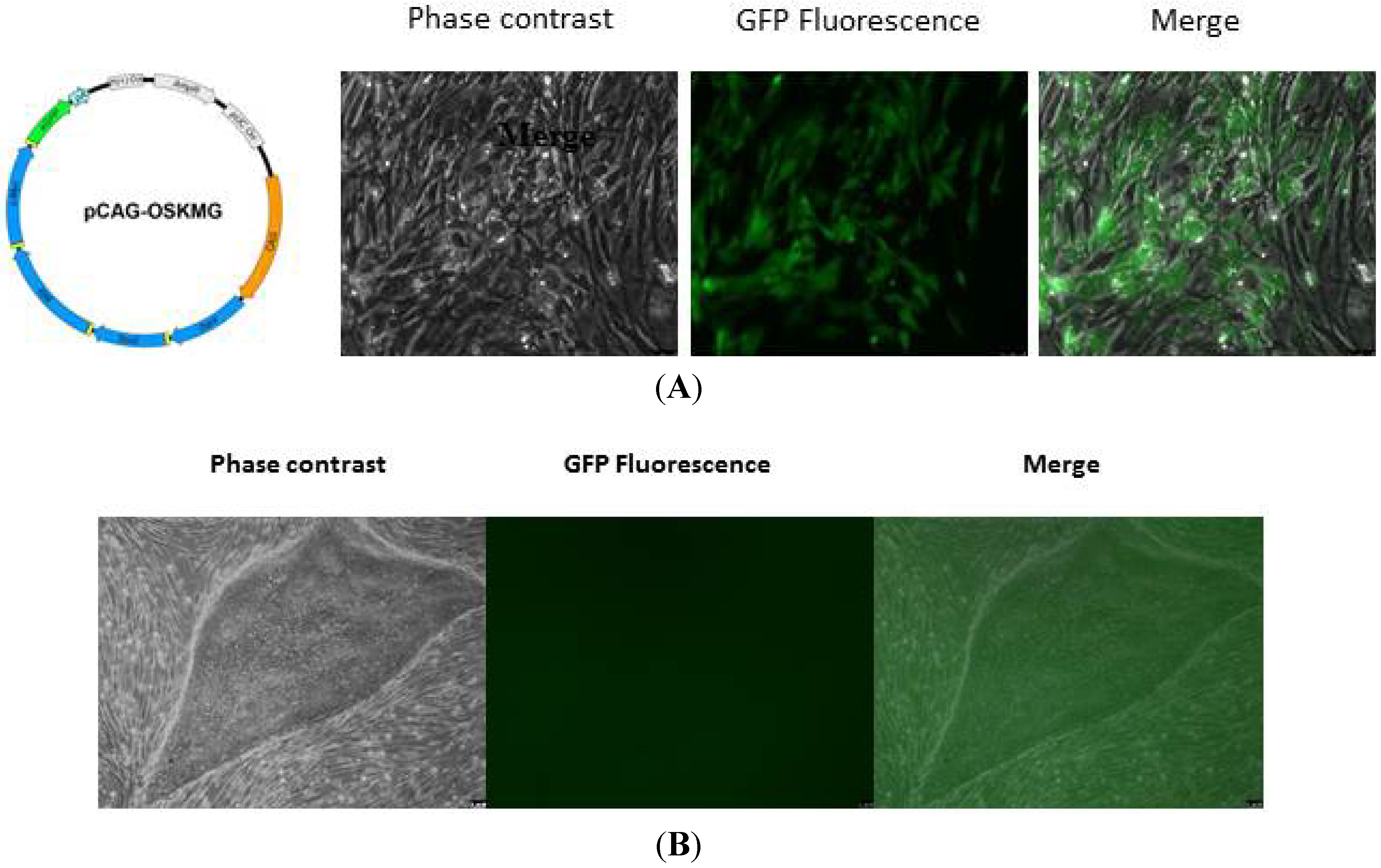
2.1. Creating iPSC from Patients
2.1.1. Using Pluripotent Stem Cells
2.1.2. Induced Pluripotent Stem Cells
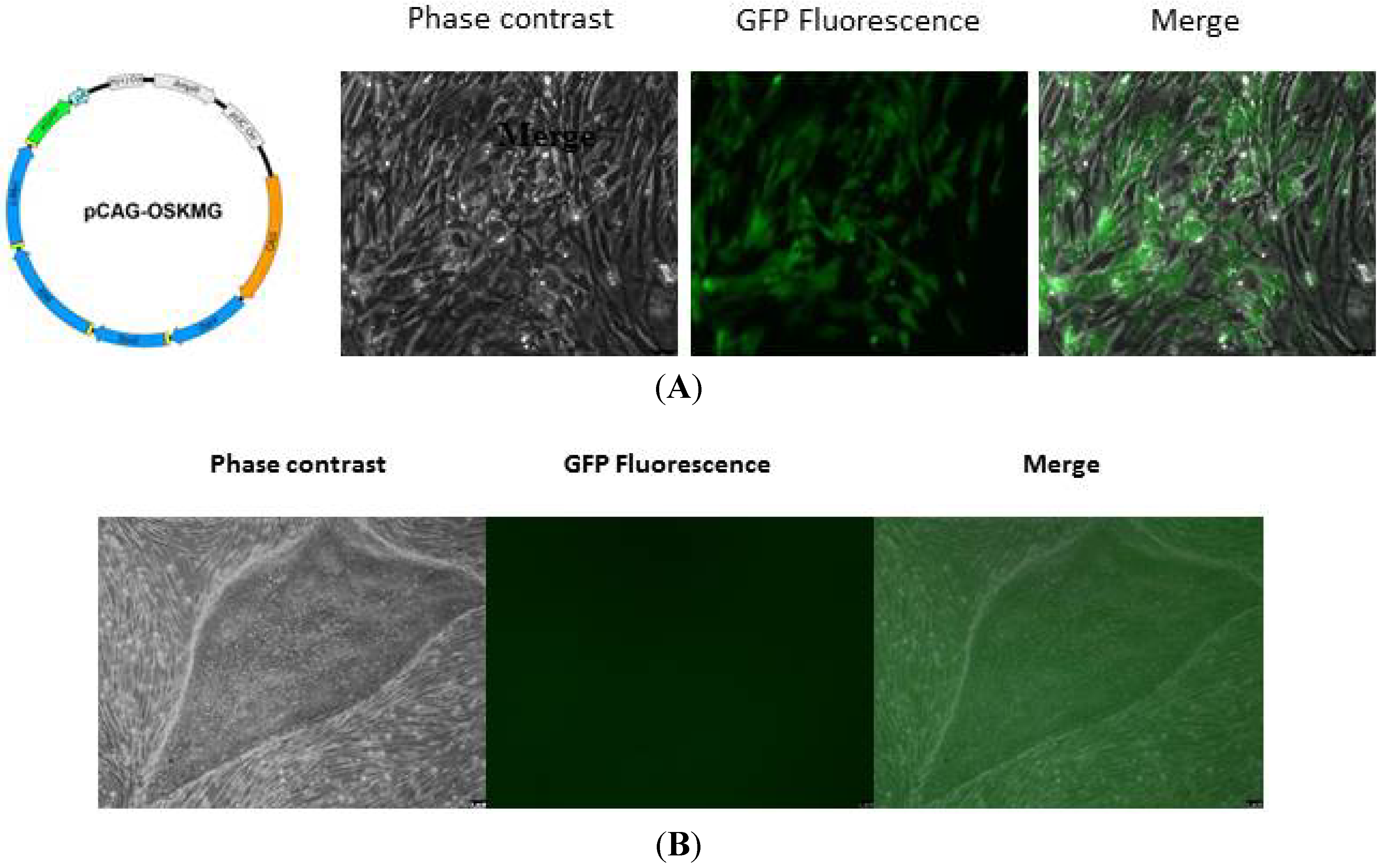
2.1.3. Validation of Human iPSC Lines
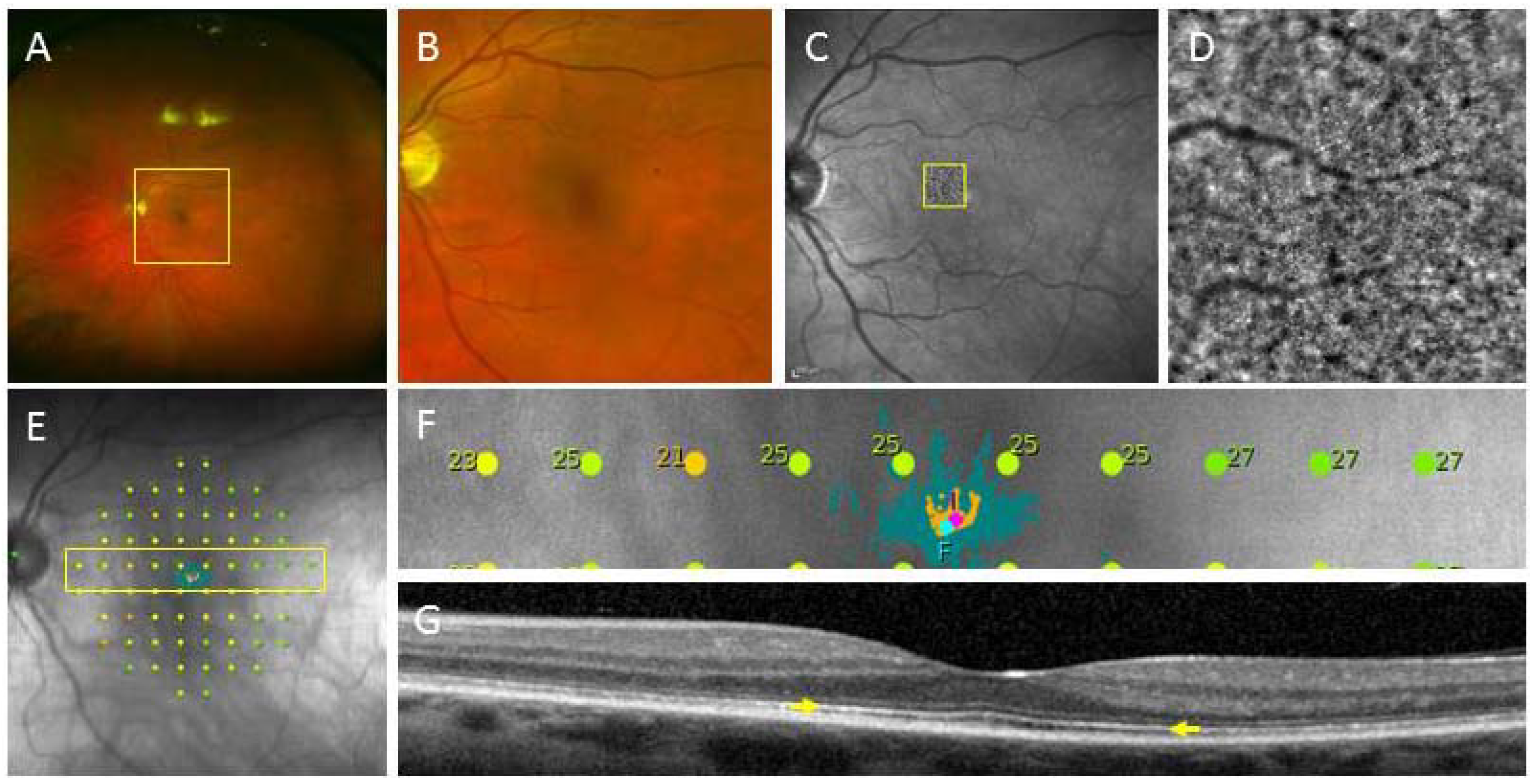
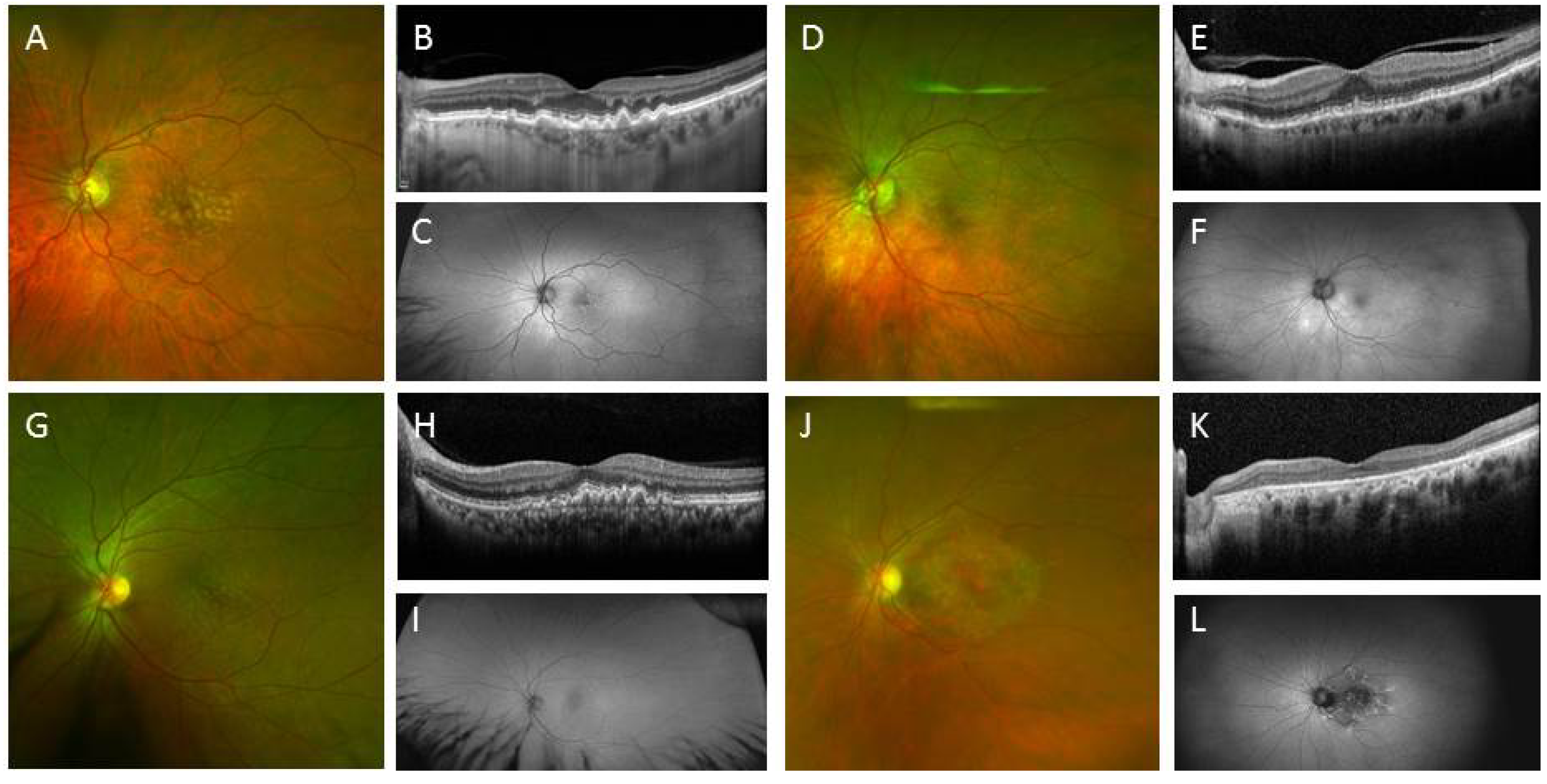
2.2. Creating Retinal Tissue from iPSC
2.2.1. Derivation of Retina Lineages

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Techniques of Characterisation | Induced Pluripotent Stem Cells | Photoreceptor Cells | Retinal Pigment Epithelium |
|---|---|---|---|
| Morphology (light microscopy) | Flat colonies; small and round cells; high nuclear to cytoplasmic ratio | Located in outer nuclear layer; cell bodies with processes; inner and outer segments | Monolayer; pigmentation; hexagonal |
| Morphology (electron microscopy) | N/A | Outer segment discs, myoid and ellipsoid segments, connecting cilia, basal body | Apical microvilli, basal infoldings, tight-junctional complexes, pigment granules |
| Cellular markers (pluripotency) | Surface: SSEA-3, TRA-1-60, TRA-1-81; Others: NANOG, SOX2, OCT4 | Loss of OCT3/4, SOX2, NANOG | Loss of OCT3/4, SOX2, NANOG |
| Cellular markers (progenitors/precursors) | N/A | PAX6, CHX10, CRX, OTX2, NRL | PAX6, MITF |
| Cellular markers (differentiated/mature) | N/A | Phototransduction: recoverin, transducing, cGMP phosphodiesterase, retinal guanylate cyclase, cyclic-nucleotide gated channel, rhodopsin, cone opsins (S or L/M), arrestin; visual cycle | Visual cycle: RPE65, RLBP1, CRALBP; phagocytosis: FAK, MERTK; pigmentation: tyrosinase; growth factor: VEGF, PEDF, PDGF; membrane: Na/K ATPase, ZO-1, BEST1 |
| Molecular | RT-PCR, bisulphite sequence analysis | RT-PCR | RT-PCR |
| Functional (in vitro) | Embryoid body formation | Patch recordings; response to white flash | Phagocytosis assay/rhodopsin clearance; fluid transport, polarised secretion of growth factors (PEGF/VEGF); transepithelial resistance |
| Functional (in vivo) | Teratoma assay in animal to identify all three germ layers | Cell transplantation to demonstrate rescue of visual function | Cell transplantation (RCS rat) to demonstrate rescue of visual function |
| Genetic | Karyotyping sequencing to look for new mutations | Sequencing to check no new mutations | Sequencing to check no new mutations |
2.2.2. iPSC to Photoreceptor Cells
| Reference | Source of iPSC | Duration | Markers to Confirm Photoreceptor Lineage | Tests to Suggest Photoreceptor Cell Function | Transplant | Disease Modelling | Therapeutics Screening |
|---|---|---|---|---|---|---|---|
| Hirami et al. [58] | Human fibroblast | 120 days | CRX, RCVRN, RHO | No | No | No | No |
| Osakada et al. [59] | Human fibroblast | 120–140 days | CRX, PDC, PDE6b, PDE6c, RHO, GRK1, SAG, RCVRN | Molecules required for photo-transduction | No | No | No |
| Jin et al. [7] | Patient fibroblast | 120 days | CRX, RCVRN, RHO, OPN1SW, OPN1LW | Patch clamp to detect voltage dependent channels 8-OHdG, caspase-3, acrolein, BiP, CHOP | No | Yes | Yes |
| Jin et al. [53] | Patient fibroblast * | 120–150 days | CRX, RCVRN | BiP, CHOP | No | Yes | No |
| Meyer et al. [51] | Human fibroblast | 80 days | CRX, RCVRN, Opsin | No | No | No | No |
| Meyer et al. [6] | Patient fibroblast | 80 days | CRX, RCVRN | No | No | No | No |
| Phillips et al. [36] | Patient T-cells | 108 days | CRX, RCVRN, S-OPSIN, RHO, CX36, SNAP-25, VGLUT1 | Molecules required for synaptic function | No | No | No |
| Phillips et al. [4] | Patient T-cells | 80 days | CRX, RCVRN, NRL, OPN1SW, PED6B | Molecules required for photo-transduction | No | Yes | No |
| Tucker et al. [3] | Patient fibroblast | 33 days | RCVRN | No | No | Yes | No |
| Tucker et al. [2] | Patient keratinocyte | 60 days | CRX, NRL, RCVRN, RHO, Acy Tubulin, OPN1SW, OPN1LW | GRP78, GRP94 | Yes | Yes | No |
| Burnight et al. [64] | Patient fibroblast | 90 days | CRX, RHO, OPN1SW, RCVRN, ROM1 | No | No | No | Yes |
| Tucker et al. [37] | Patient fibroblast, Human keratinocyte and IPE *,† | 90 days | CRX, NRL, RCVRN, RHO | No | No | No | No |
| Sridhar et al. [63] | Human fibroblast | 60 days | CRX, RCVRN | No | No | No | No |
| Mellough et al. [62] | Human fibroblast | 60 days | CRX, OPN1SW, OPN1LW, RHO, RCVRN, ARRESTIN 3 | No | No | No | No |
| Reichman et al. [54] | Human fibroblast | 49–112 days | CRX, NRL, RHO, R/G/B OPSIN, ARRESTIN 3, RECVRN | No | No | No | No |
| Zhong et al. [57] | Human fibroblast | 175 days | CRX, OPN1SW, OPN1LW, RHO, PDE6α/β, Gtα, CNGA1/B1, RetGC1 | Patch clamp-light induced response; outer segment disc formation on EM; molecules required for photo-transduction | No | No | No |
| Lambda et al. [52] | Human fibroblast | 28 days | CRX, OTX2, NRL, RECVRN, AIPL-1, RHO, S-Opsin, Arrestin, PAX6, Blimp1 | Molecules required for photo-transduction | No | No | No |
| Yoshida et al. [8] | Patient fibroblast | 35 days | NRL promoter, recoverin | BiP, CHOP, BID, NOXA LC3, ATG5, ATG7 | No | Yes | No |
2.2.3. iPSC to Retinal Pigmented Epithelial Cells

3. Clinical Use of Patient-Specific iPSC-Derived Retinal Cells
3.1. IPSC for Genetic Diagnosis and Modelling
3.1.1. Confirming Pathogenicity of Mutation
3.1.2. Modelling Developmental Diseases of the Retina
3.1.3. Modelling Degenerative Diseases of the Retina
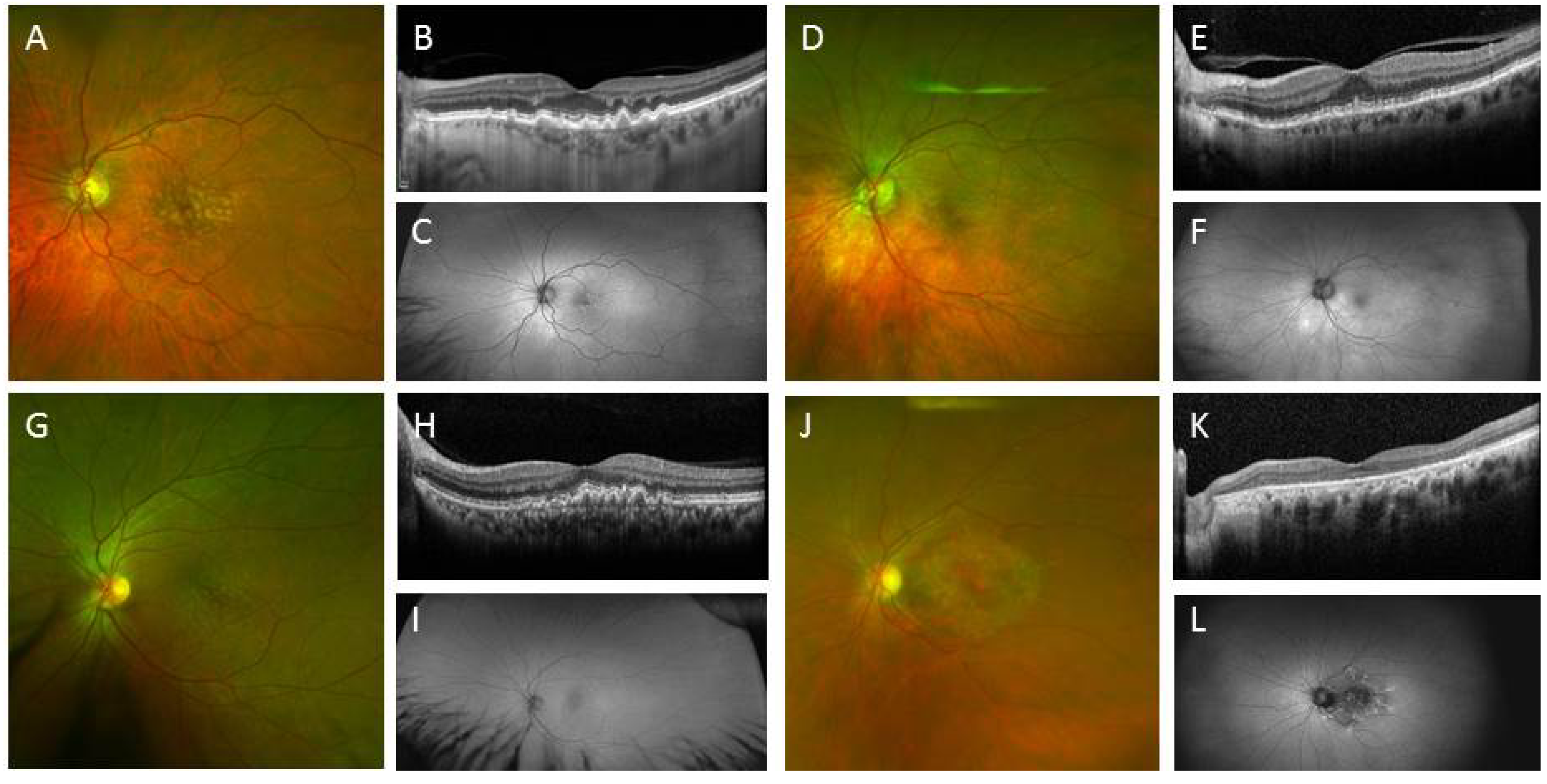
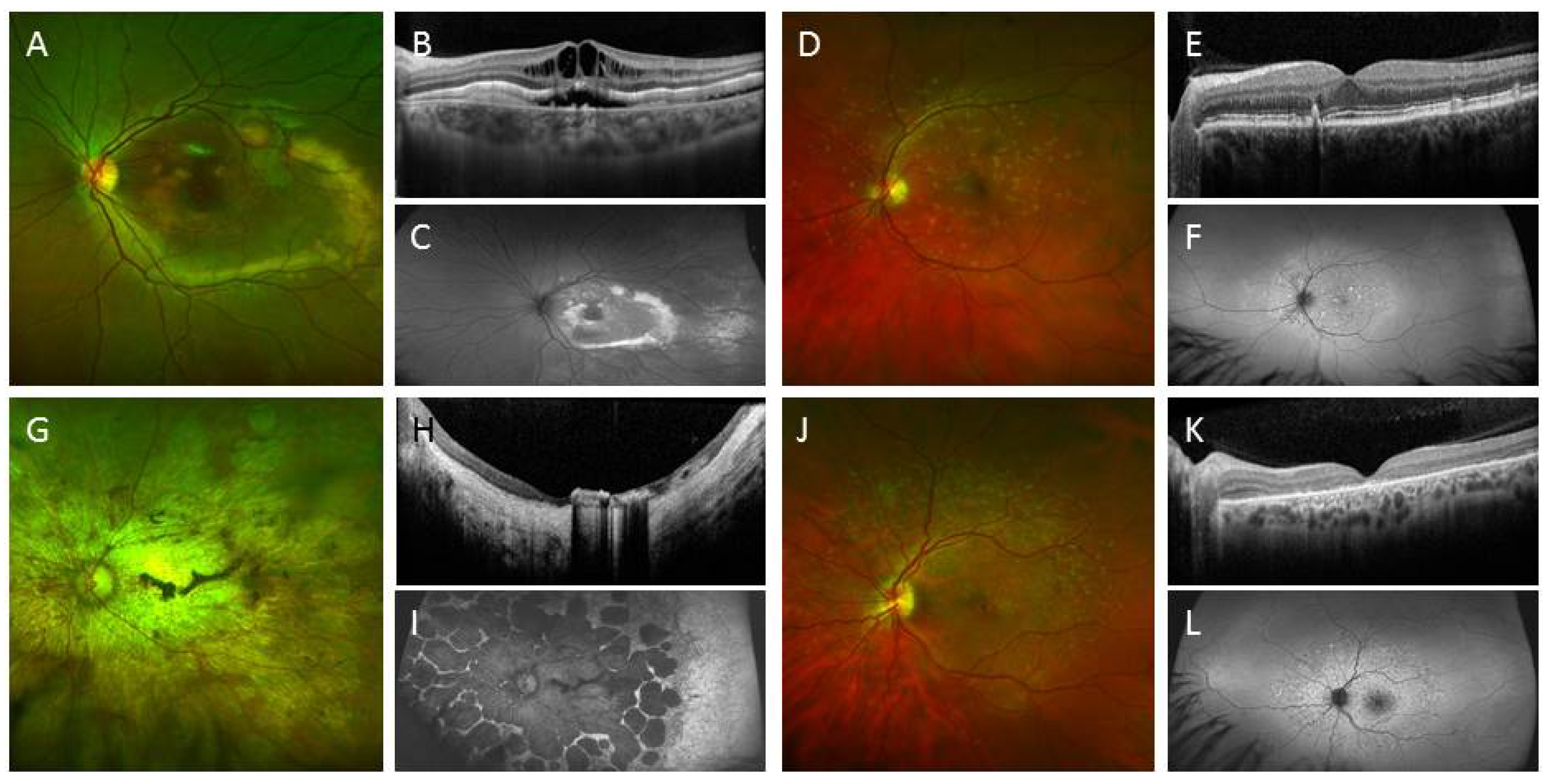
3.2. IPSC for Therapeutics Development and Treatment
3.2.1. IPSC for Drug Screening
3.2.2. IPSC for Testing Gene Therapy
3.2.3. iPSC for Cellular Therapy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ding, X.; Patel, M.; Chan, C.C. Molecular pathology of age-related macular degeneration. Prog. Retin. Eye Res. 2009, 28, 1–18. [Google Scholar] [PubMed]
- Tucker, B.A.; Mullins, R.F.; Streb, L.M.; Anfinson, K.; Eyestone, M.E.; Kaalberg, E.; Riker, M.J.; Drack, A.V.; Braun, T.A.; Stone, E.M. Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. eLife 2013, 2, e00824. [Google Scholar]
- Tucker, B.A.; Scheetz, T.E.; Mullins, R.F.; DeLuca, A.P.; Hoffmann, J.M.; Johnston, R.M.; Jacobson, S.G.; Sheffield, V.C.; Stone, E.M. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2011, 108. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Perez, E.T.; Martin, J.M.; Reshel, S.T.; Wallace, K.A.; Capowski, E.E.; Singh, R.; Wright, L.S.; Clark, E.M.; Barney, P.M.; et al. Modeling human retinal development with patient-specific induced pluripotent stem cells reveals multiple roles for visual system homeobox 2. Stem Cells 2014, 32, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Shen, W.; Kuai, D.; Martin, J.M.; Guo, X.; Smith, M.A.; Perez, E.T.; Phillips, M.J.; Simonett, J.M.; Wallace, K.A.; et al. IPS cell modeling of Best disease: Insights into the pathophysiology of an inherited macular degeneration. Hum. Mol. Genet. 2013, 22, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.S.; Howden, S.E.; Wallace, K.A.; Verhoeven, A.D.; Wright, L.S.; Capowski, E.E.; Pinilla, I.; Martin, J.M.; Tian, S.; Stewart, R.; et al. Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment. Stem Cells 2011, 29, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.B.; Okamoto, S.; Osakada, F.; Homma, K.; Assawachananont, J.; Hirami, Y.; Iwata, T.; Takahashi, M. Modeling retinal degeneration using patient-specific induced pluripotent stem cells. PLoS One 2011, 6, e17084. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ozawa, Y.; Suzuki, K.; Yuki, K.; Ohyama, M.; Akamatsu, W.; Matsuzaki, Y.; Shimmura, S.; Mitani, K.; Tsubota, K.; et al. The use of induced pluripotent stem cells to reveal pathogenic gene mutations and explore treatments for retinitis pigmentosa. Mol. Brain 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, L.; Coley, B.F.; Dorn, J.; Merlini, F.; Filley, E.; Christopher, P.; Chen, F.K.; Wuyyuru, V.; Sahel, J.; Stanga, P.; et al. The Argus II epiretinal prosthesis system allows letter and word reading and long-term function in patients with profound vision loss. Br. J. Ophthalmol. 2013, 97, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Zrenner, E.; Bartz-Schmidt, K.U.; Benav, H.; Besch, D.; Bruckmann, A.; Gabel, V.P.; Gekeler, F.; Greppmaier, U.; Harscher, A.; Kibbel, S.; et al. Subretinal electronic chips allow blind patients to read letters and combine them to words. Proc. Biol. Sci. R. Soc. 2011, 278, 1489–1497. [Google Scholar] [CrossRef]
- Villalobos, J.; Nayagam, D.A.; Allen, P.J.; McKelvie, P.; Luu, C.D.; Ayton, L.N.; Freemantle, A.L.; McPhedran, M.; Basa, M.; McGowan, C.C.; et al. A wide-field suprachoroidal retinal prosthesis is stable and well tolerated following chronic implantation. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3751–3762. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Hubschman, J.P.; Heilwell, G.; Franco-Cardenas, V.; Pan, C.K.; Ostrick, R.M.; Mickunas, E.; Gay, R.; Klimanskaya, I.; Lanza, R. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet 2012, 379, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Kauper, K.; McGovern, C.; Sherman, S.; Heatherton, P.; Rapoza, R.; Stabila, P.; Dean, B.; Lee, A.; Borges, S.; Bouchard, B.; et al. Two-year intraocular delivery of ciliary neurotrophic factor by encapsulated cell technology implants in patients with chronic retinal degenerative diseases. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7484–7491. [Google Scholar] [CrossRef]
- Carvalho, L.S.; Vandenberghe, L.H. Promising and delivering gene therapies for vision loss. Vis. Res. 2014. [Google Scholar] [CrossRef]
- Marc, R.; Pfeiffer, R.; Jones, B. Retinal Prosthetics, Optogenetics, and Chemical Photoswitches. ACS Chem. Neurosci. 2014, 5, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Mullins, R.F.; Stone, E.M. Stem cells for investigation and treatment of inherited retinal disease. Hum. Mol. Genet. 2014, 23. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.S.; Phillips, M.J.; Pinilla, I.; Hei, D.; Gamm, D.M. Induced pluripotent stem cells as custom therapeutics for retinal repair: Progress and rationale. Exp. Eye Res. 2014, 123, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Al-Shamekh, S.; Goldberg, J.L. Retinal repair with induced pluripotent stem cells. Trans. Res. J. Lab. Clin. Med. 2014, 163, 377–386. [Google Scholar] [CrossRef]
- Borooah, S.; Phillips, M.J.; Bilican, B.; Wright, A.F.; Wilmut, I.; Chandran, S.; Gamm, D.; Dhillon, B. Using human induced pluripotent stem cells to treat retinal disease. Prog. Retin. Eye Res. 2013, 37, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Davidson, K.C.; Guymer, R.H.; Pera, M.F.; Pebay, A. Human pluripotent stem cell strategies for age-related macular degeneration. Optom. Vis. Sci. 2014, 91, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Gamm, D.M.; Phillips, M.J.; Singh, R. Modeling retinal degenerative diseases with human iPS-derived cells: Current status and future implications. Exp. Rev. Ophthalmol. 2013, 8, 213–216. [Google Scholar] [CrossRef]
- Salero, E.; Blenkinsop, T.A.; Corneo, B.; Harris, A.; Rabin, D.; Stern, J.H.; Temple, S. Adult human RPE can be activated into a multipotent stem cell that produces mesenchymal derivatives. Cell Stem Cell 2012, 10, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Moshiri, A.; Close, J.; Reh, T.A. Retinal stem cells and regeneration. Int. J. Dev. Biol. 2004, 48, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Thomson, H.; Hossain, P.; Lotery, A. Characterisation of mouse limbal neurosphere cells: A potential cell source of functional neurons. Br. J. Ophthalmol. 2012, 96, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Das, A.V.; Bhattacharya, S.; Thoreson, W.B.; Sierra, J.R.; Mallya, K.B.; Ahmad, I. Derivation of neurons with functional properties from adult limbal epithelium: Implications in autologous cell therapy for photoreceptor degeneration. Stem Cells 2008, 26, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Das, A.V.; Thoreson, W.B.; James, J.; Wattnem, T.E.; Rodriguez-Sierra, J.; Ahmad, I. Adult corneal limbal epithelium: A model for studying neural potential of non-neural stem cells/progenitors. Dev. Biol. 2002, 250, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.K. Retinal stem cells and regeneration of vision system. Anat. Rec. 2014, 297, 137–160. [Google Scholar] [CrossRef]
- Pellegrini, G.; Rama, P.; di Rocco, A.; Panaras, A.; de Luca, M. Concise review: Hurdles in a successful example of limbal stem cell-based regenerative medicine. Stem Cells 2014, 32, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Miri, A.; Said, D.G.; Dua, H.S. Donor site complications in autolimbal and living-related allolimbal transplantation. Ophthalmology 2011, 118, 1265–1271. [Google Scholar] [PubMed]
- Wilmut, I.; Schnieke, A.E.; McWhir, J.; Kind, A.J.; Campbell, K.H. Viable offspring derived from fetal and adult mammalian cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.A.; Atienza, J.; Melton, D.A.; Eggan, K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science 2005, 309, 1369–1373. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S.; Blau, H.M. Nuclear reprogramming to a pluripotent state by three approaches. Nature 2010, 465, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Friedrich, A.M.; Johnson, L.V.; Clegg, D.O. Memory in induced pluripotent stem cells: Reprogrammed human retinal-pigmented epithelial cells show tendency for spontaneous redifferentiation. Stem Cells 2010, 28, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Wallace, K.A.; Dickerson, S.J.; Miller, M.J.; Verhoeven, A.D.; Martin, J.M.; Wright, L.S.; Shen, W.; Capowski, E.E.; Percin, E.F.; et al. Blood-derived human iPS cells generate optic vesicle-like structures with the capacity to form retinal laminae and develop synapses. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2007–2019. [Google Scholar] [CrossRef]
- Tucker, B.A.; Anfinson, K.R.; Mullins, R.F.; Stone, E.M.; Young, M.J. Use of a synthetic xeno-free culture substrate for induced pluripotent stem cell induction and retinal differentiation. Stem Cells Trans. Med. 2013, 2, 16–24. [Google Scholar] [CrossRef]
- Chang, Y.C.; Chang, W.C.; Hung, K.H.; Yang, D.M.; Cheng, Y.H.; Liao, Y.W.; Woung, L.C.; Tsai, C.Y.; Hsu, C.C.; Lin, T.C.; et al. The generation of induced pluripotent stem cells for macular degeneration as a drug screening platform: Identification of curcumin as a protective agent for retinal pigment epithelial cells against oxidative stress. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Babai, N.; Chaudhuri, A.; Qiu, F.; Bhattacharya, S.; Dave, B.J.; Parameswaran, S.; Carson, S.D.; Thoreson, W.B.; Sharp, J.G.; et al. Non cell-autonomous reprogramming of adult ocular progenitors: Generation of pluripotent stem cells without exogenous transcription factors. Stem Cells 2009, 27, 3053–3062. [Google Scholar] [PubMed]
- Sareen, D.; Saghizadeh, M.; Ornelas, L.; Winkler, M.A.; Narwani, K.; Sahabian, A.; Funari, V.A.; Tang, J.; Spurka, L.; Punj, V.; et al. Differentiation of Human Limbal-Derived Induced Pluripotent Stem Cells Into Limbal-Like Epithelium. Stem Cells Trans. Med. 2014, 3, 1002–1012. [Google Scholar] [CrossRef]
- Geti, I.; Ormiston, M.L.; Rouhani, F.; Toshner, M.; Movassagh, M.; Nichols, J.; Mansfield, W.; Southwood, M.; Bradley, A.; Rana, A.A.; et al. A practical and efficient cellular substrate for the generation of induced pluripotent stem cells from adults: Blood-derived endothelial progenitor cells. Stem Cells Trans. Med. 2012, 1, 855–865. [Google Scholar] [CrossRef]
- Seki, T.; Yuasa, S.; Fukuda, K. Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus. Nat. Protocols 2012, 7, 718–728. [Google Scholar] [CrossRef]
- Brown, M.E.; Rondon, E.; Rajesh, D.; Mack, A.; Lewis, R.; Feng, X.; Zitur, L.J.; Learish, R.D.; Nuwaysir, E.F. Derivation of induced pluripotent stem cells from human peripheral blood T lymphocytes. PLoS One 2010, 5, e11373. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chau, K.F.; Vodyanik, M.A.; Jiang, J.; Jiang, Y. Efficient feeder-free episomal reprogramming with small molecules. PLoS One 2011, 6, e17557. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ambasudhan, R.; Yuan, X.; Li, W.; Hilcove, S.; Abujarour, R.; Lin, X.; Hahm, H.S.; Hao, E.; Hayek, A.; et al. A chemical platform for improved induction of human iPSCs. Nat. Methods 2009, 6, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.S.; Shearer, R.L.; Capowski, E.E.; Wright, L.S.; Wallace, K.A.; McMillan, E.L.; Zhang, S.C.; Gamm, D.M. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 16698–16703. [Google Scholar] [CrossRef] [PubMed]
- Lamba, D.A.; McUsic, A.; Hirata, R.K.; Wang, P.R.; Russell, D.; Reh, T.A. Generation, purification and transplantation of photoreceptors derived from human induced pluripotent stem cells. PLoS One 2010, 5, e8763. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.B.; Okamoto, S.; Xiang, P.; Takahashi, M. Integration-free induced pluripotent stem cells derived from retinitis pigmentosa patient for disease modeling. Stem Cells Trans. Med. 2012, 1, 503–509. [Google Scholar] [CrossRef]
- Reichman, S.; Terray, A.; Slembrouck, A.; Nanteau, C.; Orieux, G.; Habeler, W.; Nandrot, E.F.; Sahel, J.A.; Monville, C.; Goureau, O. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc. Natl. Acad. Sci. USA. 2014, 111, 8518–8523. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Hirami, Y.; Osakada, F.; Takahashi, K.; Okita, K.; Yamanaka, S.; Ikeda, H.; Yoshimura, N.; Takahashi, M. Generation of retinal cells from mouse and human induced pluripotent stem cells. Neurosci. Lett. 2009, 458, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Osakada, F.; Jin, Z.B.; Hirami, Y.; Ikeda, H.; Danjyo, T.; Watanabe, K.; Sasai, Y.; Takahashi, M. In vitro differentiation of retinal cells from human pluripotent stem cells by small-molecule induction. J. Cell Sci. 2009, 122, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Lamba, D.A.; Karl, M.O.; Ware, C.B.; Reh, T.A. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2006, 103, 12769–12774. [Google Scholar] [CrossRef] [PubMed]
- Osakada, F.; Ikeda, H.; Mandai, M.; Wataya, T.; Watanabe, K.; Yoshimura, N.; Akaike, A.; Sasai, Y.; Takahashi, M. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat. Biotechnol. 2008, 26, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Mellough, C.B.; Sernagor, E.; Moreno-Gimeno, I.; Steel, D.H.; Lako, M. Efficient stage-specific differentiation of human pluripotent stem cells toward retinal photoreceptor cells. Stem Cells 2012, 30, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, A.; Steward, M.M.; Meyer, J.S. Nonxenogeneic growth and retinal differentiation of human induced pluripotent stem cells. Stem Cells Trans. Med. 2013, 2, 255–264. [Google Scholar] [CrossRef]
- Burnight, E.R.; Wiley, L.A.; Drack, A.V.; Braun, T.A.; Anfinson, K.R.; Kaalberg, E.E.; Halder, J.A.; Affatigato, L.M.; Mullins, R.F.; Stone, E.M.; et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. 2014, 21, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.J.; Vugler, A.A.; Hikita, S.T.; Lawrence, J.M.; Gias, C.; Chen, L.L.; Buchholz, D.E.; Ahmado, A.; Semo, M.; Smart, M.J.; et al. Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS One 2009, 4, e8152. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, D.E.; Hikita, S.T.; Rowland, T.J.; Friedrich, A.M.; Hinman, C.R.; Johnson, L.V.; Clegg, D.O. Derivation of functional retinal pigmented epithelium from induced pluripotent stem cells. Stem Cells 2009, 27, 2427–2434. [Google Scholar] [CrossRef] [PubMed]
- Maruotti, J.; Wahlin, K.; Gorrell, D.; Bhutto, I.; Lutty, G.; Zack, D.J. A simple and scalable process for the differentiation of retinal pigment epithelium from human pluripotent stem cells. Stem Cells Trans. Med. 2013, 2, 341–354. [Google Scholar] [CrossRef]
- Maeda, T.; Lee, M.J.; Palczewska, G.; Marsili, S.; Tesar, P.J.; Palczewski, K.; Takahashi, M.; Maeda, A. Retinal pigmented epithelial cells obtained from human induced pluripotent stem cells possess functional visual cycle enzymes in vitro and in vivo. J. Biol. Chem. 2013, 288, 34484–34493. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tsai, Y.T.; Hsu, C.W.; Erol, D.; Yang, J.; Wu, W.H.; Davis, R.J.; Egli, D.; Tsang, S.H. Long-term safety and efficacy of human-induced pluripotent stem cell (iPS) grafts in a preclinical model of retinitis pigmentosa. Mol. Med. 2012, 18, 1312–1319. [Google Scholar] [PubMed]
- Kamao, H.; Mandai, M.; Okamoto, S.; Sakai, N.; Suga, A.; Sugita, S.; Kiryu, J.; Takahashi, M. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Rep. 2014, 2, 205–218. [Google Scholar] [CrossRef]
- Kanemura, H.; Go, M.J.; Shikamura, M.; Nishishita, N.; Sakai, N.; Kamao, H.; Mandai, M.; Morinaga, C.; Takahashi, M.; Kawamata, S. Tumorigenicity studies of induced pluripotent stem cell (iPSC)-derived retinal pigment epithelium (RPE) for the treatment of age-related macular degeneration. PLoS One 2014, 9, e85336. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Cai, W.J.; Huang, X.F.; Liu, Q.; Lu, F.; Qu, J.; Wu, J.; Jin, Z.B. “RetinoGenetics”: A comprehensive mutation database for genes related to inherited retinal degeneration. Database J. Biol. Databases Curation 2014, 2014. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; High, K.A.; Auricchio, A.; Wright, J.F.; Pierce, E.A.; Testa, F.; Mingozzi, F.; Bennicelli, J.L.; Ying, G.S.; Rossi, S.; et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: A phase 1 dose-escalation trial. Lancet 2009, 374, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- MacLaren, R.E.; Groppe, M.; Barnard, A.R.; Cottriall, C.L.; Tolmachova, T.; Seymour, L.; Clark, K.R.; During, M.J.; Cremers, F.P.; Black, G.C.; et al. Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet 2014, 383, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Lustremant, C.; Habeler, W.; Plancheron, A.; Goureau, O.; Grenot, L.; de la Grange, P.; Audo, I.; Nandrot, E.F.; Monville, C. Human induced pluripotent stem cells as a tool to model a form of Leber congenital amaurosis. Cell. Reprogram. 2013, 15, 233–246. [Google Scholar] [PubMed]
- Howden, S.E.; Gore, A.; Li, Z.; Fung, H.L.; Nisler, B.S.; Nie, J.; Chen, G.; McIntosh, B.E.; Gulbranson, D.R.; Diol, N.R.; et al. Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy. Proc. Natl. Acad. Sci. USA. 2011, 108, 6537–6542. [Google Scholar] [CrossRef] [PubMed]
- Chong, N.H.; Alexander, R.A.; Gin, T.; Bird, A.C.; Luthert, P.J. TIMP-3, collagen, and elastin immunohistochemistry and histopathology of Sorsby’s fundus dystrophy. Investig. Ophthalmol. Vis. Sci. 2000, 41, 898–902. [Google Scholar]
- Chong, N.H.; Kvanta, A.; Seregard, S.; Bird, A.C.; Luthert, P.J.; Steen, B. TIMP-3 mRNA is not overexpressed in Sorsby fundus dystrophy. Am. J. Ophthalmol. 2003, 136, 954–955. [Google Scholar] [CrossRef] [PubMed]
- Roybal, C.N.; Marmorstein, L.Y.; Vander Jagt, D.L.; Abcouwer, S.F. Aberrant accumulation of fibulin-3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3973–3979. [Google Scholar] [CrossRef]
- Hayward, C.; Shu, X.; Cideciyan, A.V.; Lennon, A.; Barran, P.; Zareparsi, S.; Sawyer, L.; Hendry, G.; Dhillon, B.; Milam, A.H.; et al. Mutation in a short-chain collagen gene, CTRP5, results in extracellular deposit formation in late-onset retinal degeneration: A genetic model for age-related macular degeneration. Hum. Mol. Genet. 2003, 12, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Robson, A.G.; Holder, G.E.; Moore, A.T. The negative ERG: Clinical phenotypes and disease mechanisms of inner retinal dysfunction. Surv. Ophthalmol. 2008, 53, 16–40. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Kohl, S.; Leroy, B.P.; Munier, F.L.; Guillonneau, X.; Mohand-Said, S.; Bujakowska, K.; Nandrot, E.F.; Lorenz, B.; Preising, M.; et al. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2009, 85, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Vasireddy, V.; Mills, J.A.; Gaddameedi, R.; Basner-Tschakarjan, E.; Kohnke, M.; Black, A.D.; Alexandrov, K.; Zhou, S.; Maguire, A.M.; Chung, D.C.; et al. AAV-mediated gene therapy for choroideremia: Preclinical studies in personalized models. PLoS One 2013, 8, e61396. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Japanese woman is first recipient of next-generation stem cells. Nature 2014. [Google Scholar] [CrossRef]
- Kanemura, H.; Go, M.J.; Nishishita, N.; Sakai, N.; Kamao, H.; Sato, Y.; Takahashi, M.; Kawamata, S. Pigment epithelium-derived factor secreted from retinal pigment epithelium facilitates apoptotic cell death of iPSC. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Heller, J.P.; Martin, K.R. Enhancing RPE Cell-Based Therapy Outcomes for AMD: The Role of Bruch’s Membrane. Trans. Vis. Sci. Technol. 2014, 3. [Google Scholar] [CrossRef]
- Leveillard, T.; Fridlich, R.; Clerin, E.; Ait-Ali, N.; Millet-Puel, G.; Jaillard, C.; Yang, Y.; Zack, D.; van-Dorsselaer, A.; Sahel, J.A. Therapeutic strategy for handling inherited retinal degenerations in a gene-independent manner using rod-derived cone viability factors. C. R. Biol. 2014, 337, 207–213. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.K.; McLenachan, S.; Edel, M.; Da Cruz, L.; Coffey, P.J.; Mackey, D.A. iPS Cells for Modelling and Treatment of Retinal Diseases. J. Clin. Med. 2014, 3, 1511-1541. https://doi.org/10.3390/jcm3041511
Chen FK, McLenachan S, Edel M, Da Cruz L, Coffey PJ, Mackey DA. iPS Cells for Modelling and Treatment of Retinal Diseases. Journal of Clinical Medicine. 2014; 3(4):1511-1541. https://doi.org/10.3390/jcm3041511
Chicago/Turabian StyleChen, Fred K., Samuel McLenachan, Michael Edel, Lyndon Da Cruz, Peter J. Coffey, and David A. Mackey. 2014. "iPS Cells for Modelling and Treatment of Retinal Diseases" Journal of Clinical Medicine 3, no. 4: 1511-1541. https://doi.org/10.3390/jcm3041511
APA StyleChen, F. K., McLenachan, S., Edel, M., Da Cruz, L., Coffey, P. J., & Mackey, D. A. (2014). iPS Cells for Modelling and Treatment of Retinal Diseases. Journal of Clinical Medicine, 3(4), 1511-1541. https://doi.org/10.3390/jcm3041511