Clinical Potentials of Cardiomyocytes Derived from Patient-Specific Induced Pluripotent Stem Cells

Abstract
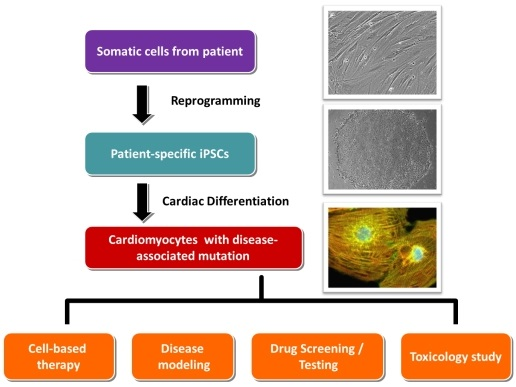
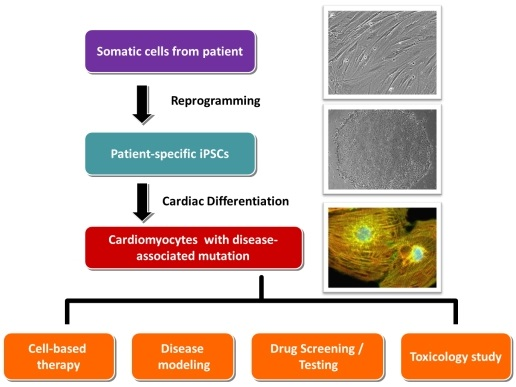
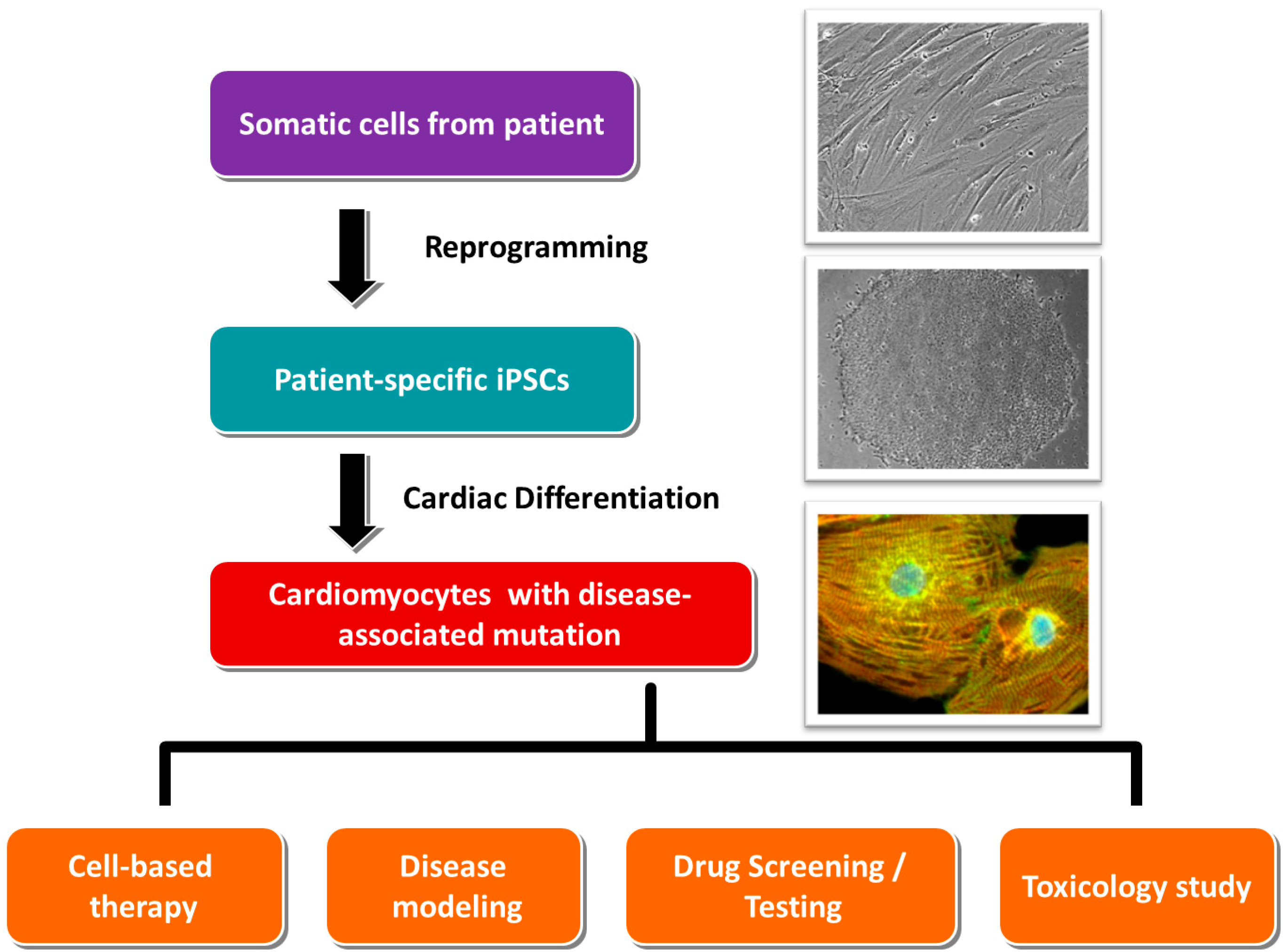
:
1. Introduction
2. Patient-Specific iPSCs and Their Cardiac Derivatives
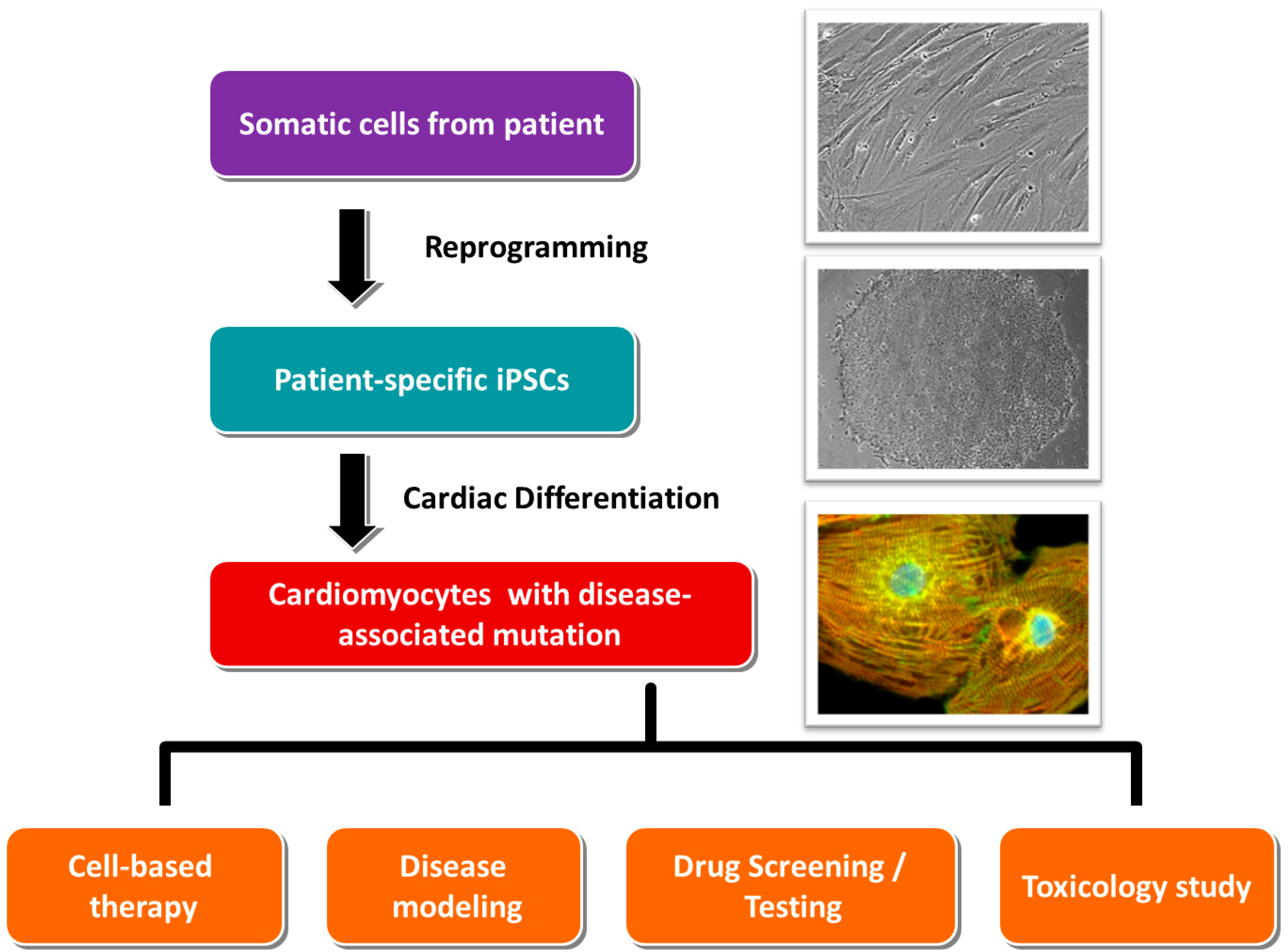
3. Standard Procedures in Generating Patient-Specific iPSCs and Their Cardiac Derivatives
3.1. Collection of Somatic Tissues/Cells
3.2. Reprogramming
3.3. Cardiac Differentiation
4. Application of Patient-Specific iPSCs in Cell Replacement Therapy/Regenerative Medicine

{kind=link}
{kind=link}
| Cell Type | Animal Model | Number of Cell | Delivery Method | Timing of the Delivery | Follow up Duration | Reference |
|---|---|---|---|---|---|---|
| iPSC | Mouse | 50,000 | IM | Immediately after MI induction | 2 weeks | [30] |
| iPSC-derived cardiac progenitors | Rat | 2 × 106 | IM | 10 min after MI induction | 10 weeks | [31] |
| Cardiosphere | Rat | - | Cell sheet | Immediately after MI induction | 3 weeks | [32] |
5. Applications of iPSCs-Derived Cardiomyocytes in Modeling Genetic Cardiomyopathies
| Disorder | Gene Involved | Details of the Mutation | References |
|---|---|---|---|
| Long QT syndrome, Type 1 | KCNQ1 | missense mutation (R190Q) leads to the production of a mutant protein | [41] |
| Long QT syndrome, Type 2 | KCNH2 | missense mutation (A614V) leads to the production of a mutant protein | [38] |
| Long QT syndrome, Type 2 | KCNH2 | missense mutation (G1618A) leads to the production of a mutant protein | [40] |
| Long QT syndrome, Type 2 | KCNH2 | missense mutation (R176W) leads to the production of mutant protein | [42] |
| Long QT syndrome, Type 3 | SCN5A | Multiple mutations (G5287A; V1763M) leads the production of a mutant protein | [43] |
| Long QT syndrome, Type 8 | CACNA1C | Missense mutation (G406R) leads to the production of a mutant protein | [44] |
| Catecholaminergic polymorphic ventricular tachycardia, Type 1 | RYR2 | Missense mutation (F2483I) leads to the production of a mutant protein with an altered FKBP12.6 binding domain | [45] |
| Catecholaminergic polymorphic ventricular tachycardia, Type 1 | RYR2 | Missense mutation (S406L) leads to the production of a mutant protein | [46] |
| Catecholaminergic polymorphic ventricular tachycardia, Type 2 | CASQ2 | Missense mutation (D307H) leads to the production of a mutant protein | [47] |
| Catecholaminergic polymorphic ventricular tachycardia, Type 2 | CASQ2 | Missense mutation (D307H) leads to the production of a mutant protein | [47] |
| Dilated cardiomyopathy | TNNT2 | missense mutation (R173W) leads to the production of a mutant protein | [48] |
| Dilated cardiomyopathy | DES | missense mutation (A285V) leads to the production of a mutant protein | [49] |
| Hypertrophic cardiomyopathy | MYH7 | Missense mutation (R663H) leads to the production of a mutant protein | [50] |
| Friedreich ataxia-associated hypertrophic cardiomyopathy | FXN | GAA repeat expansion in the first intron leads to the partial silencing of gene expression | [51] |
5.1. Modeling Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
5.2. Modeling Dilated Cardiomyopathy Associated with TNNT2 Mutation
5.3. Modeling Cardiomyopathy Associated with DES Mutation
5.4. Modeling Hypertrophic Cardiomyopathy Associated with MYH7 Mutation
5.5. Modeling Friedreich Ataxia Associated Cardiomyopathy
6. Application of Patient-Specific iPSCs-Derived Cardiomyocytes in Efficacy Testing and Drug Screening
7. Application of Patient-Specific iPSCs-Derived Cardiomyocytes in Toxicology Test
8. Limitations of iPSCs
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [PubMed]
- Goktepe, S.; Abilez, O.J.; Parker, K.K.; Kuhl, E. A multiscale model for eccentric and concentric cardiac growth through sarcomerogenesis. J. Theor. Biol. 2010, 265, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, N.; Jeon, I.; Lee, H.J.; Do, J.T.; Lee, D.R.; Oh, S.H.; Shin, D.A.; Kim, A.; Song, J. Neuronal differentiation of a human induced pluripotent stem cell line (FS-1) derived from newborn foreskin fibroblasts. Int. J. Stem. Cells 2012, 5, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Kattman, S.J.; Witty, A.D.; Gagliardi, M.; Dubois, N.C.; Niapour, M.; Hotta, A.; Ellis, J.; Keller, G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 2011, 8, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Shtrichman, R.; Germanguz, I.; Segev, H.; Zeevi-Levin, N.; Fishman, B.; Mandel, Y.E.; Barad, L.; Domev, H.; Kotton, D.; et al. Enhanced reprogramming and cardiac differentiation of human keratinocytes derived from plucked hair follicles, using a single excisable lentivirus. Cell. Reprogram. 2010, 12, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Merling, R.K.; Sweeney, C.L.; Choi, U.; De Ravin, S.S.; Myers, T.G.; Otaizo-Carrasquero, F.; Pan, J.; Linton, G.; Chen, L.; Koontz, S.; et al. Transgene-free ipscs generated from small volume peripheral blood nonmobilized CD34+ cells. Blood 2013, 121. [Google Scholar] [CrossRef] [PubMed]
- Churko, J.M.; Burridge, P.W.; Wu, J.C. Generation of human ipscs from human peripheral blood mononuclear cells using non-integrative sendai virus in chemically defined conditions. Methods Mol. Biol. 2013, 1036, 81–88. [Google Scholar] [PubMed]
- Wang, Y.; Liu, J.; Tan, X.; Li, G.; Gao, Y.; Liu, X.; Zhang, L.; Li, Y. Induced pluripotent stem cells from human hair follicle mesenchymal stem cells. Stem Cell Rev. 2013, 9, 451–460. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, B.A.; van Baaren, J.M.; Dubey, G.K.; Lee, J.M.; Cuccaro, M.L.; Vance, J.M.; Pericak-Vance, M.A.; Dykxhoorn, D.M. Derivation of autism spectrum disorder-specific induced pluripotent stem cells from peripheral blood mononuclear cells. Neurosci. Lett. 2012, 516, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Gianotti-Sommer, A.; Rozelle, S.S.; Sullivan, S.; Mills, J.A.; Park, S.M.; Smith, B.W.; Iyer, A.M.; French, D.L.; Kotton, D.N.; Gadue, P.; et al. Generation of human induced pluripotent stem cells from peripheral blood using the stemcca lentiviral vector. J. Vis. Exp. 2008, 68. [Google Scholar] [CrossRef]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, X.; Fan, W.; Zhao, P.; Chan, Y.C.; Chen, S.; Zhang, S.; Guo, X.; Zhang, Y.; Li, Y.; et al. Modeling abnormal early development with induced pluripotent stem cells from aneuploid syndromes. Human Mol. Genet. 2012, 21, 32–45. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.A.; Stadtfeld, M.; Murphy, G.J.; Hochedlinger, K.; Kotton, D.N.; Mostoslavsky, G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells 2009, 27, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P.; Tomishima, M.J.; Chambers, S.M.; Mica, Y.; Reed, E.; Menon, J.; Tabar, V.; Mo, Q.; Studer, L.; Sadelain, M. Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human ipsc induction and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 12759–12764. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.A.; Sommer, A.G.; Longmire, T.A.; Christodoulou, C.; Thomas, D.D.; Gostissa, M.; Alt, F.W.; Murphy, G.J.; Kotton, D.N.; Mostoslavsky, G. Excision of reprogramming transgenes improves the differentiation potential of iPS cells generated with a single excisable vector. Stem Cells 2010, 28, 64–74. [Google Scholar] [PubMed]
- Somers, A.; Jean, J.C.; Sommer, C.A.; Omari, A.; Ford, C.C.; Mills, J.A.; Ying, L.; Sommer, A.G.; Jean, J.M.; Smith, B.W.; et al. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells 2010, 28, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on sendai virus, an rna virus that does not integrate into the host genome. Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar]
- Mummery, C.; Ward, D.; van den Brink, C.E.; Bird, S.D.; Doevendans, P.A.; Opthof, T.; Brutel de la Riviere, A.; Tertoolen, L.; van der Heyden, M.; Pera, M. Cardiomyocyte differentiation of mouse and human embryonic stem cells. J. Anat. 2002, 200, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Passier, R.; Oostwaard, D.W.; Snapper, J.; Kloots, J.; Hassink, R.J.; Kuijk, E.; Roelen, B.; de la Riviere, A.B.; Mummery, C. Increased cardiomyocyte differentiation from human embryonic stem cells in serum-free cultures. Stem Cells 2005, 23, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Soonpaa, M.H.; Adler, E.D.; Roepke, T.K.; Kattman, S.J.; Kennedy, M.; Henckaerts, E.; Bonham, K.; Abbott, G.W.; Linden, R.M.; et al. Human cardiovascular progenitor cells develop from a Kdr+ embryonic-stem-cell-derived population. Nature 2008, 453, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical wnt signaling. In Proceeding of the National Academy of Sciences of the United States of America, Cambridge, MA, USA; 2012. [Google Scholar]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Chen, K.Y.; Naumova, A.V.; Muskheli, V.; Fugate, J.A.; Dupras, S.K.; Reinecke, H.; Xu, C.; Hassanipour, M.; Police, S.; et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 2007, 25, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Van Laake, L.W.; Passier, R.; Monshouwer-Kloots, J.; Verkleij, A.J.; Lips, D.J.; Freund, C.; den Ouden, K.; Ward-van Oostwaard, D.; Korving, J.; Tertoolen, L.G.; et al. Human embryonic stem cell-derived cardiomyocytes survive and mature in the mouse heart and transiently improve function after myocardial infarction. Stem Cell Res. 2007, 1, 9–24. [Google Scholar]
- Zakharova, L.; Mastroeni, D.; Mutlu, N.; Molina, M.; Goldman, S.; Diethrich, E.; Gaballa, M.A. Transplantation of cardiac progenitor cell sheet onto infarcted heart promotes cardiogenesis and improves function. Cardiovasc. Res. 2010, 87, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, L.; Carr, C.; Yang, C.T.; Stuckey, D.J.; Clarke, K.; Watt, S.M. Efficient differentiation of human induced pluripotent stem cells generates cardiac cells that provide protection following myocardial infarction in the rat. Stem Cells Dev. 2012, 21, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Abdelli, L.S.; Singla, D.K. Transplanted induced pluripotent stem cells improve cardiac function and induce neovascularization in the infarcted hearts of db/db mice. Mol. Pharm. 2011, 8, 1602–1610. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart, A.; et al. Contemporary definitions and classification of the cardiomyopathies. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(v)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Chen, B.; Hund, T.J.; Koval, O.M.; Purohit, A.; Song, L.S.; Mohler, P.J.; Anderson, M.E. Proarrhythmic defects in timothy syndrome require calmodulin kinase ii. Circulation 2008, 118, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.P.; Yuan, C.; Navedo, M.F.; Dixon, R.E.; Nieves-Cintron, M.; Scott, J.D.; Santana, L.F. Restoration of normal l-type Ca2+ channel function during timothy syndrome by ablation of an anchoring protein. Circ. Res. 2011, 109, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Bader, P.L.; Faizi, M.; Kim, L.H.; Owen, S.F.; Tadross, M.R.; Alfa, R.W.; Bett, G.C.; Tsien, R.W.; Rasmusson, R.L.; Shamloo, M. Mouse model of timothy syndrome recapitulates triad of autistic traits. Proc. Natl. Acad. Sci. USA 2011, 108, 15432–15437. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long qt syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef]
- Yazawa, M.; Dolmetsch, R.E. Modeling timothy syndrome with iPS cells. J. Cardiovasc. Transl. Res. 2013, 6, 1–9. [Google Scholar] [CrossRef]
- Matsa, E.; Rajamohan, D.; Dick, E.; Young, L.; Mellor, I.; Staniforth, A.; Denning, C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur. Heart J. 2011, 32, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flugel, L.; Dorn, T.; Goedel, A.; Hohnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Lahti, A.L.; Kujala, V.J.; Chapman, H.; Koivisto, A.P.; Pekkanen-Mattila, M.; Kerkela, E.; Hyttinen, J.; Kontula, K.; Swan, H.; Conklin, B.R.; et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis. Model. Mech. 2012, 5, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wei, H.; Zhao, Y.; Lu, J.; Li, G.; Sahib, N.B.; Tan, T.H.; Wong, K.Y.; Shim, W.; Wong, P.; et al. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int. J. Cardiol. 2013, 168, 5277–5286. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using induced pluripotent stem cells to investigate cardiac phenotypes in timothy syndrome. Nature 2011, 471, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Fatima, A.; Xu, G.; Shao, K.; Papadopoulos, S.; Lehmann, M.; Arnaiz-Cot, J.J.; Rosa, A.O.; Nguemo, F.; Matzkies, M.; Dittmann, S.; et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell. Physiol. Biochem. 2011, 28, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.B.; Moretti, A.; Mederos y Schnitzler, M.; Iop, L.; Storch, U.; Bellin, M.; Dorn, T.; Ruppenthal, S.; Pfeiffer, S.; Goedel, A.; et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol. Med. 2012, 4, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Barad, L.; Zeevi-Levin, N.; Shick, R.; Shtrichman, R.; Lorber, A.; Itskovitz-Eldor, J.; Binah, O. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to beta-adrenergic stimulation. J. Cell. Mol. Med. 2012, 16, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130–147. [Google Scholar] [CrossRef]
- Tse, H.F.; Ho, J.C.; Choi, S.W.; Lee, Y.K.; Butler, A.W.; Ng, K.M.; Siu, C.W.; Simpson, M.A.; Lai, W.H.; Chan, Y.C.; et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Human Mol. Genet. 2013, 22, 1395–1403. [Google Scholar] [CrossRef]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [PubMed]
- Lee, Y.K.; Ho, P.W.; Schick, R.; Lau, Y.M.; Lai, W.H.; Zhou, T.; Li, Y.; Ng, K.M.; Ho, S.L.; Esteban, M.A.; et al. Modeling of friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells. Pflug. Arch.: Eur. J. Physiol. 2013, 466, 1831–1844. [Google Scholar] [CrossRef]
- Kontula, K.; Laitinen, P.J.; Lehtonen, A.; Toivonen, L.; Viitasalo, M.; Swan, H. Catecholaminergic polymorphic ventricular tachycardia: Recent mechanistic insights. Cardiovasc. Res. 2005, 67, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Ruan, Y.; Priori, S.G. Catecholaminergic polymorphic ventricular tachycardia. Prog. Cardiovasc. Dis. 2008, 51, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in bedouin families from israel. Am. J. Human Genet. 2001, 69, 1378–1384. [Google Scholar] [CrossRef]
- Viatchenko-Karpinski, S.; Terentyev, D.; Gyorke, I.; Terentyeva, R.; Volpe, P.; Priori, S.G.; Napolitano, C.; Nori, A.; Williams, S.C.; Gyorke, S. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ. Res. 2004, 94, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Nori, A.; Santoro, M.; Viatchenko-Karpinski, S.; Kubalova, Z.; Gyorke, I.; Terentyeva, R.; Vedamoorthyrao, S.; Blom, N.A.; Valle, G.; et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ. Res. 2006, 98, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Di Barletta, M.R.; Viatchenko-Karpinski, S.; Nori, A.; Memmi, M.; Terentyev, D.; Turcato, F.; Valle, G.; Rizzi, N.; Napolitano, C.; Gyorke, S.; et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation 2006, 114, 1012–1019. [Google Scholar]
- Cerrone, M.; Colombi, B.; Santoro, M.; di Barletta, M.R.; Scelsi, M.; Villani, L.; Napolitano, C.; Priori, S.G. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ. Res. 2005, 96. [Google Scholar] [CrossRef]
- Burkett, E.L.; Hershberger, R.E. Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Grunig, E.; Tasman, J.A.; Kucherer, H.; Franz, W.; Kubler, W.; Katus, H.A. Frequency and phenotypes of familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 1998, 31, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Goerss, J.B.; Michels, V.V.; Burnett, J.; Driscoll, D.J.; Miller, F.; Rodeheffer, R.; Tajik, A.J.; Schaid, D. Frequency of familial dilated cardiomyopathy. Eur. Heart J. 1995, 16 (Suppl. O), 2–4. [Google Scholar] [CrossRef] [PubMed]
- Mahon, N.G.; Murphy, R.T.; MacRae, C.A.; Caforio, A.L.; Elliott, P.M.; McKenna, W.J. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann. Intern. Med. 2005, 143, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Banerjee, S.K.; Lage, M.L.; Huang, X.N.; Smith, S.H.; Saba, S.; Rager, J.; Conner, D.A.; Janczewski, A.M.; Tobita, K.; et al. The role of cardiac troponin t quantity and function in cardiac development and dilated cardiomyopathy. PLoS One 2008, 3, e2642. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Bell, A.; Senthil, V.; Sidhu, J.; Noseda, M.; Roberts, R.; Marian, A.J. Differential interactions of thin filament proteins in two cardiac troponin T mouse models of hypertrophic and dilated cardiomyopathies. Cardiovasc. Res. 2008, 79, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Gudkova, A.; Kostareva, A.; Sjoberg, G.; Smolina, N.; Turalchuk, M.; Kuznetsova, I.; Rybakova, M.; Edstrom, L.; Shlyakhto, E.; Sejersen, T. Diagnostic challenge in desmin cardiomyopathy with transformation of clinical phenotypes. Pediatr. Cardiol. 2013, 34, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Milner, D.J.; Taffet, G.E.; Wang, X.; Pham, T.; Tamura, T.; Hartley, C.; Gerdes, A.M.; Capetanaki, Y. The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J. Mol. Cell. Cardiol. 1999, 31, 2063–2076. [Google Scholar] [PubMed]
- Ghosh, N.; Haddad, H. Recent progress in the genetics of cardiomyopathy and its role in the clinical evaluation of patients with cardiomyopathy. Curr. Opin. Cardiol. 2011, 26, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Keren, A.; Syrris, P.; McKenna, W.J. Hypertrophic cardiomyopathy: The genetic determinants of clinical disease expression. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Geisterfer-Lowrance, A.A.; Christe, M.; Conner, D.A.; Ingwall, J.S.; Schoen, F.J.; Seidman, C.E.; Seidman, J.G. A mouse model of familial hypertrophic cardiomyopathy. Science 1996, 272, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Wu, Y.; Lim, D.S.; McCluggage, M.; Youker, K.; Yu, Q.T.; Brugada, R.; DeMayo, F.; Quinones, M.; Roberts, R. A transgenic rabbit model for human hypertrophic cardiomyopathy. J. Clin. Investig. 1999, 104, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Tardiff, J.C.; Hewett, T.E.; Palmer, B.M.; Olsson, C.; Factor, S.M.; Moore, R.L.; Robbins, J.; Leinwand, L.A. Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J. Clin. Investig. 1999, 104, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Gucev, Z.; Tasic, V.; Jancevska, A.; Popjordanova, N.; Koceva, S.; Kuturec, M.; Sabolic, V. Friedreich ataxia (FA) associated with diabetes mellitus type 1 and hyperthrophic cardiomyopathy. Bosn. J. Basic. Med. Sci. 2009, 9, 107–110. [Google Scholar] [PubMed]
- Redfern, W.S.; Carlsson, L.; Davis, A.S.; Lynch, W.G.; MacKenzie, I.; Palethorpe, S.; Siegl, P.K.; Strang, I.; Sullivan, A.T.; Wallis, R.; et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003, 58, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Gintant, G.A.; Su, Z.; Martin, R.L.; Cox, B.F. Utility of herg assays as surrogate markers of delayed cardiac repolarization and QT safety. Toxicol. Pathol. 2006, 34, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Dumotier, B.M.; Deurinck, M.; Yang, Y.; Traebert, M.; Suter, W. Relevance of in vitro screenit results for drug-induced QT interval prolongation in vivo: A database review and analysis. Pharmacol. Ther. 2008, 119, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, N.; Baba, S.; Kaichi, S.; Niwa, A.; Mima, T.; Doi, H.; Yamanaka, S.; Nakahata, T.; Heike, T. The effects of cardioactive drugs on cardiomyocytes derived from human induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 2009, 387, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Lacerda, A.E.; Kirsch, G.E.; Brown, A.M.; Bruening-Wright, A. The action potential and comparative pharmacology of stem cell-derived human cardiomyocytes. J. Pharmacol. Toxicol. Methods 2010, 61, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Reuter, H.; Schwinger, R.H. Calcium handling in human heart failure—Abnormalities and target for therapy. Wien. Med. Wochenschr. 2012, 162, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Lou, Q.; Janardhan, A.; Efimov, I.R. Remodeling of calcium handling in human heart failure. Adv. Exp. Med. Biol. 2012, 740, 1145–1174. [Google Scholar] [PubMed]
- Cerignoli, F.; Charlot, D.; Whittaker, R.; Ingermanson, R.; Gehalot, P.; Savchenko, A.; Gallacher, D.J.; Towart, R.; Price, J.H.; McDonough, P.M.; et al. High throughput measurement of Ca(2)(+) dynamics for drug risk assessment in human stem cell-derived cardiomyocytes by kinetic image cytometry. J. Pharmacol. Toxicol. Methods 2012, 66, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef]
- Andersson, H.; Steel, D.; Asp, J.; Dahlenborg, K.; Jonsson, M.; Jeppsson, A.; Lindahl, A.; Kagedal, B.; Sartipy, P.; Mandenius, C.F. Assaying cardiac biomarkers for toxicity testing using biosensing and cardiomyocytes derived from human embryonic stem cells. J. Biotechnol. 2010, 150, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Mandenius, C.F.; Steel, D.; Noor, F.; Meyer, T.; Heinzle, E.; Asp, J.; Arain, S.; Kraushaar, U.; Bremer, S.; Class, R.; et al. Cardiotoxicity testing using pluripotent stem cell-derived human cardiomyocytes and state-of-the-art bioanalytics: A review. J. Appl. Toxicol. 2011, 31, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Ng, K.M.; Lai, W.H.; Chan, Y.C.; Lau, Y.M.; Lian, Q.; Tse, H.F.; Siu, C.W. Calcium homeostasis in human induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Rev. 2011, 7, 976–986. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ng, K.-M.; Law, C.-Y.; Tse, H.-F. Clinical Potentials of Cardiomyocytes Derived from Patient-Specific Induced Pluripotent Stem Cells. J. Clin. Med. 2014, 3, 1105-1123. https://doi.org/10.3390/jcm3041105
Ng K-M, Law C-Y, Tse H-F. Clinical Potentials of Cardiomyocytes Derived from Patient-Specific Induced Pluripotent Stem Cells. Journal of Clinical Medicine. 2014; 3(4):1105-1123. https://doi.org/10.3390/jcm3041105
Chicago/Turabian StyleNg, Kwong-Man, Cheuk-Yiu Law, and Hung-Fat Tse. 2014. "Clinical Potentials of Cardiomyocytes Derived from Patient-Specific Induced Pluripotent Stem Cells" Journal of Clinical Medicine 3, no. 4: 1105-1123. https://doi.org/10.3390/jcm3041105
APA StyleNg, K.-M., Law, C.-Y., & Tse, H.-F. (2014). Clinical Potentials of Cardiomyocytes Derived from Patient-Specific Induced Pluripotent Stem Cells. Journal of Clinical Medicine, 3(4), 1105-1123. https://doi.org/10.3390/jcm3041105