Neurosurgical Hyponatremia

Abstract
:1. Introduction
2. The Clinical Effects of Hyponatremia

{kind=link}
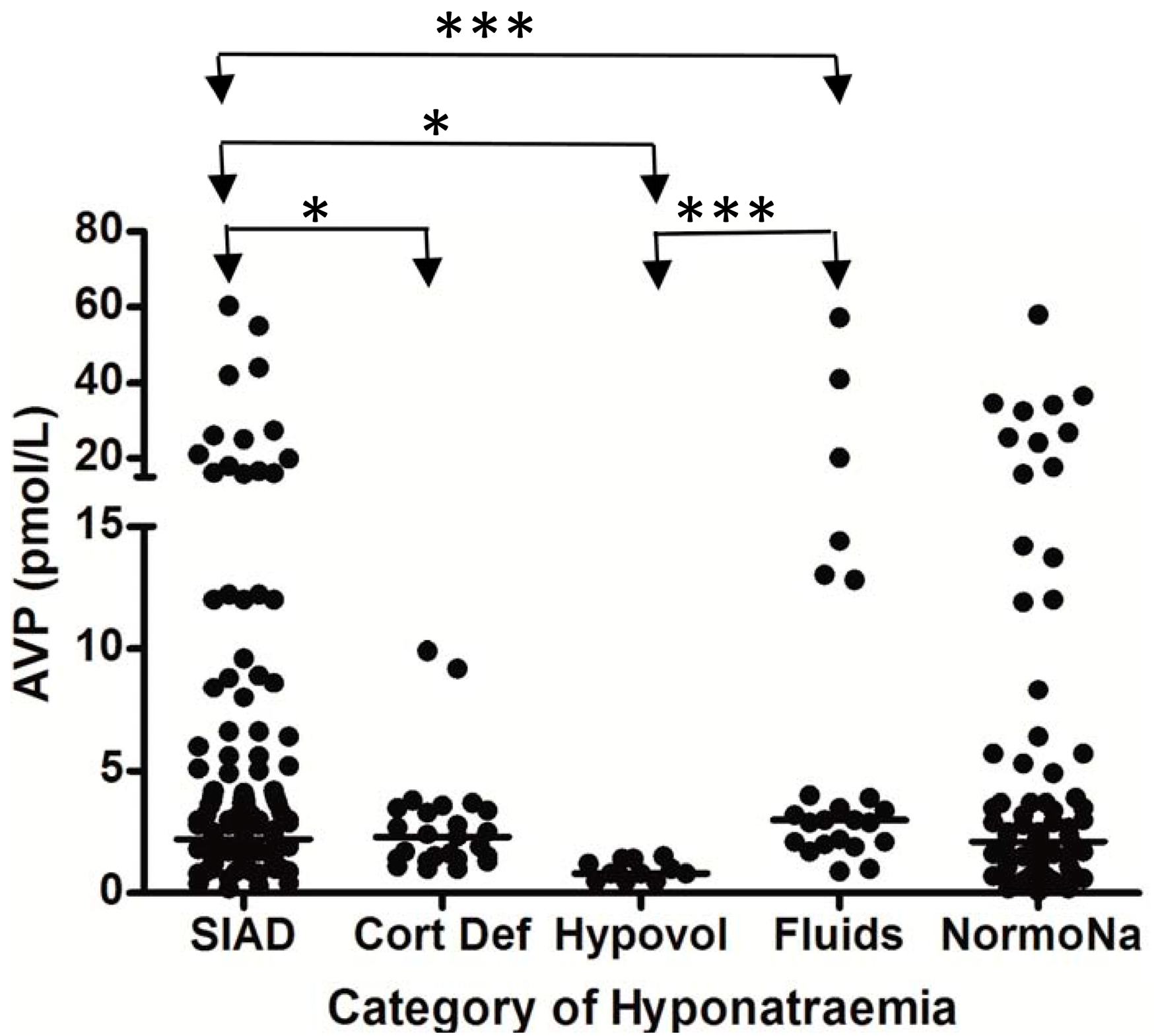
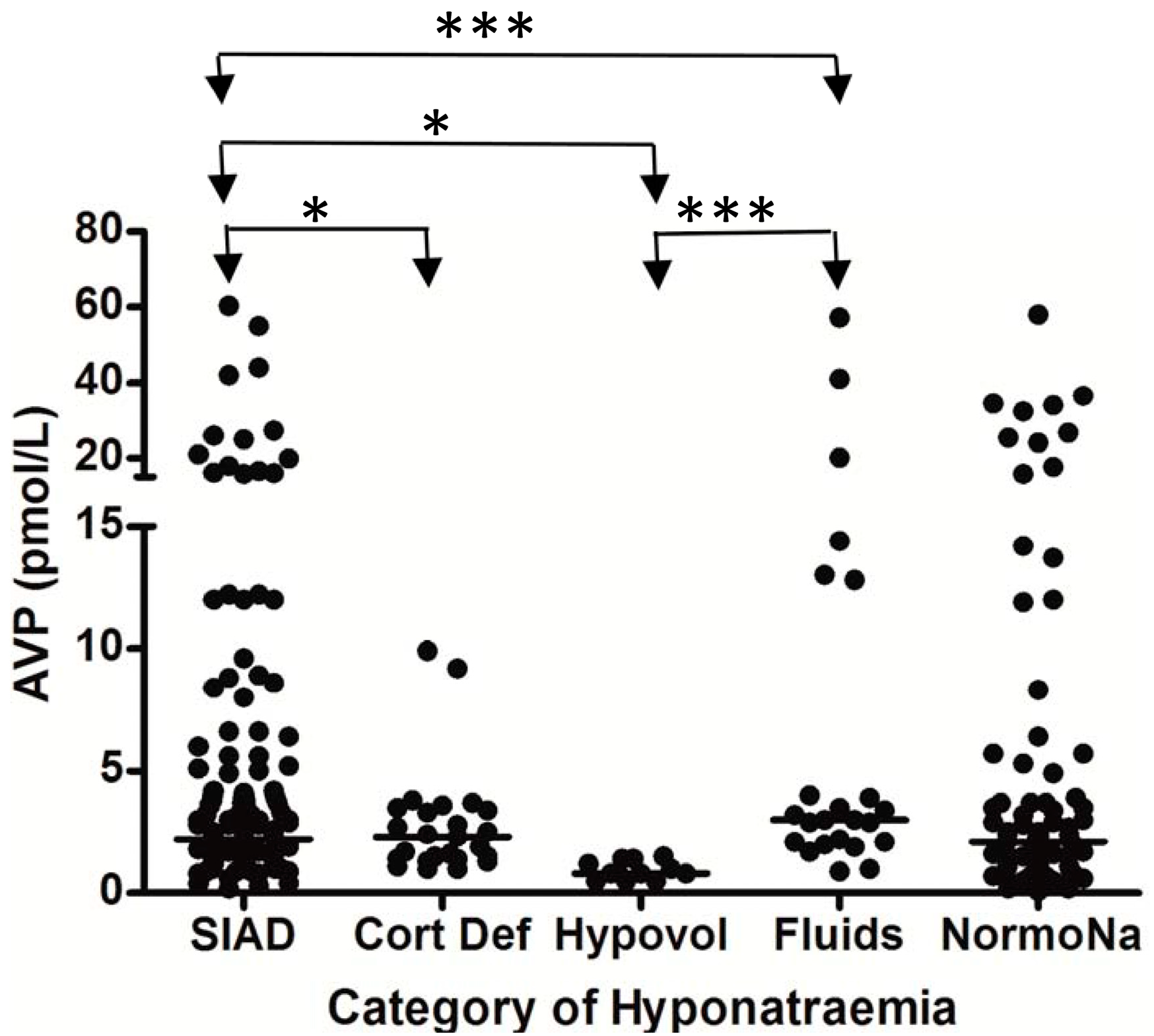{kind=link}
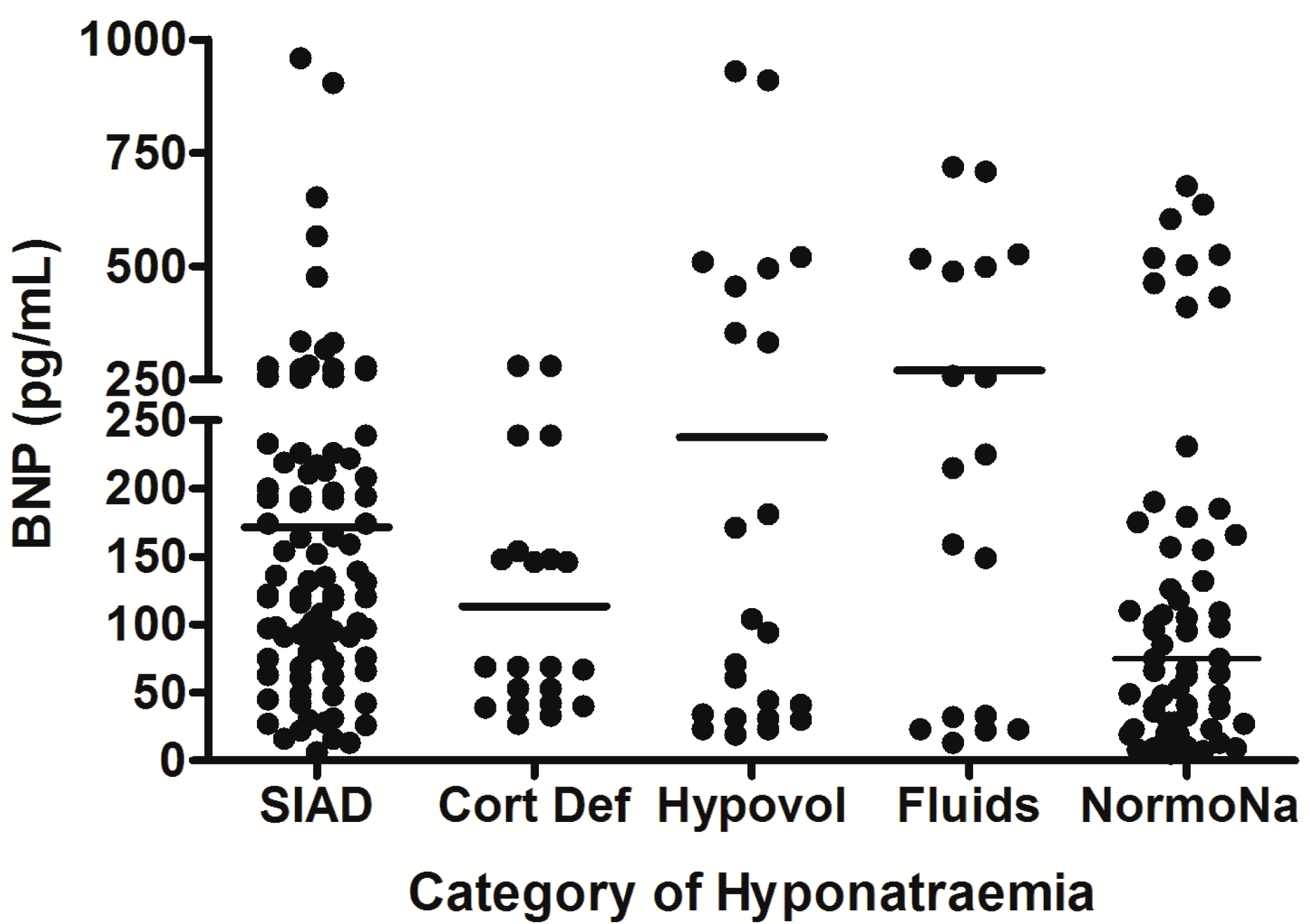{kind=link}
| Diagnosis | No. of Patients with Plasma Sodium <130 mmol/L | Total | % |
|---|---|---|---|
| All Patients | 187 | 1698 | 11 |
| SAH | 62 | 316 | 19.6 |
| Tumor | 56 | 355 | 15.8 |
| TBI | 44 | 457 | 9.6 |
| Pituitary surgery | 5 | 81 | 6.2 |
| Spinal | 4 | 489 | 0.81 |
3. The Pathophysiology of Hyponatremia in Neurosurgical Patients
| Diagnosis | Blood Volume Status | Diagnostic Criteria | Treatment |
|---|---|---|---|
| SIADH | Euvolaemic | See Table 3 | Fluid restriction Vaptan therapy |
| Acute ACTH deficiency | Euvolaemic (may be hypotensive) | 0900 h cortisol <300 nmol/L in stressed patient | Steroid replacement therapy |
| Hypovolaemia | Hypovolaemic | Negative fluid balance | IV fluids |
| CSWS | Hypovolaemic | Profound diuresis and natriuresis Low BP and CVP | Aggressive IV fluids |
| Mixed SIADH and CSWS | Variable/fluctuating | Usually SIADH initially, then progressing to CSWS | Depends on stage |
| Inappropriate IV fluids | Hypervolaemic | Positive fluid balance May have edema/LVF | Diuretics Stop IV fluids |
| 1. Hyposomolality; plasma osmolality <280 mOsm/kg |
| 2. Inappropriate urinary concentration (Uosm >100 mOsm/kg) |
| 3. Patient is clinically euvolemic |
| 4. Elevated urinary sodium (>40 mmol/L), with normal salt and water intake |
| 5. Exclude hypothyroidism and glucocorticoid deficiency―particularly in patients with neurosurgical conditions |
4. Hyponatremia Following Traumatic Brain Injury
5. Hyponatremia Following Subarachnoid Hemorrhage
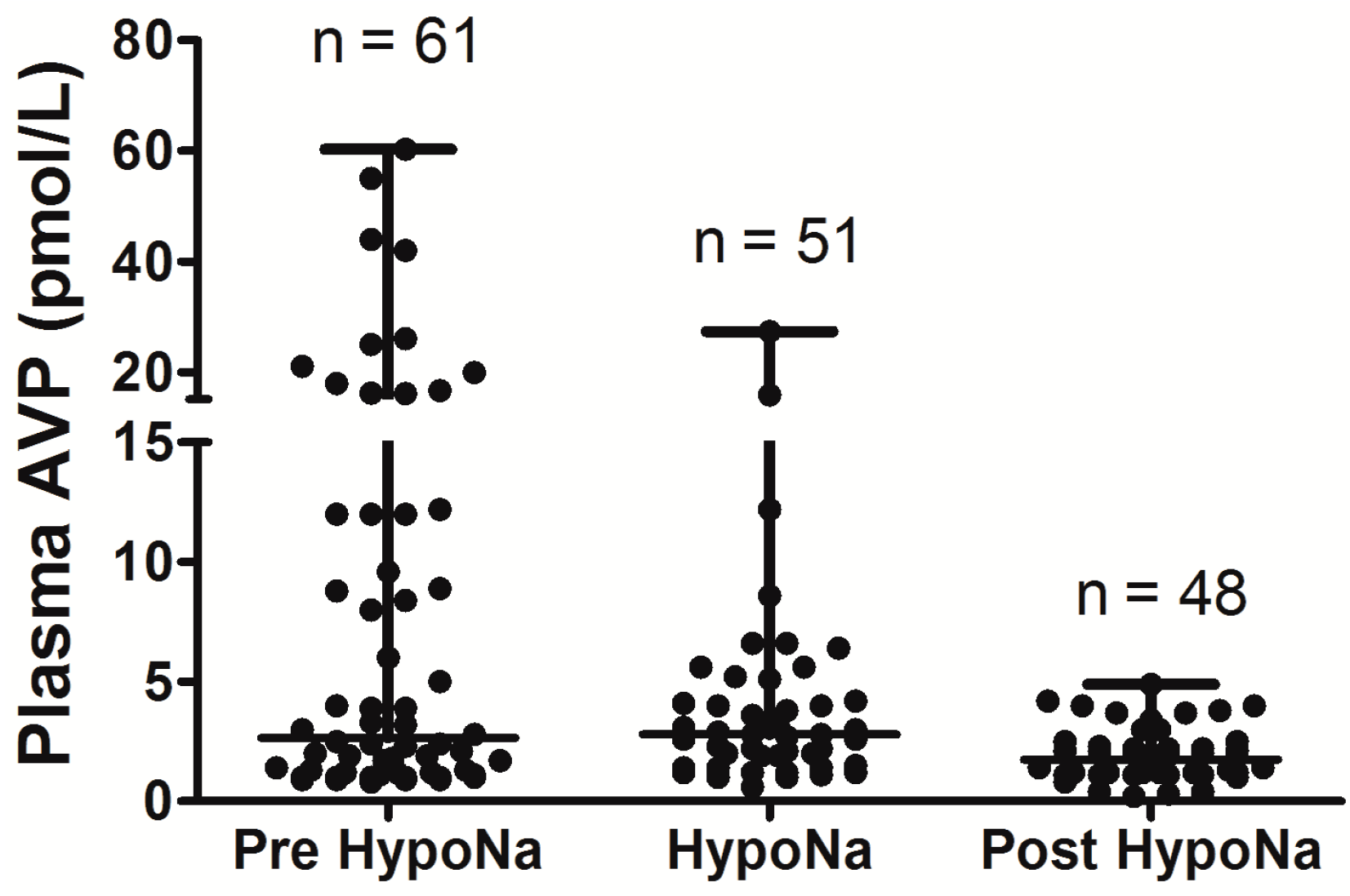
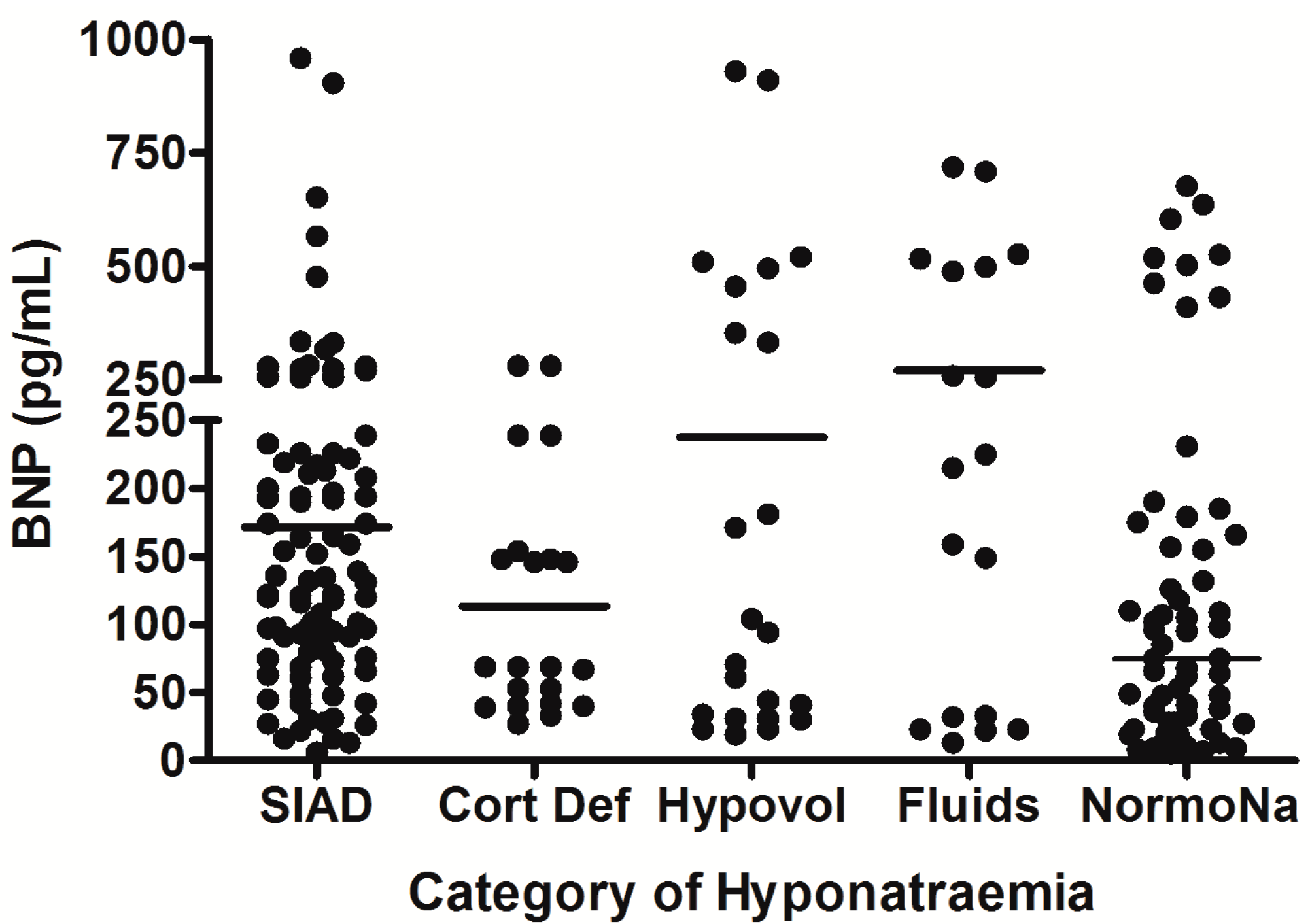
6. Hyponatremia Following Pituitary Surgery
7. Management of Hyponatremia in the Neurosurgical Patient
7.1. Management of Acute Symptomatic Hyponatremia
7.2. Management of Hyponatremia Due to Glucocorticoid Insufficiency
7.3. Management of Other Causes of Hyponatremia
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hannon, M.J.; Crowley, R.K.; Behan, L.A.; O’Sullivan, E.P.; O’Brien, M.M.; Sherlock, M.; Rawluk, D.; O’Dwyer, R.; Tormey, W.; Thompson, C.J. Acute glucocorticoid deficiency and diabetes insipidus are common after acute traumatic brain injury and predict mortality. J. Clin. Endocrinol. MeTable 2013, 98, 3229–3237. [Google Scholar] [CrossRef]
- Hannon, M.J.; Finucane, F.M.; Sherlock, M.; Agha, A.; Thompson, C.J. Clinical review: Disorders of water homeostasis in neurosurgical patients. J. Clin. Endocrinol. MeTable 2012, 97, 1423–1433. [Google Scholar] [CrossRef]
- Moro, N.; Katayama, Y.; Igarashi, T.; Mori, T.; Kawamata, T.; Kojima, J. Hyponatremia in patients with traumatic brain injury: Incidence, mechanism, and response to sodium supplementation or retention therapy with hydrocortisone. Surg. Neurol. 2007, 68, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, M.; O’Sullivan, E.; Agha, A.; Behan, L.A.; Rawluk, D.; Brennan, P.; Tormey, W.; Thompson, C.J. The incidence and pathophysiology of hyponatraemia after subarachnoid haemorrhage. Clin. Endocrinol. 2006, 64, 250–254. [Google Scholar] [CrossRef]
- Kao, L.; Al-Lawati, Z.; Vavao, J.; Steinberg, G.K.; Katznelson, L. Prevalence and clinical demographics of cerebral salt wasting in patients with aneurysmal subarachnoid hemorrhage. Pituitary 2009, 12, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Hannon, M.J.; Behan, L.A.; O’Brien, M.M.; Tormey, W.; Ball, S.G.; Javadpur, M.; Sherlock, M.; Thompson, C.J. Hyponatremia following mild/moderate subarachnoid hemorrhage is due to SIAD and glucocorticoid deficiency and not cerebral salt wasting. J. Clin. Endocrinol. MeTable 2014, 99, 291–298. [Google Scholar] [CrossRef]
- Sherlock, M.; O’Sullivan, E.; Agha, A.; Behan, L.A.; Owens, D.; Finucane, F.; Rawluk, D.; Tormey, W.; Thompson, C.J. Incidence and pathophysiology of severe hyponatraemia in neurosurgical patients. Postgrad. Med. J. 2009, 85, 171–175. [Google Scholar] [CrossRef]
- Gill, G.; Huda, B.; Boyd, A.; Skagen, K.; Wile, D.; Watson, I.; van Heyningen, C. Characteristics and mortality of severe hyponatraemia—A hospital—based study. Clin. Endocrinol. 2006, 65, 246–249. [Google Scholar] [CrossRef]
- Clayton, J.A.; le Jeune, I.R.; Hall, I.P. Severe hyponatraemia in medical in-patients: Aetiology, assessment and outcome. QJM 2006, 99, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Hoorn, E.J.; Lindemans, J.; Zietse, R. Development of severe hyponatraemia in hospitalized patients: treatment-related risk factors and inadequate management. Nephrol. Dial. Transpl. 2006, 21, 70–76. [Google Scholar] [CrossRef]
- Sturdik, I.; Adamcova, M.; Kollerova, J.; Koller, T.; Zelinkova, Z.; Payer, J. Hyponatraemia is an independent predictor of in-hospital mortality. Eur. J. Intern. Med. 2014, 25, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, P.; Bagkeris, E.; Bouloux, P.M. A case-control study of hyponatraemia as an independent risk factor for inpatient mortality. Clin. Endocrinol. 2014, 81, 401–407. [Google Scholar] [CrossRef]
- Zilberberg, M.D.; Exuzides, A.; Spalding, J.; Foreman, A.; Jones, A.G.; Colby, C.; Shorr, A.F. Hyponatremia and hospital outcomes among patients with pneumonia: A retrospective cohort study. BMC Pulm. Med. 2008, 8. [Google Scholar] [CrossRef]
- Stelfox, H.T.; Ahmed, S.B.; Khandwala, F.; Zygun, D.; Shahpori, R.; Laupland, K. The epidemiology of intensive care unit-acquired hyponatraemia and hypernatraemia in medical-surgical intensive care units. Crit. Care 2008, 12. [Google Scholar] [CrossRef] [PubMed]
- Sajadieh, A.; Binici, Z.; Mouridsen, M.R.; Nielsen, O.W.; Hansen, J.F.; Haugaard, S.B. Mild hyponatremia carries a poor prognosis in community subjects. Am. J. Med. 2009, 122, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Gu, S.; Parikh, A.; Radhakrishnan, J. Prevalence of hyponatremia and association with mortality: Results from NHANES. Am. J. Med. 2013, 126, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Waikar, S.S.; Mount, D.B.; Curhan, G.C. Mortality after hospitalization with mild, moderate, and severe hyponatremia. Am. J. Med. 2009, 122, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, M.D.; Exuzides, A.; Spalding, J.; Foreman, A.; Jones, A.G.; Colby, C.; Shorr, A.F. Epidemiology, clinical and economic outcomes of admission hyponatremia among hospitalized patients. Curr. Med. Res. Opin. 2008, 24, 1601–1608. [Google Scholar] [CrossRef]
- Amin, A.; Deitelzweig, S.; Christian, R.; Friend, K.; Lin, J.; Lowe, T.J. Healthcare resource burden associated with hyponatremia among patients hospitalized for heart failure in the US. J. Med. Econ. 2013, 16, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Deitelzweig, S.; Amin, A.; Christian, R.; Friend, K.; Lin, J.; Lowe, T.J. Hyponatremia-associated healthcare burden among US patients hospitalized for cirrhosis. Adv. Ther. 2013, 30, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Deitelzweig, S.; Christian, R.; Friend, K.; Lin, J.; Belk, K.; Baumer, D.; Lowe, T.J. Evaluation of incremental healthcare resource burden and readmission rates associated with hospitalized hyponatremic patients in the US. J. Hosp. Med. 2012, 7, 634–639. [Google Scholar] [CrossRef]
- Verbalis, J.G.; Barsony, J.; Sugimura, Y.; Tian, Y.; Adams, D.J.; Carter, E.A.; Resnick, H.E. Hyponatremia-induced osteoporosis. J. Bone Miner. Res. 2010, 25, 554–563. [Google Scholar] [CrossRef]
- Arieff, A.I.; Llach, F.; Massry, S.G. Neurological manifestations and morbidity of hyponatremia: Correlation with brain water and electrolytes. Medicine 1976, 55, 121–129. [Google Scholar] [CrossRef]
- Rojiani, A.M.; Prineas, J.W.; Cho, E.S. Electrolyte-induced demyelination in rats. 1. Role of the blood-brain barrier and edema. Acta Neuropathol. 1994, 88, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Sterns, R.H.; Baer, J.; Ebersol, S.; Thomas, D.; Lohr, J.W.; Kamm, D.E. Organic osmolytes in acute hyponatremia. Am. J. Physiol. 1993, 264, F833–F836. [Google Scholar] [PubMed]
- Gullans, S.R.; Verbalis, J.G. Control of brain volume during hyperosmolar and hypoosmolar conditions. Annu. Rev. Med. 1993, 44, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Verbalis, J.G. Adaptation to acute and chronic hyponatremia: implications for symptomatology, diagnosis, and therapy. Semin. Nephrol. 1998, 18, 3–19. [Google Scholar] [PubMed]
- Fraser, J.F.; Stieg, P.E. Hyponatremia in the neurosurgical patient: Epidemiology, pathophysiology, diagnosis, and management. Neurosurgery 2006, 59, 222–229. [Google Scholar] [CrossRef]
- Kuramatsu, J.B.; Bobinger, T.; Volbers, B.; Staykov, D.; Lucking, H.; Kloska, S.P.; Köhrmann, M.; Huttner, H.B. Hyponatremia is an independent predictor of in-hospital mortality in spontaneous intracerebral hemorrhage. Stroke 2014, 45, 1285–1291. [Google Scholar] [CrossRef]
- Rodrigues, B.; Staff, I.; Fortunato, G.; McCullough, L.D. Hyponatremia in the prognosis of acute ischemic stroke. J. Stroke Cerebrovasc. Dis. 2014, 23, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Lindner, G.; Funk, G.C. Hypernatremia in critically ill patients. J. Crit. Care 2013, 28. [Google Scholar] [CrossRef]
- Hannon, M.J.; Thompson, C.J. The syndrome of inappropriate antidiuretic hormone: Prevalence, causes and consequences. Eur. J. Endocrinol. 2010, 162 (Suppl. 1), 5–12. [Google Scholar] [CrossRef]
- Verbalis, J.G. Hyponatraemia. Balliere’s Clin. Endocrinol. Metab. 1989, 3, 499–530. [Google Scholar] [CrossRef]
- Ishikawa, S.; Saito, T.; Kasono, K. Pathological role of aquaporin-2 in impaired water excretion and hyponatremia. J. Neuroendocrinol. 2004, 16, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Higashiyama, M.; Nagasaka, S.; Sasaki, S.; Ishikawa, S.E. Role of aquaporin-2 gene expression in hyponatremic rats with chronic vasopressin-induced antidiuresis. Kidney Int. 2001, 60, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Chen, Y.; Spatz, M. Involvement of non-neuronal brain cells in AVP-mediated regulation of water space at the cellular, organ, and whole-body level. J. Neurosci. Res. 2000, 62, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Oelkers, W. Hyponatremia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism. N. Engl. J. Med. 1989, 321, 492–496. [Google Scholar] [PubMed]
- Robertson, G.L. Syndrome of inappropriate antidiuresis. N. Engl. J. Med. 1989, 321, 538–539. [Google Scholar] [CrossRef] [PubMed]
- Linas, S.L.; Berl, T.; Robertson, G.L.; Aisenbrey, G.A.; Schrier, R.W.; Anderson, R.J. Role of vasopressin in the impaired water excretion of glucocorticoid deficiency. Kidney Int. 1980, 18, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Erkut, Z.A.; Pool, C.; Swaab, D.F. Glucocorticoids suppress corticotropin-releasing hormone and vasopressin expression in human hypothalamic neurons. J. Clin. Endocrinol. MeTable 1998, 83, 2066–2073. [Google Scholar]
- Andrioli, M.; Pecori Giraldi, F.; Cavagnini, F. Isolated corticotrophin deficiency. Pituitary 2006, 9, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.P.; Welt, L.G.; Sims, E.A.H.; Orloff, J.; Needham, J. A salt wasting syndrome associated with cerebral disease. Trans. Assoc. Am. Phys. 1950, 63, 57–64. [Google Scholar] [PubMed]
- Nelson, P.B.; Seif, S.M.; Maroon, J.C.; Robinson, A.G. Hyponatremia in intracranial disease: Perhaps not the syndrome of inappropriate secretion of antidiuretic hormone (SIADH). J. Neurosurg. 1981, 55, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Citerio, G.; Gaini, S.M.; Tomei, G.; Stocchetti, N. Management of 350 aneurysmal subarachnoid hemorrhages in 22 Italian neurosurgical centers. Intensive Care Med. 2007, 33, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Thornton, E.; O’Kelly, P.; Tormey, W.; Phillips, J.; Thompson, C.J. Posterior pituitary dysfunction after traumatic brain injury. J. Clin. Endocrinol. MeTable 2004, 89, 5987–5992. [Google Scholar] [CrossRef]
- Sivakumar, V.; Rajshekhar, V.; Chandy, M.J. Management of neurosurgical patients with hyponatremia and natriuresis. Neurosurgery 1994, 34, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Wijdicks, E.F.; Vermeulen, M.; Hijdra, A.; van Gijn, J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: Is fluid restriction harmful? Ann. Neurol. 1985, 17, 137–140. [Google Scholar]
- Oh, M.S.; Carroll, H.J. Cerebral salt-wasting syndrome. Nephron 1999, 82, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Lohani, S.; Devkota, U.P. Hyponatremia in patients with traumatic brain injury: Etiology, incidence, and severity correlation. World Neurosurg. 2011, 76, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Hackl, J.M.; Gottardis, M.; Wieser, C.; Rumpl, E.; Stadler, C.; Schwarz, S.; Monkayo, R. Endocrine abnormalities in severe traumatic brain injury—A cue to prognosis in severe craniocerebral trauma? Intensive Care Med. 1991, 17, 25–29. [Google Scholar]
- Della Corte, F.; Mancini, A.; Valle, D.; Gallizzi, F.; Carducci, P.; Mignani, V.; de Marinis, L. Provocative hypothalamopituitary axis tests in severe head injury: Correlations with severity and prognosis. Crit. Care Med. 1998, 26, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Cernak, I.; Savic, V.J.; Lazarov, A.; Joksimovic, M.; Markovic, S. Neuroendocrine responses following graded traumatic brain injury in male adults. Brain Inj. 1999, 13, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Rogers, B.; Mylotte, D.; Taleb, F.; Tormey, W.; Phillips, J.; Thompson, C.J. Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clin. Endocrinol. 2004, 60, 584–591. [Google Scholar] [CrossRef]
- Cohan, P.; Wang, C.; McArthur, D.L.; Cook, S.W.; Dusick, J.R.; Armin, B.; Swerdloff, R.; Vespa, P.; Muizelaar, J.P.; Cryer, H.G.; et al. Acute secondary adrenal insufficiency after traumatic brain injury: A prospective study. Crit. Care Med. 2005, 33, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; Senyurek, H.; Unluhizarci, K.; Selcuklu, A.; Casanueva, F.F.; Kelestimur, F. High risk of hypopituitarism after traumatic brain injury: A prospective investigation of anterior pituitary function in the acute phase and 12 months after trauma. J. Clin. Endocrinol. MeTable 2006, 91, 2105–2111. [Google Scholar] [CrossRef]
- Klose, M.; Juul, A.; Struck, J.; Morgenthaler, N.G.; Kosteljanetz, M.; Feldt-Rasmussen, U. Acute and long-term pituitary insufficiency in traumatic brain injury: A prospective single-centre study. Clin. Endocrinol. 2007, 67, 598–606. [Google Scholar] [CrossRef]
- Hannon, M.J.; Sherlock, M.; Thompson, C.J. Pituitary dysfunction following traumatic brain injury or subarachnoid haemorrhage—In “Endocrine Management in the Intensive Care Unit”. Best Pract. Res. Clin. Endocrinol. MeTable 2011, 25, 783–798. [Google Scholar] [CrossRef]
- Agha, A.; Sherlock, M.; Thompson, C.J. Post-Traumatic hyponatraemia due to acute hypopituitarism. QJM 2005, 98, 463–464. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, Y.; Uede, T.; Ishiguro, M.; Honda, O.; Honmou, O.; Kato, T.; Wanibuchi, M. Pathogenesis of hyponatremia following subarachnoid hemorrhage due to ruptured cerebral aneurysm. Surg. Neurol. 1996, 46, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Isotani, E.; Suzuki, R.; Tomita, K.; Hokari, M.; Monma, S.; Marumo, F.; Hirakawa, K. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994, 25, 2198–2203. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F. Hyponatraemia in a neurosurgical patient: Syndrome of inappropriate antidiuretic hormone secretion versus cerebral salt wasting. Nephrol. Dial. Transpl. 2000, 15, 262–268. [Google Scholar] [CrossRef]
- Berendes, E.; Walter, M.; Cullen, P.; Prien, T.; van Aken, H.; Horsthemke, J.; Schulte, M.; von Wild, K.; Scherer, R. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997, 349, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Dorhout Mees, S.M.; Hoff, R.G.; Rinkel, G.J.; Algra, A.; van den Bergh, W.M. Brain natriuretic peptide concentrations after aneurysmal subarachnoid hemorrhage: Relationship with hypovolemia and hyponatremia. Neurocrit. Care 2011, 14, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Brennum, J.; Poulsgaard, L.; Kosteljanetz, M.; Wagner, A.; Feldt-Rasmussen, U. Hypopituitarism is uncommon after aneurysmal subarachnoid haemorrhage. Clin. Endocrinol. 2010, 73, 95–101. [Google Scholar]
- Parenti, G.; Cecchi, P.C.; Ragghianti, B.; Schwarz, A.; Ammannati, F.; Mennonna, P.; di Rita, A.; Gallina, P.; di Lorenzo, N.; Innocenti, P.; et al. Evaluation of the anterior pituitary function in the acute phase after spontaneous subarachnoid hemorrhage. J. Endocrinol. Invest. 2010, 34, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Lanterna, L.A.; Spreafico, V.; Gritti, P.; Prodam, F.; Signorelli, A.; Biroli, F.; Aimaretti, G. Hypocortisolism in noncomatose patients during the acute phase of subarachnoid hemorrhage. J. Stroke Cerebrovasc. Dis. 2012, 22. [Google Scholar] [CrossRef]
- Zerbe, R.; Stropes, L.; Robertson, G. Vasopressin function in the syndrome of inappropriate antidiuresis. Annu. Rev. Med. 1980, 31, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Janneck, M.; Burkhardt, T.; Rotermund, R.; Sauer, N.; Flitsch, J.; Aberle, J. Hyponatremia after trans-sphenoidal surgery. Minerva Endocrinol. 2014, 39, 27–31. [Google Scholar] [PubMed]
- Jahangiri, A.; Wagner, J.; Tran, M.T.; Miller, L.M.; Tom, M.W.; Kunwar, S.; Blevins, L., Jr.; Aghi, M.K. Factors predicting postoperative hyponatremia and efficacy of hyponatremia management strategies after more than 1000 pituitary operations. J. Neurosurg. 2013, 119, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.J. Severe hyponatraemia: Complications and treatment. QJM 1995, 88, 905–909. [Google Scholar] [PubMed]
- Ayus, J.C.; Varon, J.; Arieff, A.I. Hyponatremia, cerebral edema, and noncardiogenic pulmonary edema in marathon runners. Ann. Intern. Med. 2000, 132, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Sterns, R.H.; Silver, S.M. Brain volume regulation in response to hypo-osmolality and its correction. Am. J. Med. 2006, 119 (Suppl. 1), 12–16. [Google Scholar] [CrossRef]
- Ayus, J.C.; Arieff, A.I. Chronic hyponatremic encephalopathy in postmenopausal women: Association of therapies with morbidity and mortality. JAMA 1999, 281, 2299–2304. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.G.; Laureno, R.; Victor, M. Pontine and extrapontine myelinolysis. Brain 1979, 102, 361–385. [Google Scholar] [PubMed]
- Cluitmans, F.H.; Meinders, A.E. Management of severe hyponatremia: Rapid or slow correction? Am. J. Med. 1990, 88, 161–166. [Google Scholar] [CrossRef]
- Mickel, H.S.; Oliver, C.N.; Starke-Reed, P.E. Protein oxidation and myelinolysis occur in brain following rapid correction of hyponatremia. Biochem. Biophys. Res. Commun. 1990, 172, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Verbalis, J.G.; Goldsmith, S.R.; Greenberg, A.; Korzelius, C.; Schrier, R.W.; Sterns, R.H.; Thompson, C.J. Diagnosis, evaluation, and treatment of hyponatremia: Expert panel recommendations. Am. J. Med. 2013, 126 (Suppl. 1), 1–42. [Google Scholar]
- Koenig, M.A.; Bryan, M.; Lewin, J.L., III; Mirski, M.A.; Geocadin, R.G.; Stevens, R.D. Reversal of transtentorial herniation with hypertonic saline. Neurology 2008, 70, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Rogers, I.R.; Hook, G.; Stuempfle, K.J.; Hoffman, M.D.; Hew-Butler, T. An intervention study of oral versus intravenous hypertonic saline administration in ultramarathon runners with exercise-associated hyponatremia: A preliminary randomized trial. Clin. J. Sport Med. 2011, 21, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Gankam Kengne, F.; Soupart, A.; Pochet, R.; Brion, J.P.; Decaux, G. Re-Induction of hyponatremia after rapid overcorrection of hyponatremia reduces mortality in rats. Kidney Int. 2009, 76, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Arafah, B.M. Hypothalamic pituitary adrenal function during critical illness: Limitations of current assessment methods. J. Clin. Endocrinol. MeTable 2006, 91, 3725–3745. [Google Scholar] [CrossRef]
- Marik, P.E.; Pastores, S.M.; Annane, D.; Meduri, G.U.; Sprung, C.L.; Arlt, W.; Keh, D.; Briegel, J.; Beishuizen, A.; Dimopoulou, I.; et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: Consensus statements from an international task force by the American College of Critical Care Medicine. Crit. Care Med. 2008, 36, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Phillips, J.; O’Kelly, P.; Tormey, W.; Thompson, C.J. The natural history of post-traumatic hypopituitarism: Implications for assessment and treatment. Am. J. Med. 2005, 118. [Google Scholar] [CrossRef]
- Agha, A.; Sherlock, M.; Phillips, J.; Tormey, W.; Thompson, C.J. The natural history of post-traumatic neurohypophysial dysfunction. Eur. J. Endocrinol. 2005, 152, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Soupart, A.; Schroeder, B.; Decaux, G. Treatment of hyponatraemia by urea decreases risks of brain complications in rats. Brain osmolyte contents analysis. Nephrol. Dial. Transpl. 2007, 22, 1856–1863. [Google Scholar] [CrossRef]
- Decaux, G.; Andres, C.; Gankam Kengne, F.; Soupart, A. Treatment of euvolemic hyponatremia in the intensive care unit by urea. Crit. Care 2010, 14. [Google Scholar] [CrossRef]
- Pierrakos, C.; Taccone, F.S.; Decaux, G.; Vincent, J.L.; Brimioulle, S. Urea for treatment of acute SIADH in patients with subarachnoid hemorrhage: A single-center experience. Ann. Intensive Care 2012, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, M.; Thompson, C.J. The syndrome of inappropriate antidiuretic hormone: Current and future management options. Eur. J. Endocrinol. 2010, 162 (Suppl. 1), 13–18. [Google Scholar] [CrossRef]
- Schrier, R.W.; Gross, P.; Gheorghiade, M.; Berl, T.; Verbalis, J.G.; Czerwiec, F.S.; Cesare Orlandi, M.D. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N. Engl. J. Med. 2006, 355, 2099–2112. [Google Scholar] [CrossRef] [PubMed]
- Berl, T.; Quittnat-Pelletier, F.; Verbalis, J.G.; Schrier, R.W.; Bichet, D.G.; Ouyang, J.; Czerwiec, F.S.; SALTWATER Investigators. Oral tolvaptan is safe and effective in chronic hyponatremia. J. Am. Soc. Nephrol. 2010, 21, 705–712. [Google Scholar] [PubMed]
- Peri, A. Clinical review: The use of vaptans in clinical endocrinology. J. Clin. Endocrinol. MeTable 2013, 98, 1321–1332. [Google Scholar] [CrossRef]
- Buckley, M.S.; Patel, S.A.; Hattrup, A.E.; Kazem, N.H.; Jacobs, S.C.; Culver, M.A. Conivaptan for treatment of hyponatremia in neurologic and neurosurgical adults. Ann. Pharmacother. 2013, 47, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.B.; Choi, H.A.; Lesch, C.; Kim, M.C.; Badjatia, N.; Claassen, J.; Mayer, S.A.; Lee, K. Use of oral vasopressin V2 receptor antagonist for hyponatremia in acute brain injury. Eur. Neurol. 2013, 70, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Human, T.; Onuoha, A.; Diringer, M.; Dhar, R. Response to a bolus of conivaptan in patients with acute hyponatremia after brain injury. J. Crit. Care 2012, 27. [Google Scholar] [CrossRef]
- Higashihara, E.; Torres, V.E.; Chapman, A.B.; Grantham, J.J.; Bae, K.; Watnick, T.J.; Horie, S.; Nutahara, K.; Ouyang, J.; Krasa, H.B. Tolvaptan in autosomal dominant polycystic kidney disease: Three years’ experience. Clin. J. Am. Soc. Nephrol. 2011, 6, 2499–2507. [Google Scholar] [CrossRef] [PubMed]
- Revilla-Pacheco, F.R.; Herrada-Pineda, T.; Loyo-Varela, M.; Modiano-Esquenazi, M. Cerebral salt wasting syndrome in patients with aneurysmal subarachnoid hemorrhage. Neurol. Res. 2005, 27, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Cerda-Esteve, M.; Cuadrado-Godia, E.; Chillaron, J.J.; Pont-Sunyer, C.; Cucurella, G.; Fernandez, M.; Goday, A.; Cano-Pérez, J.F.; Rodríguez-Campello, A.; Roquer, J. Cerebral salt wasting syndrome: Review. Eur. J. Intern. Med. 2008, 19, 249–254. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannon, M.J.; Thompson, C.J. Neurosurgical Hyponatremia. J. Clin. Med. 2014, 3, 1084-1104. https://doi.org/10.3390/jcm3041084
Hannon MJ, Thompson CJ. Neurosurgical Hyponatremia. Journal of Clinical Medicine. 2014; 3(4):1084-1104. https://doi.org/10.3390/jcm3041084
Chicago/Turabian StyleHannon, Mark J., and Christopher J. Thompson. 2014. "Neurosurgical Hyponatremia" Journal of Clinical Medicine 3, no. 4: 1084-1104. https://doi.org/10.3390/jcm3041084
APA StyleHannon, M. J., & Thompson, C. J. (2014). Neurosurgical Hyponatremia. Journal of Clinical Medicine, 3(4), 1084-1104. https://doi.org/10.3390/jcm3041084