Roles of Sphingolipid Metabolism in Pancreatic β Cell Dysfunction Induced by Lipotoxicity



Abstract
: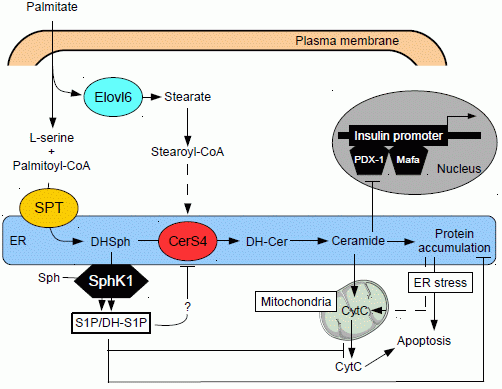
{kind=link}
{kind=link}
{kind=link}
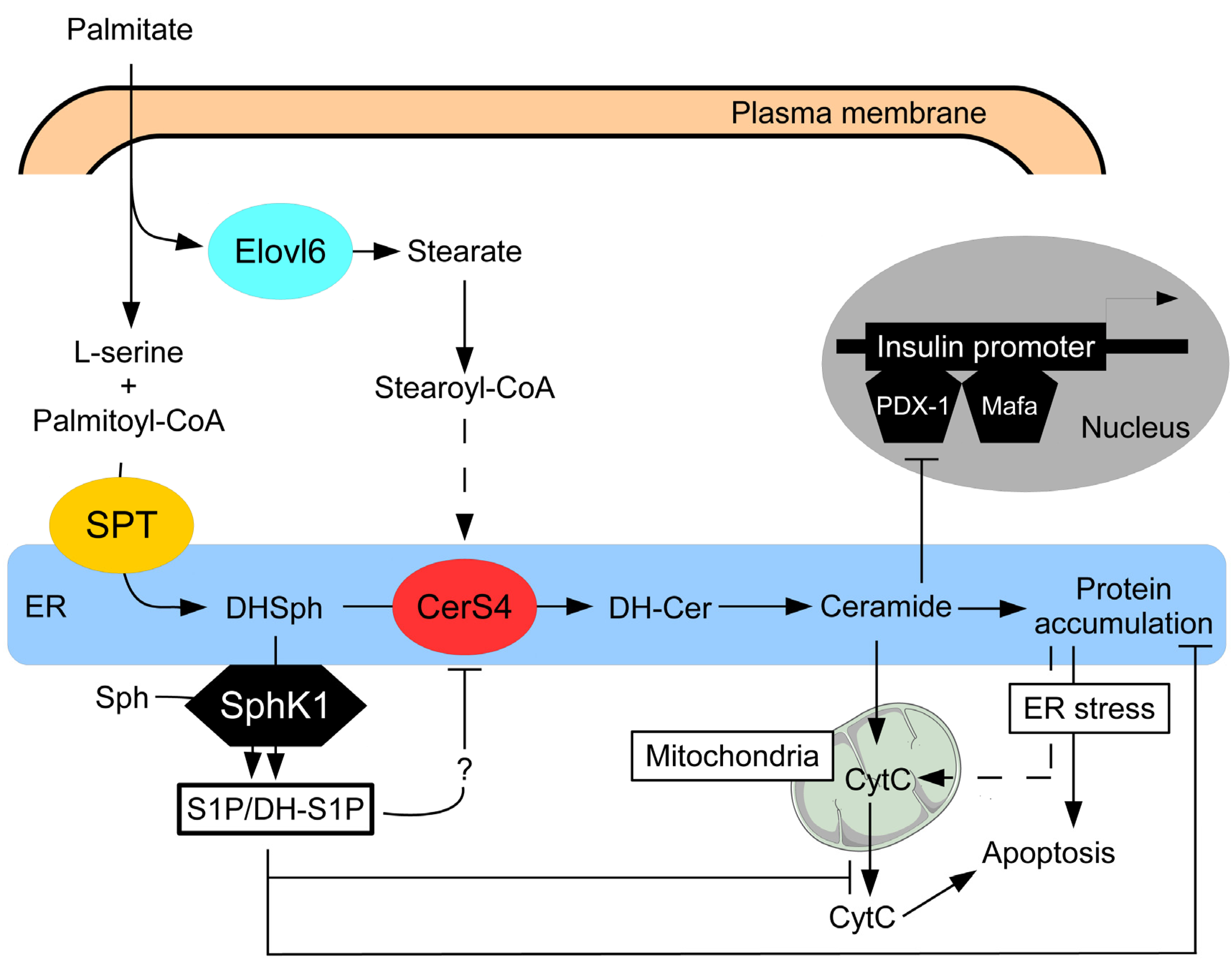{kind=link}
1. Introduction
2. Lipid Metabolism in Pancreatic β Cells
3. The Phenomenon of Pancreatic β Cell Gluco-Lipotoxicity
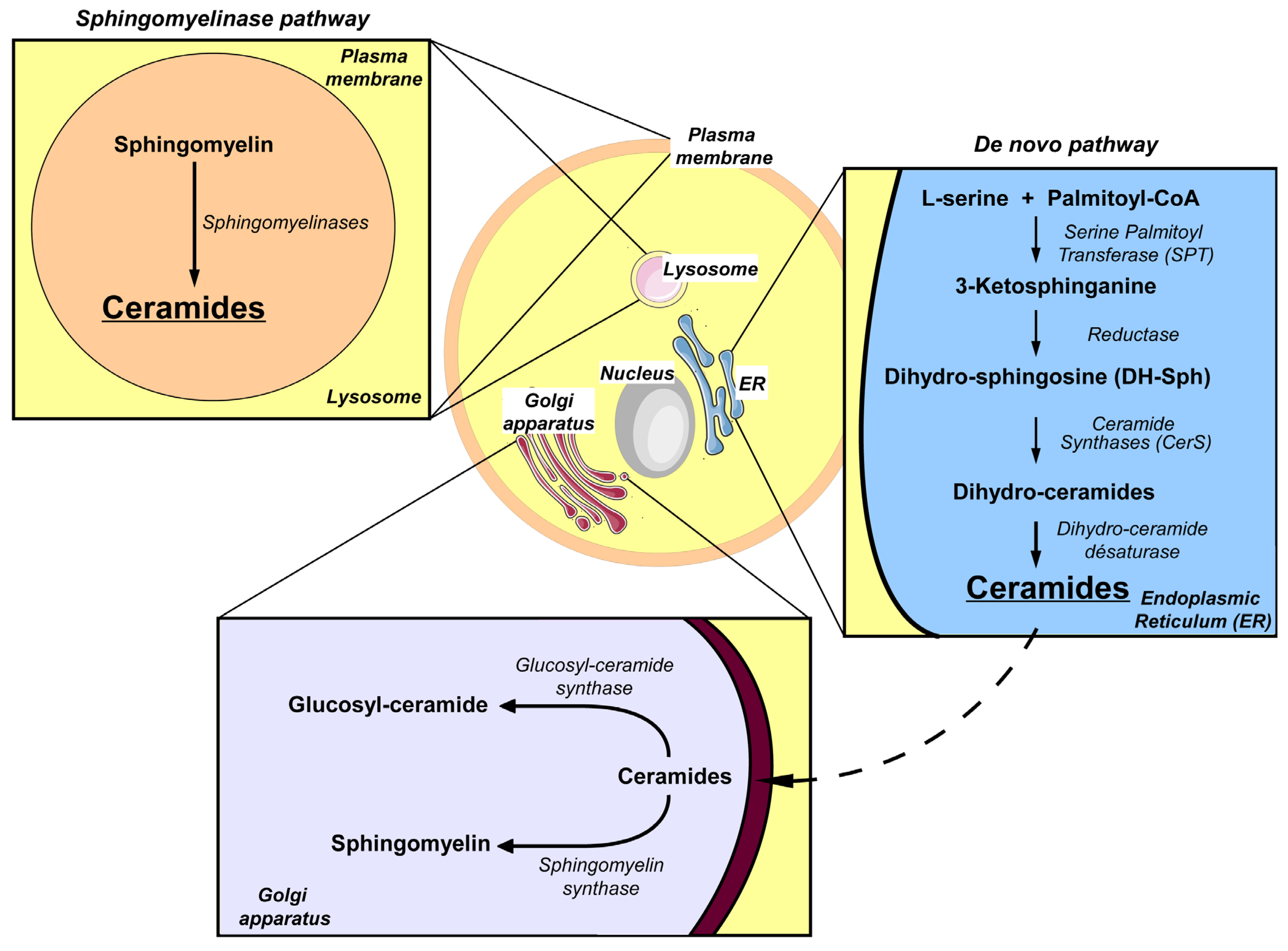
4. Sphingolipid Metabolism
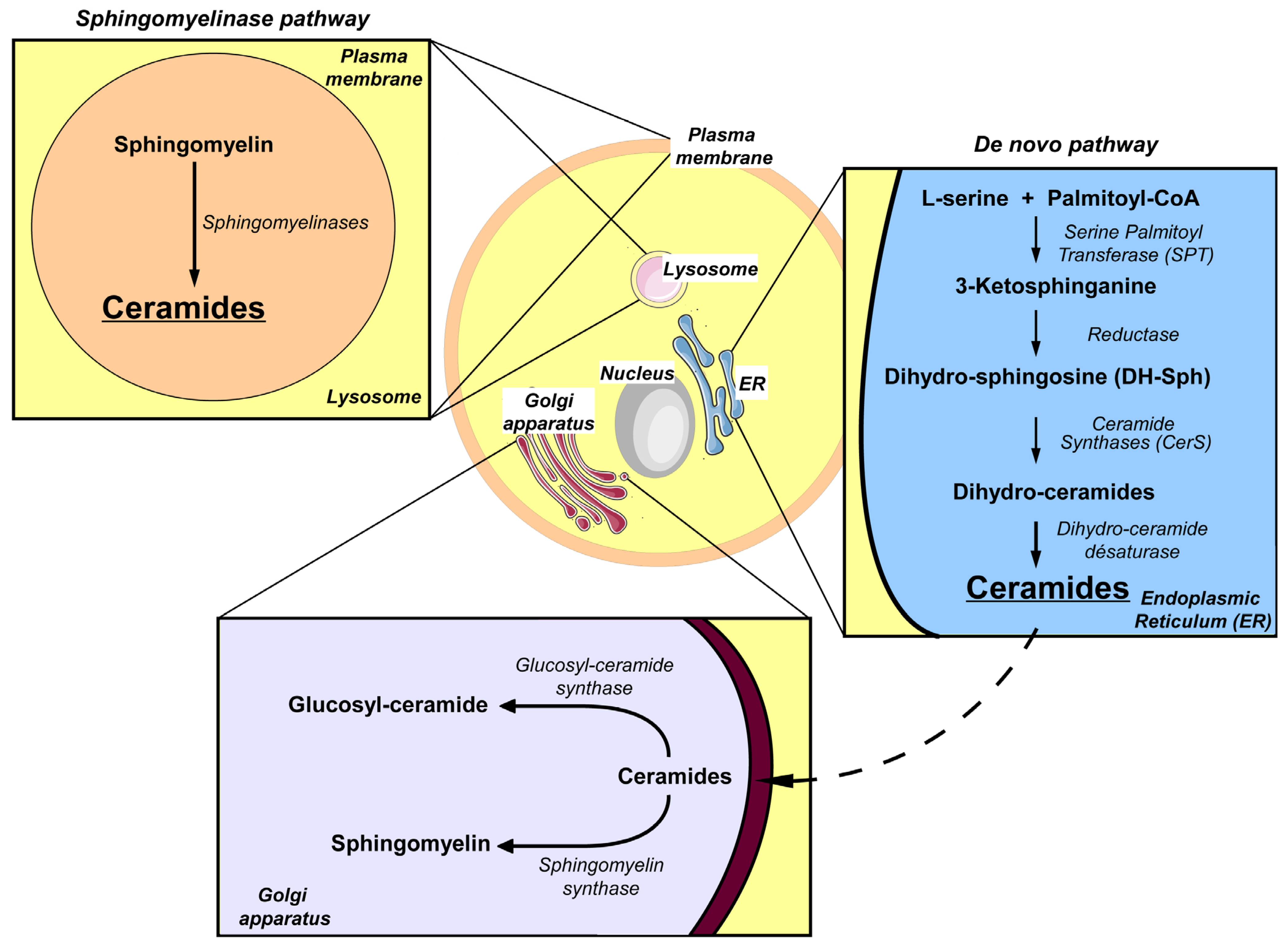
5. Ceramide and Pancreatic β Cell Gluco-Lipotoxicity
5.1. Effect of Ceramide on Pancreatic β Cell Function
5.2. Effect of Ceramide on Pancreatic β Cell Survival
5.3. Role of Different Ceramide Species in Gluco-Lipotoxicity Induced Pancreatic β Cell Apoptosis
6. Role of Sphingoid Base Phosphates in T2D
6.1. Sphingoid Base Phosphates and Insulin Secretion
6.2. Sphingoid Base Phosphates Pancreatic β Cell Survival
7. Conclusions

Acknowledgements
Abbreviations
| FFA | free fatty acid |
| LC-CoA | long chain acyl-CoA |
| S1P | shingosine-1-phosphate |
| CPT-1 | carnitine palmitoyl-CoA transferase 1 |
| ACC | acetyl-CoA carboxylase |
| FAS | fatty acid synthase |
| ER | endoplasmic reticulum |
| SPT | serine palmitoyl-transferase |
| DH-Sph | dihydrosphingosine |
| CerS | ceramide synthases |
| PASK | serine/threonine Per-Arnt-Sim domain-containing kinase |
| SMS1 | sphingomyelin synthase 1 |
| SphK | sphingosine kinase |
References
- Klöppel, G.; Löhr, M.; Habich, K.; Oberholzer, M.; Heitz, P.U. Islet Pathology and the Pathogenesis of Type 1 and Type 2 Diabetes Mellitus Revisited. Surv. Synth. Pathol. Res. 1985, 4, 110–125. [Google Scholar]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell Deficit and Increased Beta-cell Apoptosis in Humans with Type 2 Diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Khan, S.R.; Ayub, N.; Nawab, S.; Shamsi, T.S. Triglyceride Profile in Dyslipidaemia of Type 2 Diabetes Mellitus. J. Coll. Physicians Surg. Pak. 2008, 18, 270–273. [Google Scholar]
- Carey, D.G.; Jenkins, A.B.; Campbell, L.V.; Freund, J.; Chisholm, D.J. Abdominal fat and insulin resistance in normal and overweight women: Direct measurements reveal a strong relationship in subjects at both low and high risk of NIDDM. Diabetes 1996, 45, 633–638. [Google Scholar] [CrossRef]
- World Health Organisation. Available online: http://www.who.int/fr/ (accessed on 26 February 2014).
- Crespin, S.R.; Greenough, W.B., III; Steinberg, D. Stimulation of Insulin Secretion by Infusion of Free Fatty Acids. J. Clin. Invest. 1969, 48, 1934–1943. [Google Scholar] [CrossRef]
- Stein, D.T.; Stevenson, B.E.; Chester, M.W.; Basit, M.; Daniels, M.B.; Turley, S.D.; McGarry, J.D. The Insulinotropic Potency of Fatty Acids Is Influenced Profoundly by Their Chain Length and Degree of Saturation. J. Clin. Invest. 1997, 100, 398–403. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel Excess and Beta-cell Dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef]
- Boden, G.; Shulman, G.I. Free Fatty Acids in Obesity and Type 2 Diabetes: Defining Their Role in the Development of Insulin Resistance and Beta-cell Dysfunction. Eur. J. Clin. Invest. 2002, 32, 14–23. [Google Scholar] [CrossRef]
- Prentki, M.; Corkey, B.E. Are the Beta-cell Signaling Molecules Malonyl-CoA and Cystolic Long-chain Acyl-CoA Implicated in Multiple Tissue Defects of Obesity and NIDDM? Diabetes 1996, 45, 273–283. [Google Scholar] [CrossRef]
- Marshall, B.A.; Tordjman, K.; Host, H.H.; Ensor, N.J.; Kwon, G.; Marshall, C.A.; Coleman, T.; McDaniel, M.L.; Semenkovich, C.F. Relative Hypoglycemia and Hyperinsulinemia in Mice with Heterozygous Lipoprotein Lipase (LPL) Deficiency. Islet LPL Regulates Insulin Secretion. J. Biol. Chem. 1999, 274, 27426–27432. [Google Scholar] [CrossRef]
- Kim, K.H. Regulation of Mammalian Acetyl-coenzyme A Carboxylase. Annu. Rev. Nutr. 1997, 17, 77–99. [Google Scholar] [CrossRef]
- Tamarit-Rodriguez, J.; Vara, E.; Tamarit, J. Starvation-induced Secretory Changes of Insulin, Somatostatin, and Glucagon and Their Modification by 2-Bromostearate. Horm. Metab. Res. 1984, 16, 115–119. [Google Scholar] [CrossRef]
- Corkey, B.E.; Glennon, M.C.; Chen, K.S.; Deeney, J.T.; Matschinsky, F.M.; Prentki, M. A Role for Malonyl-CoA in Glucose-stimulated Insulin Secretion from Clonal Pancreatic Beta-cells. J. Biol. Chem. 1989, 264, 21608–21612. [Google Scholar]
- Liang, Y.; Matschinsky, F.M. Content of CoA-esters in Perifused Rat Islets Stimulated by Glucose and Other Fuels. Diabetes 1991, 40, 327–333. [Google Scholar] [CrossRef]
- Prentki, M.; Vischer, S.; Glennon, M.C.; Regazzi, R.; Deeney, J.T.; Corkey, B.E. Malonyl-CoA and Long Chain Acyl-CoA Esters as Metabolic Coupling Factors in Nutrient-induced Insulin Secretion. J. Biol. Chem. 1992, 267, 5802–5810. [Google Scholar]
- Brun, T.; Roche, E.; Assimacopoulos-Jeannet, F.; Corkey, B.E.; Kim, K.H.; Prentki, M. Evidence for an Anaplerotic/Malonyl-CoA Pathway in Pancreatic Beta-cell Nutrient Signaling. Diabetes 1996, 45, 190–198. [Google Scholar] [CrossRef]
- Wolf, B.A.; Easom, R.A.; McDaniel, M.L.; Turk, J. Diacylglycerol Synthesis De Novo from Glucose by Pancreatic Islets Isolated from Rats and Humans. J. Clin. Invest. 1990, 85, 482–490. [Google Scholar] [CrossRef]
- Dunlop, M.E.; Larkins, R.G. Pancreatic Islets Synthesize Phospholipids De Novo from Glucose via Acyl-dihydroxyacetone Phosphate. Biochem. Biophys. Res. Commun. 1985, 132, 467–473. [Google Scholar] [CrossRef]
- Farese, R.V.; DiMarco, P.E.; Barnes, D.E.; Sabir, M.A.; Larson, R.E.; Davis, J.S.; Morrison, A.D. Rapid Glucose-dependent Increases in Phosphatidic Acid and Phosphoinositides in Rat Pancreatic Islets. Endocrinology 1986, 118, 1498–1503. [Google Scholar] [CrossRef]
- Salt, I.P.; Johnson, G.; Ashcroft, S.J.; Hardie, D.G. AMP-Activated Protein Kinase Is Activated by Low Glucose in Cell Lines Derived from Pancreatic Beta Cells, and May Regulate Insulin Release. Biochem. J. 1998, 335, 533–539. [Google Scholar]
- Mittra, S.; Bansal, V.S.; Bhatnagar, P.K. From a Glucocentric to a Lipocentric Approach towards Metabolic Syndrome. Drug Discov. Today 2008, 13, 211–218. [Google Scholar] [CrossRef]
- Unger, R.H.; Orci, L. Diseases of Liporegulation: New Perspective on Obesity and Related Disorders. FASEB J. 2001, 15, 312–321. [Google Scholar] [CrossRef]
- Van Herpen, N.A.; Schrauwen-Hinderling, V.B. Lipid Accumulation in Non-adipose Tissue and Lipotoxicity. Physiol. Behav. 2008, 94, 231–241. [Google Scholar] [CrossRef]
- Kotronen, A.; Seppänen-Laakso, T.; Westerbacka, J.; Kiviluoto, T.; Arola, J.; Ruskeepää, A.L.; Yki-Järvinen, H.; Oresic, M. Comparison of Lipid and Fatty Acid Composition of the Liver, Subcutaneous and Intra-abdominal Adipose Tissue, and Serum. Obesity (Silver Spring) 2010, 18, 937–944. [Google Scholar] [CrossRef]
- Yetukuri, L.; Katajamaa, M.; Medina-Gomez, G.; Seppänen-Laakso, T.; Vidal-Puig, A.; Oresic, M. Bioinformatics Strategies for Lipidomics Analysis: Characterization of Obesity Related Hepatic Steatosis. BMC Syst. Biol. 2007, 1. [Google Scholar] [CrossRef]
- Sako, Y.; Grill, V.E. A 48-Hour Lipid Infusion in the Rat Time-dependently Inhibits Glucose-induced Insulin Secretion and B Cell Oxidation through a Process Likely Coupled to Fatty Acid Oxidation. Endocrinology 1990, 127, 1580–1589. [Google Scholar] [CrossRef]
- Zhou, Y.P.; Grill, V.E. Long-term Exposure of Rat Pancreatic Islets to Fatty Acids Inhibits Glucose-induced Insulin Secretion and Biosynthesis through a Glucose Fatty Acid Cycle. J. Clin. Invest. 1994, 93, 870–876. [Google Scholar] [CrossRef]
- Kelpe, C.L.; Moore, P.C.; Parazzoli, S.D.; Wicksteed, B.; Rhodes, C.J.; Poitout, V. Palmitate Inhibition of Insulin Gene Expression Is Mediated at the Transcriptional Level via Ceramide Synthesis. J. Biol. Chem. 2003, 278, 30015–30021. [Google Scholar]
- McGarry, J.D.; Dobbins, R.L. Fatty Acids, Lipotoxicity and Insulin Secretion. Diabetologia 1999, 42, 128–138. [Google Scholar] [CrossRef]
- Ritz-Laser, B.; Meda, P.; Constant, I.; Klages, N.; Charollais, A.; Morales, A.; Magnan, C.; Ktorza, A.; Philippe, J. Glucose-induced Preproinsulin Gene Expression Is Inhibited by the Free Fatty Acid Palmitate. Endocrinology 1999, 140, 4005–4014. [Google Scholar]
- El-Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated Fatty Acids Synergize with Elevated Glucose to Cause Pancreatic Beta-cell Death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef]
- Lupi, R.; Dotta, F.; Marselli, L.; Del Guerra, S.; Masini, M.; Santangelo, C.; Patané, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged Exposure to Free Fatty Acids Has Cytostatic and Pro-apoptotic Effects on Human Pancreatic Islets: Evidence that Beta-cell Death is Caspase Mediated, Partially Dependent on Ceramide Pathway, and Bcl-2 Regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef]
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct Effects of Saturated and Monounsaturated Fatty Acids on Beta-cell Turnover and Function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef]
- Shimabukuro, M.; Higa, M.; Zhou, Y.T.; Wang, M.Y.; Newgard, C.B.; Unger, R.H. Lipoapoptosis in Beta-cells of Obese Prediabetic fa/fa Rats. Role of Serine Palmitoyltransferase Overexpression. J. Biol. Chem. 1998, 273, 32487–32490. [Google Scholar]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty Acid-induced Beta Cell Apoptosis: A Link between Obesity and Diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef]
- Prentki, M.; Joly, E.; El-Assaad, W.; Roduit, R. Malonyl-CoA Signaling, Lipid Partitioning, and Glucolipotoxicity: Role in Beta-cell Adaptation and Failure in the Etiology of Diabetes. Diabetes 2002, 51, S405–S413. [Google Scholar] [CrossRef]
- Weir, G.C.; Laybutt, D.R.; Kaneto, H.; Bonner-Weir, S.; Sharma, A. Beta-cell Adaptation and Decompensation during the Progression of Diabetes. Diabetes 2001, 50, S154–S159. [Google Scholar] [CrossRef]
- Véret, J.; Coant, N.; Berdyshev, E.V.; Skobeleva, A.; Therville, N.; Bailbé, D.; Gorshkova, I.; Natarajan, V.; Portha, B.; Le Stunff, H. Ceramide Synthase 4 and De Novo Production of Ceramides with Specific N-Acyl Chain Lengths Are Involved in Glucolipotoxicity-induced Apoptosis of INS-1 β-Cells. Biochem. J. 2011, 438, 177–189. [Google Scholar] [CrossRef]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An Enigmatic Signalling Lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef]
- Kihara, A.; Mitsutake, S.; Mizutani, Y.; Igarashi, Y. Metabolism and Biological Functions of Two Phosphorylated Sphingolipids, Sphingosine 1-Phosphate and Ceramide 1-Phosphate. Prog. Lipid Res. 2007, 46, 126–144. [Google Scholar] [CrossRef]
- Taha, T.A.; Hannun, Y.A.; Obeid, L.M. Sphingosine Kinase: Biochemical and Cellular Regulation and Role in Disease. J. Biochem. Mol. Biol. 2006, 39, 113–131. [Google Scholar] [CrossRef]
- Le Stunff, H.; Milstien, S.; Spiegel, S. Generation and Metabolism of Bioactive Sphingosine-1-Phosphate. J. Cell. Biochem. 2004, 92, 882–899. [Google Scholar] [CrossRef]
- Causeret, C.; Geeraert, L.; van der Hoeven, G.; Mannaerts, G.P.; van Veldhoven, P.P. Further Characterization of Rat Dihydroceramide Desaturase: Tissue Distribution, Subcellular Localization, and Substrate Specificity. Lipids 2000, 35, 1117–1125. [Google Scholar] [CrossRef]
- Pewzner-Jung, Y.; Ben-Dor, S.; Futerman, A.H. When Do Lasses (Longevity Assurance Genes) Become CerS (Ceramide Synthases)? Insights into the Regulation of Ceramide Synthesis. J. Biol. Chem. 2006, 281, 25001–25005. [Google Scholar] [CrossRef]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular Machinery for Non-vesicular Trafficking of Ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma Ceramides Are Elevated in Obese Subjects with Type 2 Diabetes and Correlate with the Severity of Insulin Resistance. Diabetes 2009, 58, 337–343. [Google Scholar]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of Ceramide Synthesis Ameliorates Glucocorticoid-, Saturated-fat-, and Obesity-induced Insulin Resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef]
- Park, T.S.; Hu, Y.; Noh, H.L.; Drosatos, K.; Okajima, K.; Buchanan, J.; Tuinei, J.; Homma, S.; Jiang, X.C.; Abel, E.D.; Goldberg, I.J. Ceramide Is a Cardiotoxin in Lipotoxic Cardiomyopathy. J. Lipid Res. 2008, 49, 2101–2112. [Google Scholar] [CrossRef]
- Merrill, A.H., Jr.; Wang, E.; Mullins, R.E. Kinetics of Long-chain (Sphingoid) Base Biosynthesis in Intact LM Cells: Effects of Varying the Extracellular Concentrations of Serine and Fatty Acid Precursors of this Pathway. Biochemistry 1988, 27, 340–345. [Google Scholar] [CrossRef]
- Williams, R.D.; Nixon, D.W.; Merrill, A.H., Jr. Comparison of Serine Palmitoyltransferase in Morris Hepatoma 7777 and Rat Liver. Cancer Res. 1984, 44, 1918–1923. [Google Scholar]
- Perry, D.K. Serine Palmitoyltransferase: Role in Apoptotic De Novo Ceramide Synthesis and other Stress Responses. Biochim. Biophys. Acta 2002, 1585, 146–152. [Google Scholar] [CrossRef]
- Sjöholm, A. Ceramide Inhibits Pancreatic Beta-cell Insulin Production and Mitogenesis and Mimics the Actions of Interleukin-1 Beta. FEBS Lett. 1995, 367, 283–286. [Google Scholar] [CrossRef]
- Fontés, G.; Semache, M.; Hagman, D.K.; Tremblay, C.; Shah, R.; Rhodes, C.J.; Rutter, J.; Poitout, V. Involvement of Per-Arnt-Sim Kinase and Extracellular-regulated Kinases-1/2 in Palmitate Inhibition of Insulin Gene Expression in Pancreatic Beta-cells. Diabetes 2009, 58, 2048–2058. [Google Scholar] [CrossRef]
- Guo, J.; Qian, Y.; Xi, X.; Hu, X.; Zhu, J.; Han, X. Blockage of Ceramide Metabolism Exacerbates Palmitate Inhibition of Pro-insulin Gene Expression in Pancreatic Beta-cells. Mol. Cell. Biochem. 2010, 338, 283–290. [Google Scholar] [CrossRef]
- Yano, M.; Watanabe, K.; Yamamoto, T.; Ikeda, K.; Senokuchi, T.; Lu, M.; Kadomatsu, T.; Tsukano, H.; Ikawa, M.; Okabe, M.; et al. Mitochondrial Dysfunction and Increased Reactive Oxygen Species Impair Insulin Secretion in Sphingomyelin Synthase 1-Null Mice. J. Biol. Chem. 2011, 286, 3992–4002. [Google Scholar] [CrossRef]
- Guo, J.; Zhu, J.X.; Deng, X.H.; Hu, X.H.; Zhao, J.; Sun, Y.J.; Han, X. Palmitate-induced Inhibition of Insulin Gene Expression in Rat Islet β-Cells Involves the Ceramide Transport Protein. Cell. Physiol. Biochem. 2010, 26, 717–728. [Google Scholar] [CrossRef]
- Veluthakal, R.; Palanivel, R.; Zhao, Y.; McDonald, P.; Gruber, S.; Kowluru, A. Ceramide Induces Mitochondrial Abnormalities in Insulin-secreting INS-1 Cells: Potential Mechanisms Underlying Ceramide-mediated Metabolic Dysfunction of the Beta Cell. Apoptosis 2005, 10, 841–850. [Google Scholar] [CrossRef]
- Syed, I.; Szulc, Z.M.; Ogretmen, B.; Kowluru, A. l-Threo-C6-pyridinium-ceramide Bromide, a Novel Cationic Ceramide, Induces NADPH Oxidase Activation, Mitochondrial Dysfunction and Loss in Cell Viability in INS 832/13 β-Cells. Cell. Physiol. Biochem. 2012, 30, 1051–1058. [Google Scholar] [CrossRef]
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated Fatty Acids Prevent the Deleterious Effects of Palmitate and High Glucose on Human Pancreatic Beta-cell Turnover and Function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef]
- Boslem, E.; Weir, J.M.; MacIntosh, G.; Sue, N.; Cantley, J.; Meikle, P.J.; Biden, T.J. Alteration of Endoplasmic Reticulum Lipid Rafts Contributes to Lipotoxicity in Pancreatic β-cells. J. Biol. Chem. 2013, 288, 26569–26582. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The Role for Endoplasmic Reticulum Stress in Diabetes Mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef]
- Kogot-Levin, A.; Saada, A. Ceramide and the Mitochondrial Respiratory Chain. Biochimie 2014. [Google Scholar] [CrossRef]
- Moore, P.C.; Ugas, M.A.; Hagman, D.K.; Parazzoli, S.D.; Poitout, V. Evidence against the Involvement of Oxidative Stress in Fatty Acid Inhibition of Insulin Secretion. Diabetes 2004, 53, 2610–2616. [Google Scholar] [CrossRef]
- Lang, F.; Ullrich, S.; Gulbins, E. Ceramide formation as a target in beta-cell survival and function. Expert Opin. Ther. Targets 2011, 15, 1061–1071. [Google Scholar] [CrossRef]
- Blouin, C.M.; Prado, C.; Takane, K.K.; Lasnier, F.; Garcia-Ocana, A.; Ferré, P.; Dugail, I.; Hajduch, E. Plasma Membrane Subdomain Compartmentalization Contributes to Distinct Mechanisms of Ceramide Action on Insulin Signaling. Diabetes 2010, 59, 600–610. [Google Scholar] [CrossRef]
- Merril, A.H., Jr. SphinGOMAP—A Web-based Biosynthetic Pathway Map of Sphingolipids and Glycosphingolipids. Glycobiology 2005, 15, 15G. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. Mammalian Lass6 and Its Related Family Members Regulate Synthesis of Specific Ceramides. Biochem. J. 2005, 390, 263–271. [Google Scholar] [CrossRef]
- Green, C.D.; Olson, L.K. Modulation of Palmitate-induced Endoplasmic Reticulum Stress and Apoptosis in Pancreatic β-Cells by Stearoyl-CoA Desaturase and Elovl6. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E640–E649. [Google Scholar] [CrossRef]
- Guillou, H.; Zadravec, D.; Martin, P.G.P.; Jacobsson, A. The Key Roles of Elongases and Desaturases in Mammalian Fatty Acid Metabolism: Insights from Transgenic Mice. Prog. Lipid Res. 2010, 49, 186–199. [Google Scholar] [CrossRef]
- El-Assaad, W.; Joly, E.; Barbeau, A.; Sladek, R.; Buteau, J.; Maestre, I.; Pepin, E.; Zhao, S.; Iglesias, J.; Roche, E.; Prentki, M. Glucolipotoxicity alters lipid partitioning and causes mitochondrial dysfunction, cholesterol, and ceramide deposition and reactive oxygen species production in INS832/13 ss-cells. Endocrinology 2010, 151, 3061–3073. [Google Scholar] [CrossRef]
- Park, J.W.; Park, W.J.; Kuperman, Y.; Boura-Halfon, S.; Pewzner-Jung, Y.; Futerman, A.H. Ablation of very long acyl chain sphingolipids causes hepatic insulin resistance in mice due to altered detergent-resistant membranes. Hepatology 2013, 57, 525–532. [Google Scholar] [CrossRef]
- Baranowski, M.; Blachnio-Zabielska, A.; Hirnle, T.; Harasiuk, D.; Matlak, K.; Knapp, M.; Zabielski, P.; Gorski, J. Myocardium of Type 2 Diabetic and Obese Patients Is Characterized by Alterations in Sphingolipid Metabolic Enzymes but Not by Accumulation of Ceramide. J. Lipid Res. 2010, 51, 74–80. [Google Scholar] [CrossRef]
- Hu, W.; Bielawski, J.; Samad, F.; Merrill, A.H., Jr.; Cowart, L.A. Palmitate Increases Sphingosine-1-phosphate in C2C12 Myotubes via Upregulation of Sphingosine Kinase Message and Activity. J. Lipid Res. 2009, 50, 1852–1862. [Google Scholar] [CrossRef]
- Mastrandrea, L.D.; Sessanna, S.M.; Del Toro, A.; Laychock, S.G. ATP-Independent Glucose Stimulation of Sphingosine Kinase in Rat Pancreatic Islets. J. Lipid Res. 2010, 51, 2171–2180. [Google Scholar] [CrossRef]
- Wang, L.; Xing, X.P.; Holmes, A.; Wadham, C.; Gamble, J.R.; Vadas, M.A.; Xia, P. Activation of the Sphingosine Kinase-signaling Pathway by High Glucose Mediates the Proinflammatory Phenotype of Endothelial Cells. Circ. Res. 2005, 97, 891–899. [Google Scholar] [CrossRef]
- Whetzel, A.M.; Bolick, D.T.; Hedrick, C.C. Sphingosine-1-phosphate Inhibits High Glucose-mediated ERK1/2 Action in Endothelium through Induction of MAP Kinase Phosphatase-3. Am. J. Physiol. Cell Physiol. 2009, 296, C339–C345. [Google Scholar] [CrossRef]
- You, B.; Ren, A.; Yan, G.; Sun, J. Activation of Sphingosine Kinase-1 Mediates Inhibition of Vascular Smooth Muscle Cell Apoptosis by Hyperglycemia. Diabetes 2007, 56, 1445–1453. [Google Scholar] [CrossRef]
- Ma, M.M.; Chen, J.L.; Wang, G.G.; Wang, H.; Lu, Y.; Li, J.F.; Yi, J.; Yuan, Y.J.; Zhang, Q.W.; Mi, J.; Wang, L.S.; Duan, H.F.; Wu, C.T. Sphingosine Kinase 1 Participates in Insulin Signalling and Regulates Glucose Metabolism and Homeostasis in KK/Ay Diabetic Mice. Diabetologia 2007, 50, 891–900. [Google Scholar] [CrossRef]
- Kendall, M.R.; Hupfeld, C.J. FTY720, a Sphingosine-1-phosphate Receptor Modulator, Reverses High-fat Diet-induced Weight Gain, Insulin Resistance and Adipose Tissue Inflammation in C57BL/6 Mice. Diabetes Obes. Metab. 2008, 10, 802–805. [Google Scholar] [CrossRef]
- Maines, L.W.; French, K.J.; Wolpert, E.B.; Antonetti, D.A.; Smith, C.D. Pharmacologic Manipulation of Sphingosine Kinase in Retinal Endothelial Cells: Implications for Angiogenic Ocular Diseases. Invest. Ophthalmol. Vis. Sci. 2006, 47, 5022–5031. [Google Scholar] [CrossRef]
- Samad, F.; Hester, K.D.; Yang, G.; Hannun, Y.A.; Bielawski, J. Altered Adipose and Plasma Sphingolipid Metabolism in Obesity: A Potential Mechanism for Cardiovascular and Metabolic Risk. Diabetes 2006, 55, 2579–2587. [Google Scholar] [CrossRef]
- Wang, J.; Badeanlou, L.; Bielawski, J.; Ciaraldi, T.P.; Samad, F. Sphingosine Kinase 1 Regulates Adipose Proinflammatory Responses and Insulin Resistance. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E756–E768. [Google Scholar] [CrossRef]
- Laychock, S.G.; Tian, Y.; Sessanna, S.M. Endothelial Differentiation Gene Receptors in Pancreatic Islets and INS-1 Cells. Diabetes 2003, 52, 1986–1993. [Google Scholar] [CrossRef]
- Mastrandrea, L.D.; Sessanna, S.M.; Laychock, S.G. Sphingosine Kinase Activity and Sphingosine-1 Phosphate Production in Rat Pancreatic Islets and INS-1 Cells: Response to Cytokines. Diabetes 2005, 54, 1429–1436. [Google Scholar] [CrossRef]
- Véret, J.; Coant, N.; Gorshkova, I.A.; Giussani, P.; Fradet, M.; Riccitelli, E.; Skobeleva, A.; Goya, J.; Kassis, N.; Natarajan, V.; et al. Role of Palmitate-induced Sphingoid Base-1-phosphate Biosynthesis in INS-1 β-Cell Survival. Biochim. Biophys. Acta 2013, 1831, 251–262. [Google Scholar] [CrossRef]
- Shimizu, H.; Okajima, F.; Kimura, T.; Ohtani, K.; Tsuchiya, T.; Takahashi, H.; Kuwabara, A.; Tomura, H.; Sato, K.; Mori, M. Sphingosine 1-Phosphate Stimulates Insulin Secretion in HIT-T 15 Cells and Mouse Islets. Endocr. J. 2000, 47, 261–269. [Google Scholar] [CrossRef]
- Cantrell Stanford, J.; Morris, A.J.; Sunkara, M.; Popa, G.J.; Larson, K.L.; Özcan, S. Sphingosine 1-Phosphate (S1P) Regulates Glucose-stimulated Insulin Secretion in Pancreatic Beta Cells. J. Biol. Chem. 2012, 287, 13457–13464. [Google Scholar] [CrossRef]
- Allende, M.L.; Sasaki, T.; Kawai, H.; Olivera, A.; Mi, Y.; van Echten-Deckert, G.; Hajdu, R.; Rosenbach, M.; Keohane, C.A.; Mandala, S.; Spiegel, S.; Proia, R.L. Mice Deficient in Sphingosine Kinase 1 Are Rendered Lymphopenic by FTY720. J. Biol. Chem. 2004, 279, 52487–52492. [Google Scholar] [CrossRef]
- Kharel, Y.; Lee, S.; Snyder, A.H.; Sheasley-O’neill, S.L.; Morris, M.A.; Setiady, Y.; Zhu, R.; Zigler, M.A.; Burcin, T.L.; Ley, K.; et al. Sphingosine Kinase 2 Is Required for Modulation of Lymphocyte Traffic by FTY720. J. Biol. Chem. 2005, 280, 36865–36872. [Google Scholar] [CrossRef]
- Laychock, S.G.; Sessanna, S.M.; Lin, M.H.; Mastrandrea, L.D. Sphingosine 1-Phosphate Affects Cytokine-induced Apoptosis in Rat Pancreatic Islet Beta-cells. Endocrinology 2006, 147, 4705–4712. [Google Scholar] [CrossRef]
- Imasawa, T.; Koike, K.; Ishii, I.; Chun, J.; Yatomi, Y. Blockade of Sphingosine 1-Phosphate Receptor 2 Signaling Attenuates Streptozotocin-induced Apoptosis of Pancreatic Beta-cells. Biochem. Biophys. Res. Commun. 2010, 392, 207–211. [Google Scholar] [CrossRef]
- Qi, Y.; Chen, J.; Lay, A.; Don, A.; Vadas, M.; Xia, P. Loss of Sphingosine Kinase 1 Predisposes to the Onset of Diabetes via Promoting Pancreatic β-Cell Death in Diet-induced Obese Mice. FASEB J. 2013, 27, 4294–4304. [Google Scholar] [CrossRef]
- Levy, M.; Futerman, A.H. Mammalian Ceramide Synthases. IUBMB Life 2010, 62, 347–356. [Google Scholar]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated Activation of Ceramidase Activity Initiates the Pleiotropic Actions of Adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Véret, J.; Bellini, L.; Giussani, P.; Ng, C.; Magnan, C.; Stunff, H.L. Roles of Sphingolipid Metabolism in Pancreatic β Cell Dysfunction Induced by Lipotoxicity. J. Clin. Med. 2014, 3, 646-662. https://doi.org/10.3390/jcm3020646
Véret J, Bellini L, Giussani P, Ng C, Magnan C, Stunff HL. Roles of Sphingolipid Metabolism in Pancreatic β Cell Dysfunction Induced by Lipotoxicity. Journal of Clinical Medicine. 2014; 3(2):646-662. https://doi.org/10.3390/jcm3020646
Chicago/Turabian StyleVéret, Julien, Lara Bellini, Paola Giussani, Carl Ng, Christophe Magnan, and Hervé Le Stunff. 2014. "Roles of Sphingolipid Metabolism in Pancreatic β Cell Dysfunction Induced by Lipotoxicity" Journal of Clinical Medicine 3, no. 2: 646-662. https://doi.org/10.3390/jcm3020646
APA StyleVéret, J., Bellini, L., Giussani, P., Ng, C., Magnan, C., & Stunff, H. L. (2014). Roles of Sphingolipid Metabolism in Pancreatic β Cell Dysfunction Induced by Lipotoxicity. Journal of Clinical Medicine, 3(2), 646-662. https://doi.org/10.3390/jcm3020646