Where Do Bone-Targeted Agents RANK in Breast Cancer Treatment?

Abstract
:
1. Introduction
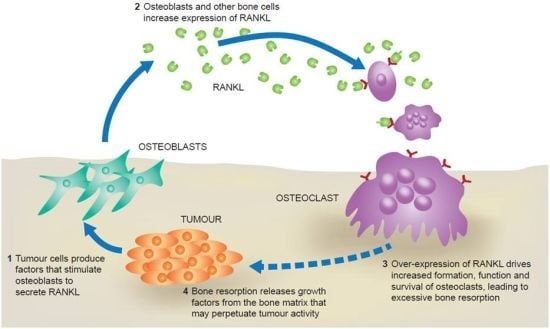
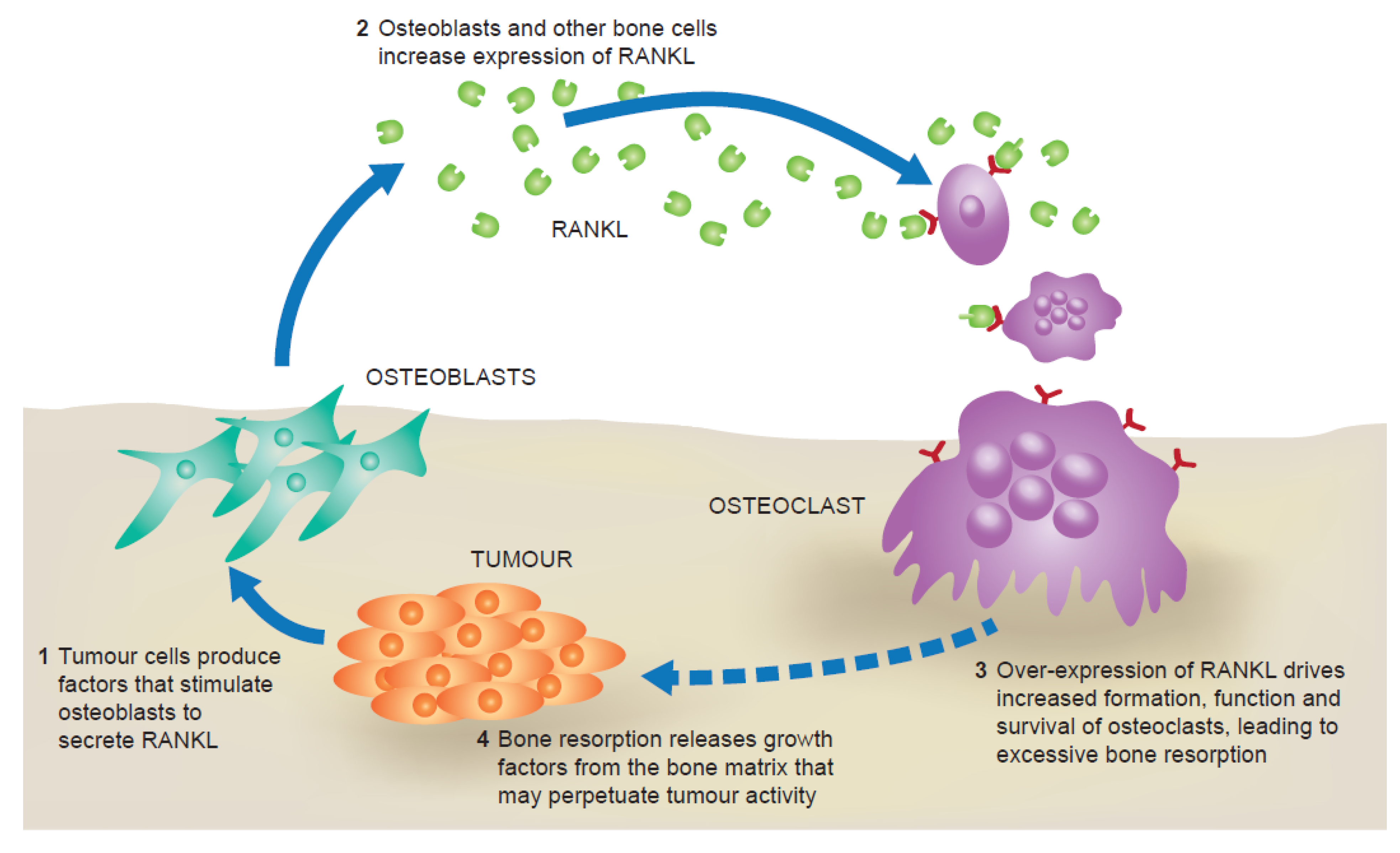
2. The RANK Pathway in Bone Metastasis
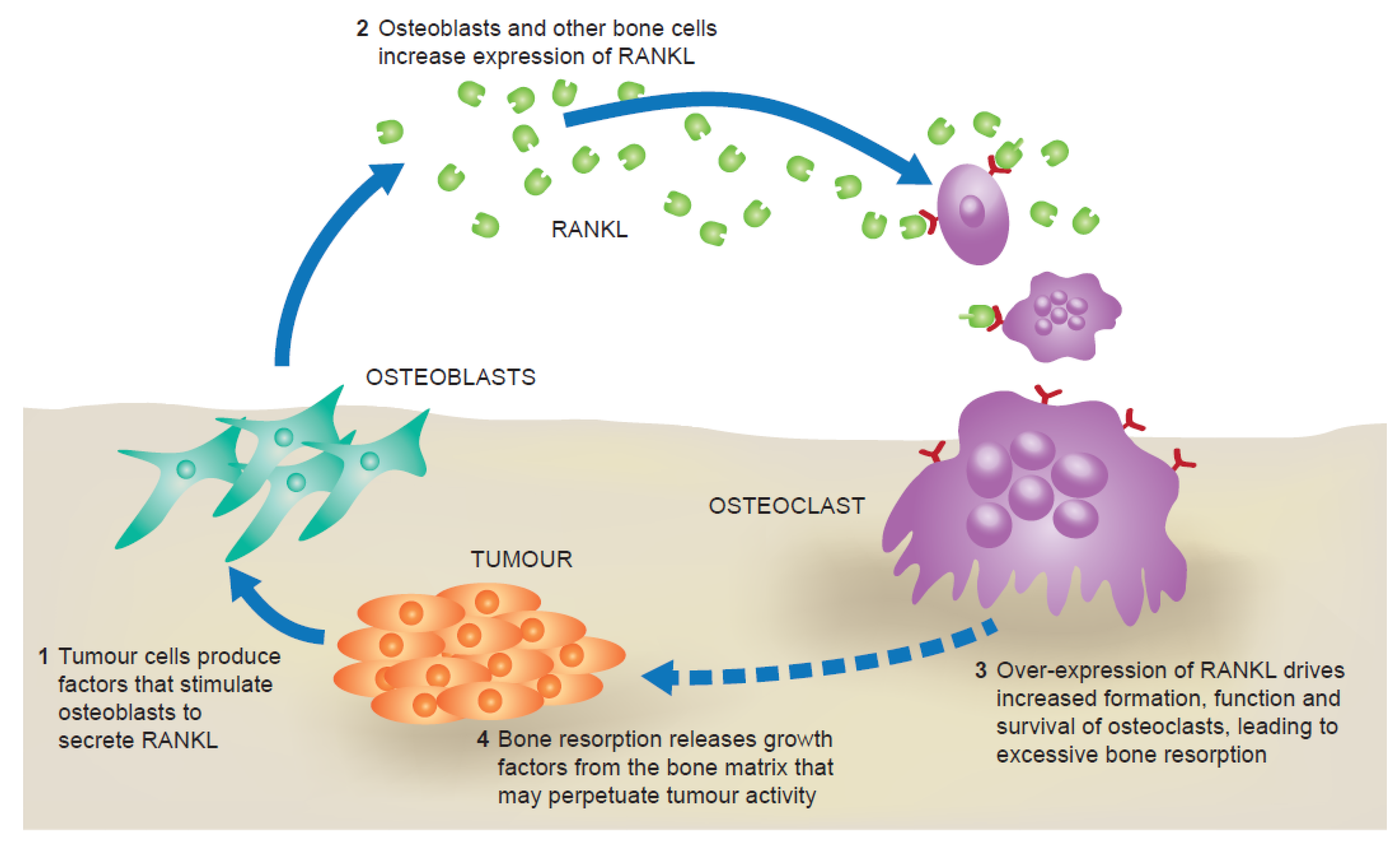

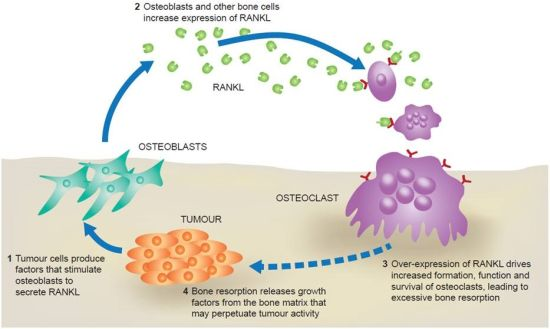{kind=link}
{kind=link}
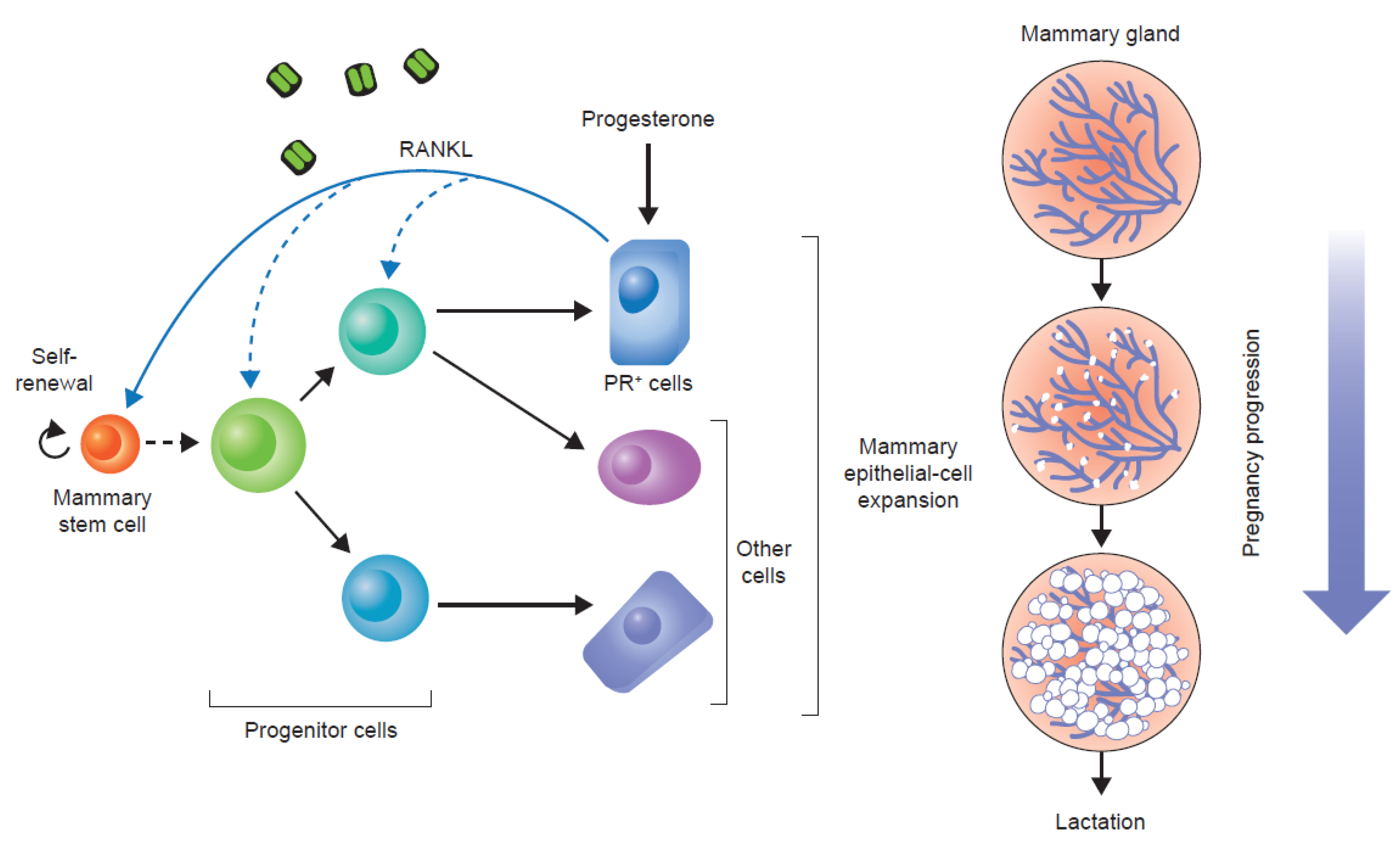{kind=link}
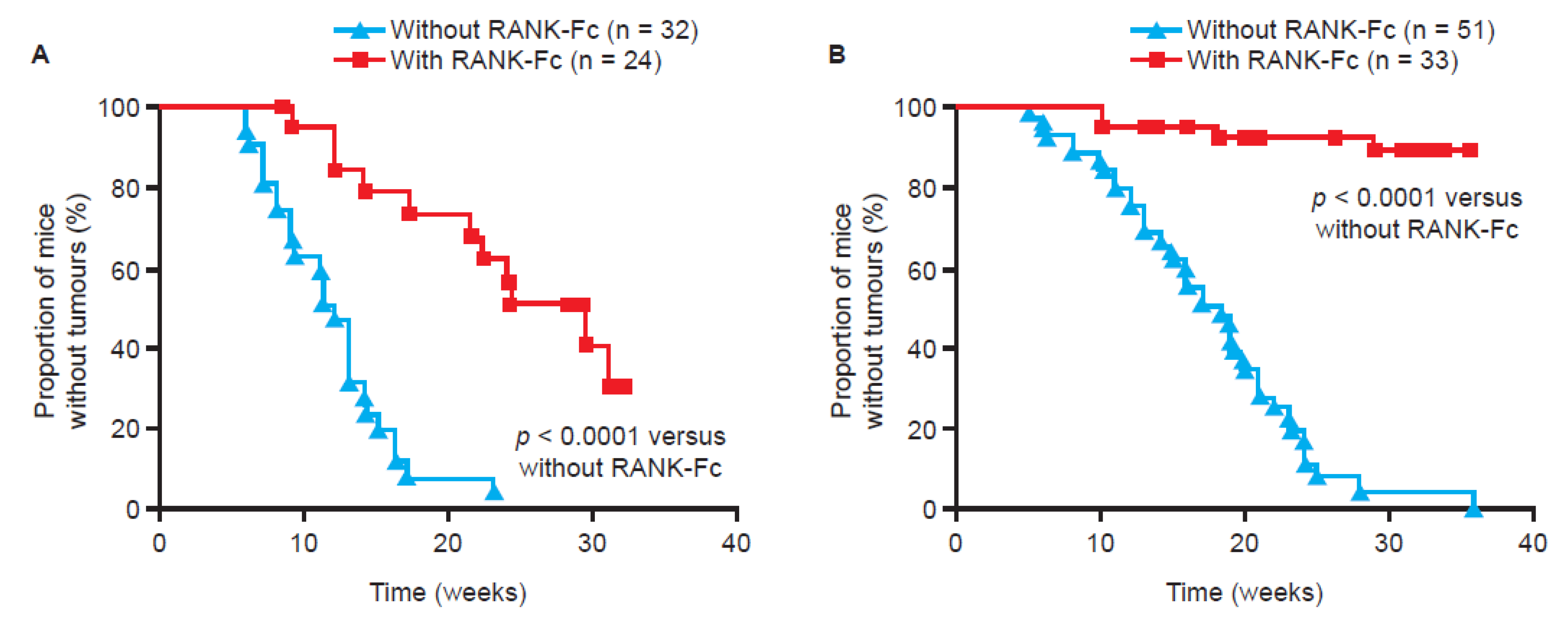{kind=link}
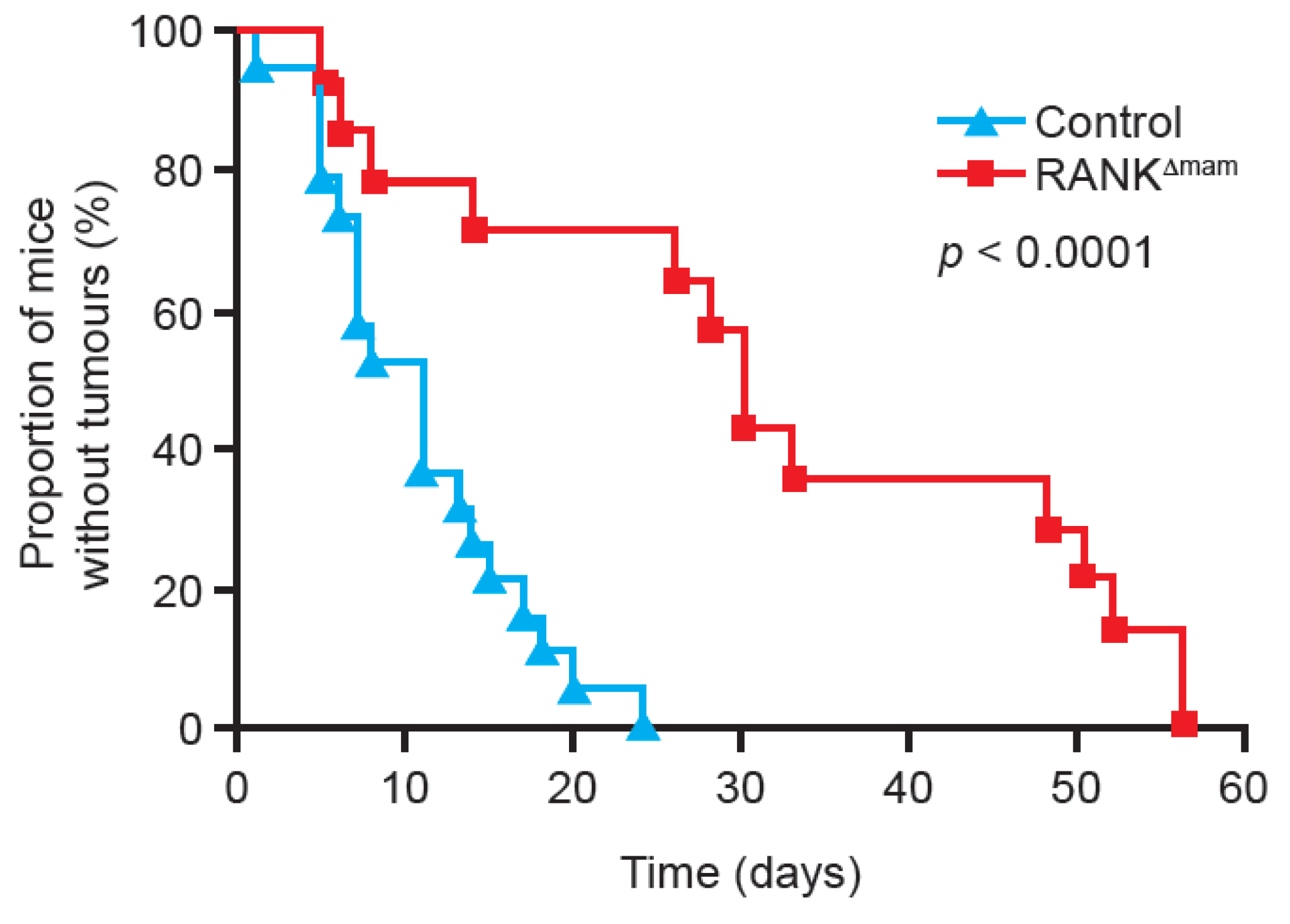{kind=link}
| Study arms | Zoledronic acid 4 mg i.v. Q4W | Denosumab 120 mg s.c. Q4W |
|---|---|---|
| Number of patients | 1020 | 1026 |
| Time to first SRE, months | 26.4 | NR |
| HR (95% CI) | 0.82 (0.71–0.95) | |
| p Value | <0.001 (non-inferiority) 0.01 (superiority) | |
| Time to first and subsequent SRE, RR (95% CI) | 0.77 (0.66–0.89) | |
| p Value | 0.001 (superiority) | |
3. RANKL and Tumour Progression
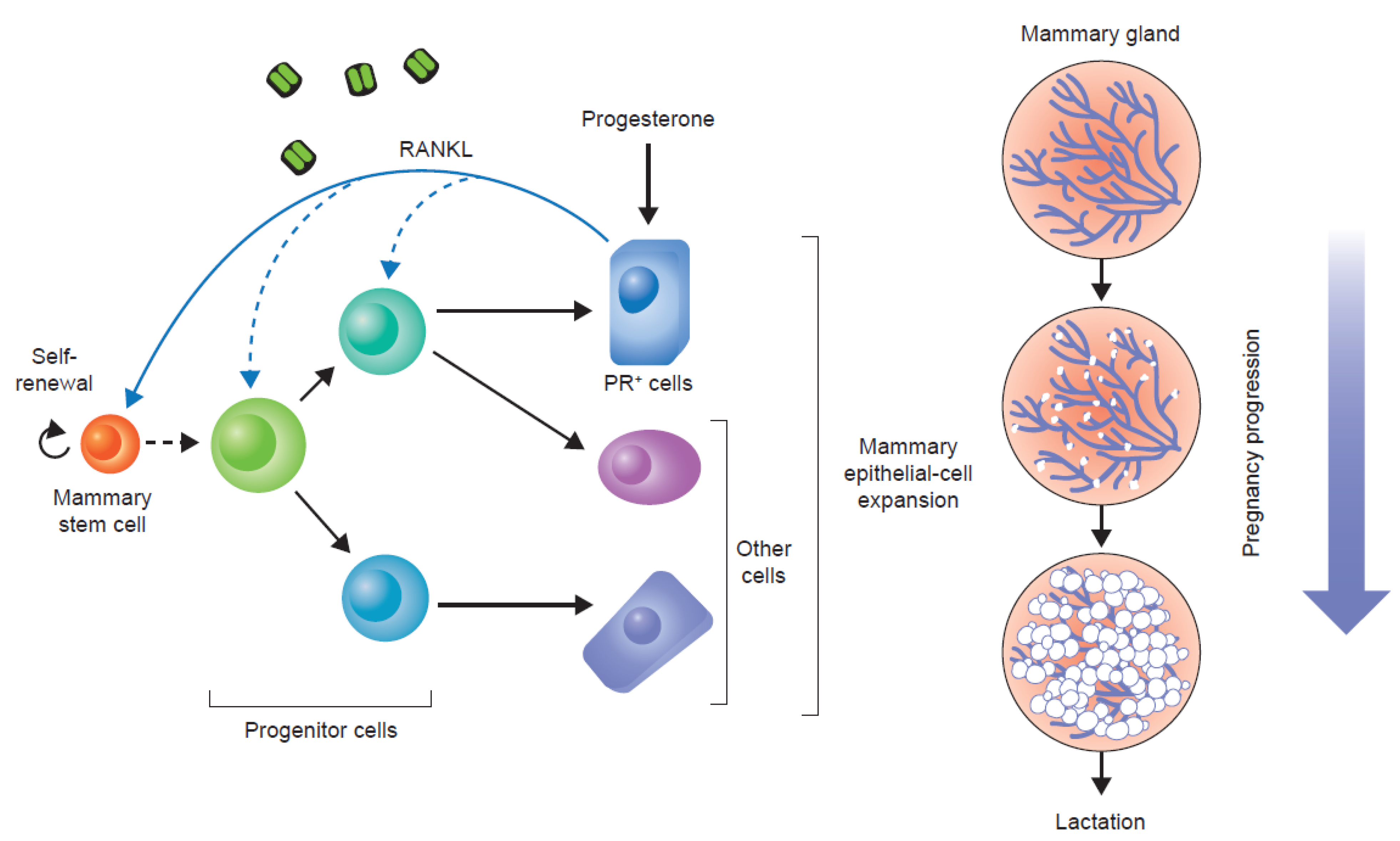
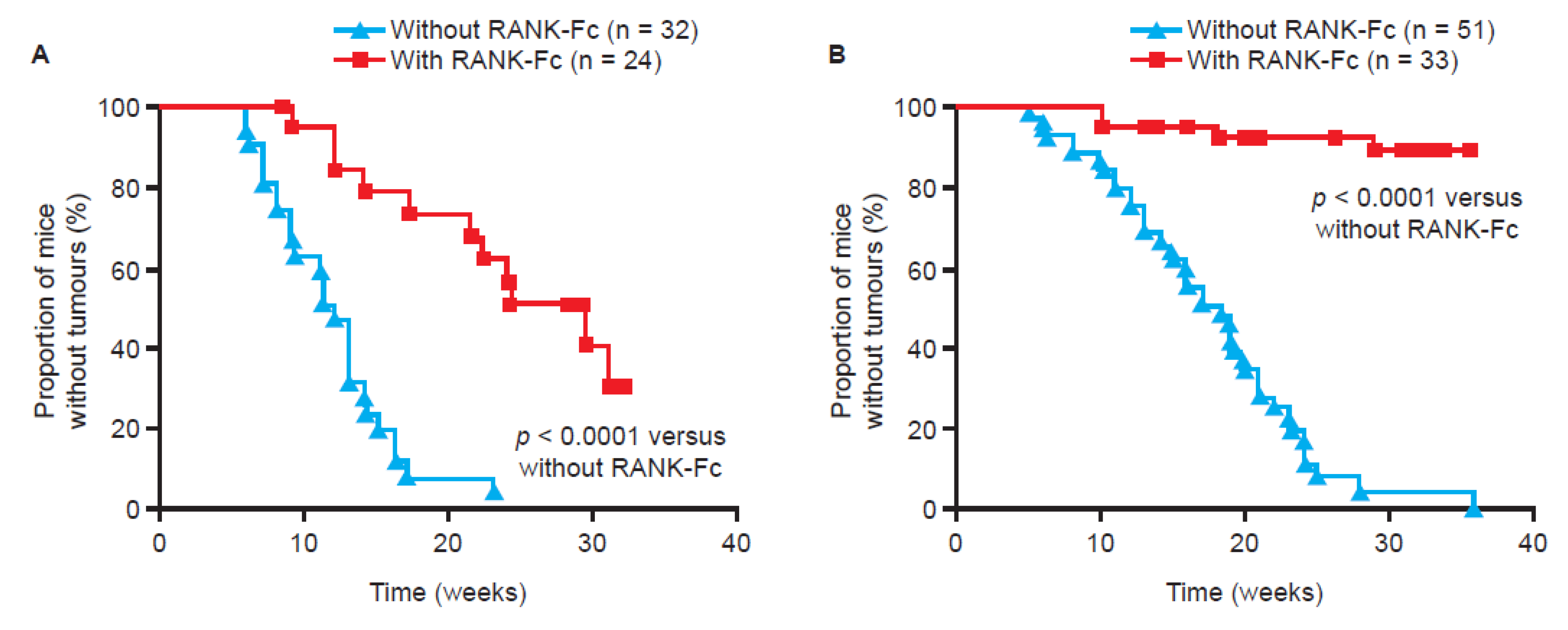
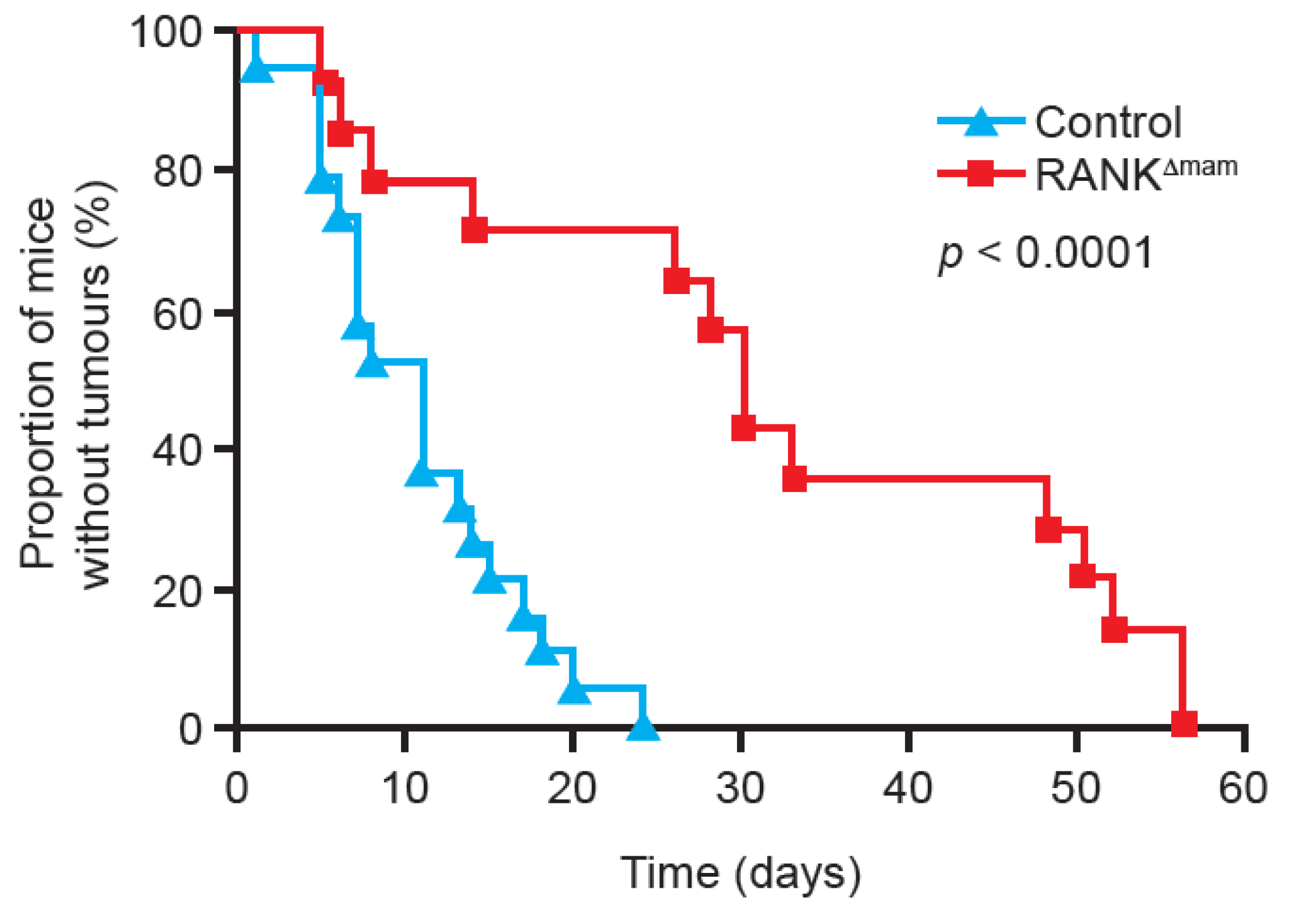
4. Anti-Tumour Effects of Bone-Targeted Agents in Clinical Trials
| Study | ABCSG-12 [36] | AZURE [37] | E-ZO-FAST [38] | Z-FAST [38] | ZO-FAST [39] |
|---|---|---|---|---|---|
| Study design | Goserelin with tamoxifen or anastrozole ± zoledronic acid (4 mg Q6M) | SOC ± zoledronic acid (6 doses 4 mg Q3W–Q4W, then 4 mg Q3M–Q6M) | Upfront zoledronic acid (4 mg Q6M) vs. delayed treatment | Upfront zoledronic acid (4 mg Q6M) vs. delayed treatment | Upfront zoledronic acid (4 mg Q6M) vs. delayed treatment |
| Number of patients | 1803 | 3360 | 527 | 602 | 1065 |
| Primary endpoint | Disease-free survival | Disease-free survival | LS BMD at 12 months | LS BMD at 12 months | LS BMD at 12 months |
| Median length of follow up, months | 76 | 59 | 36 | 54 | 60 |
| Disease-free survival, HR (95% CI) | 0.73 (NR) | 0.98 (0.85, 1.13) | 1.76 (0.83, 3.69) | 0.80 (0.45, 1.41) | 0.66 (0.44, 0.97) |
| p Value | 0.022 | 0.79 | NS | NS | 0.0375 |
| Overall survival, HR (95% CI) | 0.59 (NR) | 0.85 (0.72, 1.01) | NR | NR | 0.69 (0.42, 1.14) |
| p Value | 0.027 | 0.07 | NR | NR | 0.1463 |
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243–6249. [Google Scholar] [CrossRef]
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef]
- DePuy, V.; Anstrom, K.J.; Castel, L.D.; Schulman, K.A.; Weinfurt, K.P.; Saad, F. Effects of skeletal morbidities on longitudinal patient-reported outcomes and survival in patients with metastatic prostate cancer. Support. Care Cancer 2007, 15, 869–876. [Google Scholar] [CrossRef]
- Sathiakumar, N.; Delzell, E.; Morrisey, M.A.; Falkson, C.; Yong, M.; Chia, V.; Blackburn, J.; Arora, T.; Brill, I.; Kilgore, M.L. Mortality following bone metastasis and skeletal-related events among women with breast cancer: A population-based analysis of U. S. medicare beneficiaries, 1999–2006. Breast Cancer Res. Treat. 2012, 131, 231–238. [Google Scholar] [CrossRef]
- Costa, L.; Badia, X.; Chow, E.; Lipton, A.; Wardley, A. Impact of skeletal complications on patients’ quality of life, mobility, and functional independence. Support. Care Cancer 2008, 16, 879–889. [Google Scholar] [CrossRef]
- Briasoulis, E.; Karavasilis, V.; Kostadima, L.; Ignatiadis, M.; Fountzilas, G.; Pavlidis, N. Metastatic breast carcinoma confined to bone: Portrait of a clinical entity. Cancer 2004, 101, 1524–1528. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wu, G.Y.; Shen, M.J.; Cui, K.W.; Shen, Y. Comparison of distribution characteristics of metastatic bone lesions between breast and prostate carcinomas. Oncol. Lett. 2013, 5, 391–397. [Google Scholar]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Kingsley, L.A.; Fournier, P.G.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Dougall, W.C. Molecular pathways: Osteoclast-dependent and osteoclast-independent roles of the RANKL/RANK/OPG pathway in tumorigenesis and metastasis. Clin. Cancer Res. 2012, 18, 326–335. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Guise, T.A.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Invest. 1996, 98, 1544–1549. [Google Scholar] [CrossRef]
- Karaplis, A.C.; Goltzman, D. PTH and PTHrP effects on the skeleton. Rev. Endocr. Metab. Disord. 2000, 1, 331–341. [Google Scholar] [CrossRef]
- Canon, J.R.; Roudier, M.; Bryant, R.; Morony, S.; Stolina, M.; Kostenuik, P.J.; Dougall, W.C. Inhibition of RANKL blocks skeletal tumor progression and improves survival in a mouse model of breast cancer bone metastasis. Clin. Exp. Metastasis 2008, 25, 119–129. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhou, H.; Modzelewski, J.R.; Kalak, R.; Blair, J.M.; Seibel, M.J.; Dunstan, C.R. Accelerated bone resorption, due to dietary calcium deficiency, promotes breast cancer tumor growth in bone. Cancer Res. 2007, 67, 9542–9548. [Google Scholar] [CrossRef]
- Stopeck, A.T.; Lipton, A.; Body, J.J.; Steger, G.G.; Tonkin, K.; de Boer, R.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.; Viniegra, M.; et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: A randomized, double-blind study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef]
- Fizazi, K.; Carducci, M.; Smith, M.; Damião, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef]
- Henry, D.H.; Costa, L.; Goldwasser, F.; Hirsh, V.; Hungria, V.; Prausova, J.; Scagliotti, G.V.; Sleeboom, H.; Spencer, A.; Vadhan-Raj, S.; et al. Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J. Clin. Oncol. 2011, 29, 1125–1132. [Google Scholar] [CrossRef]
- Lipton, A.; Fizazi, K.; Stopeck, A.T.; Henry, D.H.; Brown, J.E.; Yardley, D.A.; Richardson, G.E.; Siena, S.; Maroto, P.; Clemens, M.; et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: A combined analysis of 3 pivotal, randomised, phase 3 trials. Eur. J. Cancer 2012, 48, 3082–3092. [Google Scholar] [CrossRef]
- Cleeland, C.S.; Body, J.J.; Stopeck, A.; von Moos, R.; Fallowfield, L.; Mathias, S.D.; Patrick, D.L.; Clemons, M.; Tonkin, K.; Masuda, N.; et al. Pain outcomes in patients with advanced breast cancer and bone metastases: Results from a randomized, double-blind study of denosumab and zoledronic acid. Cancer 2013, 119, 832–838. [Google Scholar] [CrossRef]
- Martin, M.; Bell, R.; Bourgeois, H.; Brufsky, A.; Diel, I.; Eniu, A.; Fallowfield, L.; Fujiwara, Y.; Jassem, J.; Paterson, A.H.; et al. Bone-related complications and quality of life in advanced breast cancer: Results from a randomized phase III trial of denosumab versus zoledronic acid. Clin. Cancer Res. 2012, 18, 4841–4849. [Google Scholar] [CrossRef]
- Kearns, A.E.; Khosla, S.; Kostenuik, P.J. Receptor activator of nuclear factor kappa B ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr. Rev. 2008, 29, 155–192. [Google Scholar]
- Beleut, M.; Rajaram, R.D.; Caikovski, M.; Ayyanan, A.; Germano, D.; Choi, Y.; Schneider, P.; Brisken, C. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc. Natl. Acad. Sci. USA 2010, 107, 2989–2994. [Google Scholar] [CrossRef]
- Tanos, T.; Sflomos, G.; Echeverria, P.C.; Ayyanan, A.; Gutierrez, M.; Delaloye, J.F.; Raffoul, W.; Fiche, M.; Dougall, W.; Schneider, P.; et al. Progesterone/RANKL is a major regulatory axis in the human breast. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Fata, J.E.; Kong, Y.Y.; Li, J.; Sasaki, T.; Irie-Sasaki, J.; Moorehead, R.A.; Elliott, R.; Scully, S.; Voura, E.B.; Lacey, D.L.; et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell 2000, 103, 41–50. [Google Scholar] [CrossRef]
- Fernandez-Valdivia, R.; Lydon, J.P. From the ranks of mammary progesterone mediators, RANKL takes the spotlight. Mol. Cell. Endocrinol. 2012, 357, 91–100. [Google Scholar] [CrossRef]
- Gonzalez-Suarez, E.; Jacob, A.P.; Jones, J.; Miller, R.; Roudier-Meyer, M.P.; Erwert, R.; Pinkas, J.; Branstetter, D.; Dougall, W.C. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010, 468, 103–107. [Google Scholar] [CrossRef]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef]
- Schramek, D.; Leibbrandt, A.; Sigl, V.; Kenner, L.; Pospisilik, J.A.; Lee, H.J.; Hanada, R.; Joshi, P.A.; Aliprantis, A.; Glimcher, L.; et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010, 468, 98–102. [Google Scholar] [CrossRef]
- Baron, R.; Ferrari, S.; Russell, R.G. Denosumab and bisphosphonates: Different mechanisms of action and effects. Bone 2011, 48, 677–692. [Google Scholar] [CrossRef]
- Santini, D.; Schiavon, G.; Vincenzi, B.; Gaeta, L.; Pantano, F.; Russo, A.; Ortega, C.; Porta, C.; Galluzzo, S.; Armento, G.; et al. Receptor activator of NF-kB (RANK) expression in primary tumors associates with bone metastasis occurrence in breast cancer patients. PLoS One 2011, 6, e19234. [Google Scholar] [CrossRef]
- Palafox, M.; Ferrer, I.; Pellegrini, P.; Vila, S.; Hernandez-Ortega, S.; Urruticoechea, A.; Climent, F.; Soler, M.T.; Munoz, P.; Vinals, F.; et al. RANK induces epithelial-mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res. 2012, 72, 2879–2888. [Google Scholar] [CrossRef]
- Gnant, M.; Mlineritsch, B.; Luschin-Ebengreuth, G.; Stoeger, H.; Dubsky, P.; Jakesz, R.; Singer, C.; Eidtmann, H.; Fesl, C.; Eiermann, W.; et al. Long-term follow-up in ABCSG-12: Significantly improved overall survival with adjuvant zoledronic acid in premenopausal patients with endocrine-receptor—Positive early breast cancer. Cancer Res. 2011, 71. [Google Scholar] [CrossRef]
- Coleman, R.E.; Marshall, H.; Cameron, D.; Dodwell, D.; Burkinshaw, R.; Keane, M.; Gil, M.; Houston, S.J.; Grieve, R.J.; Barrett-Lee, P.J.; et al. Breast-cancer adjuvant therapy with zoledronic acid. N. Engl. J. Med. 2011, 365, 1396–1405. [Google Scholar] [CrossRef]
- Coleman, R.; Bundred, N.; de Boer, R.; Llombarto, A.; Campbell, I.; Neven, P.; Barrios, C.; Dias, R.; Miller, J.; Brufsky, A. Impact of zoledronic acid in postmenopausal women with early breast cancer receiving adjuvant letrozole: Z-FAST, ZO-FAST, and E-ZO-FAST. Cancer Res. 2009, 69. [Google Scholar] [CrossRef]
- Coleman, R.; de Boer, R.; Eidtmann, H.; Llombart, A.; Davidson, N.; Neven, P.; von Minckwitz, G.; Sleeboom, H.P.; Forbes, J.; Barrios, C.; et al. Zoledronic acid (zoledronate) for postmenopausal women with early breast cancer receiving adjuvant letrozole (ZO-FAST study): Final 60-month results. Ann. Oncol. 2013, 24, 398–405. [Google Scholar] [CrossRef]
- Wong, M.H.F.; Stockler, M.R.; Pavlakis, N. Bisphosphonates and other bone agents for breast cancer. Cochrane Database Syst. Rev. 2012, 15. [Google Scholar] [CrossRef]
- Paterson, A.H.; Anderson, S.J.; Lembersky, B.C.; Fehrenbacher, L.; Falkson, C.I.; King, K.M.; Weir, L.M.; Brufsky, A.M.; Dakhil, S.; Lad, T.; et al. Oral clodronate for adjuvant treatment of operable breast cancer (National Surgical Adjuvant Breast and Bowel Project protocol B-34): A multicentre, placebo-controlled, randomised trial. Lancet Oncol. 2012, 13, 734–742. [Google Scholar] [CrossRef]
- Saarto, T.; Blomqvist, C.; Virkkunen, P.; Elomaa, I. Adjuvant clodronate treatment does not reduce the frequency of skeletal metastases in node-positive breast cancer patients: Five-year results of a randomized controlled trial. J. Clin. Oncol. 2001, 19, 10–17. [Google Scholar]
- Hadji, P.; Coleman, R.; Gnant, M.; Green, J. The impact of menopause on bone, zoledronic acid, and implications for breast cancer growth and metastasis. Ann. Oncol. 2012, 23, 2782–2790. [Google Scholar] [CrossRef]
- Steinman, R.A.; Brufsky, A.M.; Oesterreich, S. Zoledronic acid effectiveness against breast cancer metastases—A role for estrogen in the microenvironment? Breast Cancer Res. 2012, 14, 213. [Google Scholar] [CrossRef]
- Azim, H.A., Jr.; Michiels, S.; Bedard, P.L.; Singhal, S.K.; Criscitiello, C.; Ignatiadis, M.; Haibe-Kains, B.; Piccart, M.J.; Sotiriou, C.; Loi, S. Elucidating prognosis and biology of breast cancer arising in young women using gene expression profiling. Clin. Cancer Res. 2012, 18, 1341–1351. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Coleman, R.; Shore, N.; Fizazi, K.; Tombal, B.; Miller, K.; Sieber, P.; Karsh, L.; Damiao, R.; et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial. Lancet 2012, 379, 39–46. [Google Scholar] [CrossRef]
- Todenhöfer, T.; Hennenlotter, J.; Wald, A.; Leidenberger, P.; Blumenstock, G.; Aafderklamm, S.; Mundhenk, J.; Gakis, G.; Kühs, U.; Hohneder, A.; et al. RANKL pathway proteins as risk parameters for biochemical recurrence in patients undergoing radical prostatectomy. Eur. Urol. Suppl. 2013, 12. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Hirsh, V.; Siena, S.; Henry, D.H.; Woll, P.J.; Manegold, C.; Solal-Celigny, P.; Rodriguez, G.; Krzakowski, M.; Mehta, N.D.; et al. Overall survival improvement in patients with lung cancer and bone metastases treated with denosumab versus zoledronic acid: Subgroup analysis from a randomized phase 3 study. J. Thorac. Oncol. 2012, 7, 1823–1829. [Google Scholar] [CrossRef]
- European Association of Urology (EAU) Press release, 19 March 2013. Zoledronic acid does not prevent bone metastases in high-risk PCa patients. Available online: http://www.eaumilan2013.org/press/press-releases/ (accessed on 28 May 2013).
- Scagliotti, G.V.; Kosmidis, P.; de Marinis, F.; Schreurs, A.J.; Albert, I.; Engel-Riedel, W.; Schallier, D.; Barbera, S.; Kuo, H.P.; Sallo, V.; et al. Zoledronic acid in patients with stage IIIA/B NSCLC: Results of a randomized, phase III study. Ann. Oncol. 2012, 23, 2082–2087. [Google Scholar] [CrossRef]
- Study of Denosumab as Adjuvant Treatment for Women With High Risk Early Breast Cancer Receiving Neoadjuvant or Adjuvant Therapy (D-CARE). Available online: http://www.clinicaltrials.gov/ct2/show/NCT01077154?term=d-care&rank=1 (accessed on 20 April 2013).
- Coleman, R.E.; Barrios, C.; Bell, R.; Finkelstein, D.M.; Iwata, H.; Martin, M.; Braun, A.; Ke, C.; Maniar, T.; Goss, P.E. Denosumab versus placebo as adjuvant treatment for women with early-stage breast cancer at high risk of disease recurrence (D-CARE): An in progress, phase 3 clinical trial. Ann. Oncol. 2012, 23, 95–115. [Google Scholar]
- Study to Determine Treatment Effects of Denosumab in Patients with Breast Cancer Receiving Aromatase Inhibitor Therapy. Available online: http://clinicaltrials.gov/show/NCT00556374 (accessed on 20 April 2013).
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Von Moos, R.; Haynes, I. Where Do Bone-Targeted Agents RANK in Breast Cancer Treatment? J. Clin. Med. 2013, 2, 89-102. https://doi.org/10.3390/jcm2030089
Von Moos R, Haynes I. Where Do Bone-Targeted Agents RANK in Breast Cancer Treatment? Journal of Clinical Medicine. 2013; 2(3):89-102. https://doi.org/10.3390/jcm2030089
Chicago/Turabian StyleVon Moos, Roger, and Ian Haynes. 2013. "Where Do Bone-Targeted Agents RANK in Breast Cancer Treatment?" Journal of Clinical Medicine 2, no. 3: 89-102. https://doi.org/10.3390/jcm2030089
APA StyleVon Moos, R., & Haynes, I. (2013). Where Do Bone-Targeted Agents RANK in Breast Cancer Treatment? Journal of Clinical Medicine, 2(3), 89-102. https://doi.org/10.3390/jcm2030089