Mechanisms of Metastatic Tumor Dormancy

Abstract
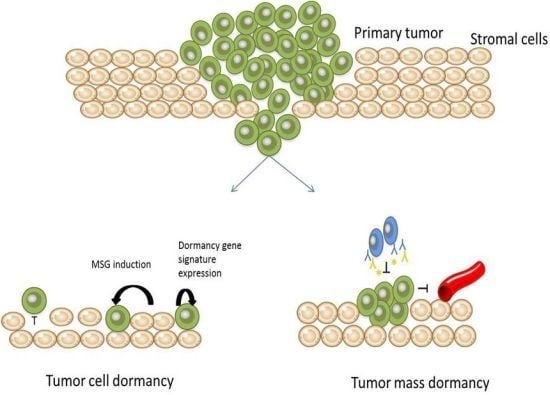
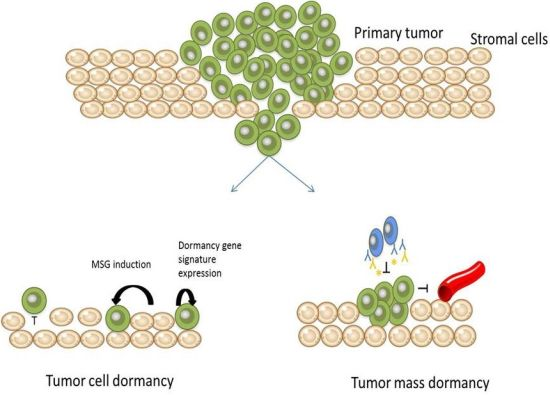
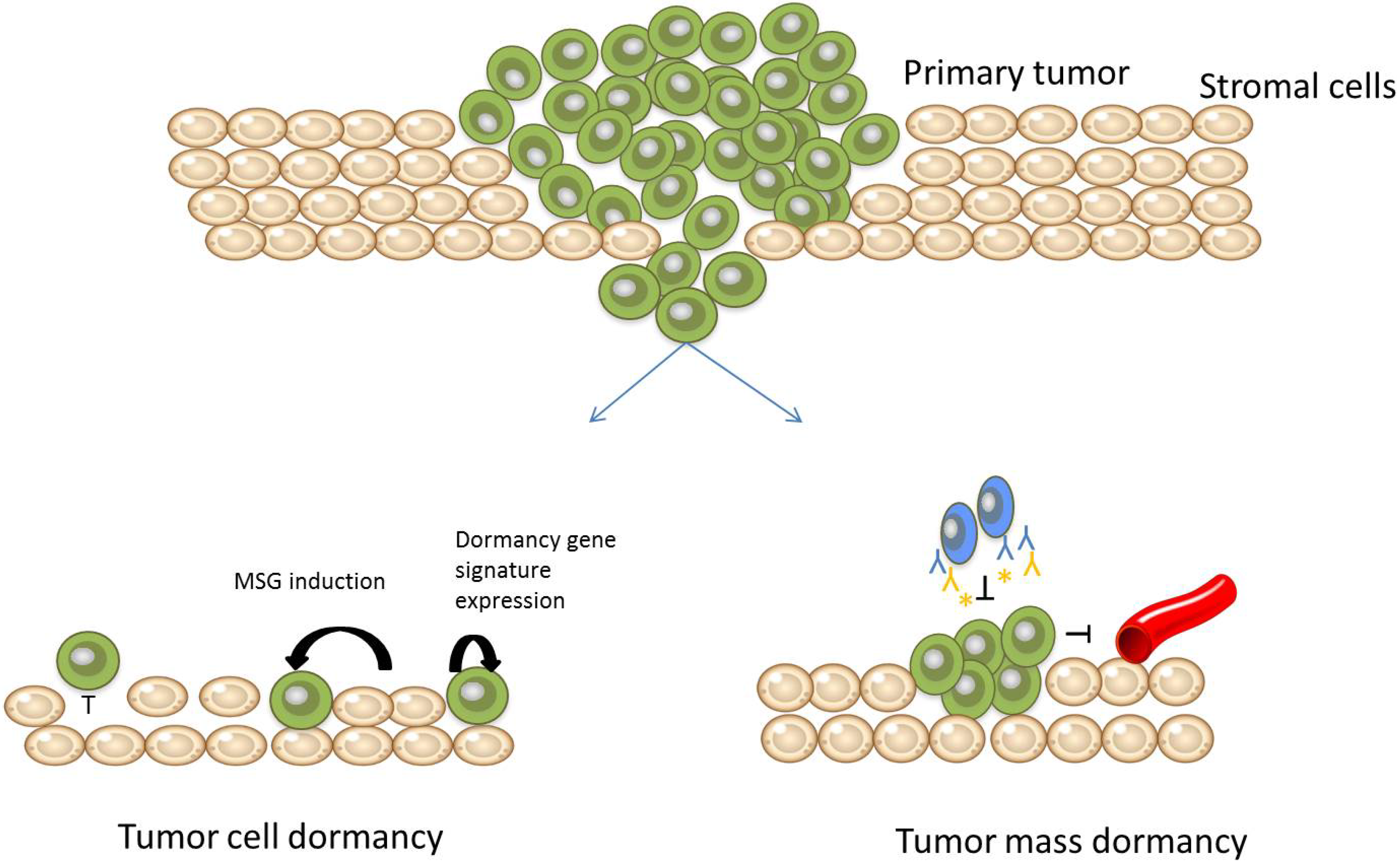
:
{kind=link}
{kind=link}
1. Introduction
2. Tumor Cell Dormancy
3. Microenvironment Induced Dormancy
4. Treatment-Induced Dormancy
5. Cancer Stem Cells
6. Tumor Mass Dormancy
7. Immune System and Tumor Dormancy
8. Angiogenesis and Tumor Dormancy
9. Escape from Tumor Dormancy
10. Conclusions and Future Directions
Conflicts of Interest
References
- Ben-Baruch, A. Site-specific metastasis formation: Chemokines as regulators of tumor cell adhesion, motility and invasion. Cell Adhes. Migr. 2009, 3, 328–333. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Sugarbaker, P.H. Metastatic inefficiency: The scientific basis for resection of liver metastases from colorectal cancer. J. Surg. Oncol. Suppl. 1993, 3, 158–160. [Google Scholar] [CrossRef]
- Weiss, L. Metastatic inefficiency. Adv. Cancer Res. 1990, 54, 159–211. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef]
- Uhr, J.W.; Pantel, K. Controversies in clinical cancer dormancy. Proc. Natl. Acad. Sci. USA 2011, 108, 12396–12400. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell cycle arrest is not senescence. Aging 2011, 3, 94–101. [Google Scholar]
- Paget, S. The distribution of secondary growths in cancer of the breast, 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar]
- Boudreau, N.; Bissell, M.J. Extracellular matrix signaling: Integration of form and function in normal and malignant cells. Curr. Opin. Cell Biol. 1998, 10, 640–646. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Hess, K.R.; Varadhachary, G.R.; Taylor, S.H.; Wei, W.; Raber, M.N.; Lenzi, R.; Abbruzzese, J.L. Metastatic patterns in adenocarcinoma. Cancer 2006, 106, 1624–1633. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Minn, A.J.; Kang, Y.; Serganova, I.; Gupta, G.P.; Giri, D.D.; Doubrovin, M.; Ponomarev, V.; Gerald, W.L.; Blasberg, R.; Massague, J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Investig. 2005, 115, 44–55. [Google Scholar]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef]
- Aguirre Ghiso, J.A.; Kovalski, K.; Ossowski, L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol. 1999, 147, 89–104. [Google Scholar] [CrossRef]
- Bragado, P.; Sosa, M.S.; Keely, P.; Condeelis, J.; Aguirre-Ghiso, J.A. Microenvironments dictating tumor cell dormancy. Recent Results Cancer Res. 2012, 195, 25–39. [Google Scholar] [CrossRef]
- Barkan, D.; Chambers, A.F. Beta1-Integrin: A potential therapeutic target in the battle against cancer recurrence. Clin. Cancer Res. 2011, 17, 7219–7223. [Google Scholar]
- Adam, A.P.; George, A.; Schewe, D.; Bragado, P.; Iglesias, B.V.; Ranganathan, A.C.; Kourtidis, A.; Conklin, D.S.; Aguirre-Ghiso, J.A. Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Res. 2009, 69, 5664–5672. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A.; Estrada, Y.; Liu, D.; Ossowski, L. ERK (MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38 (SAPK). Cancer Res. 2003, 63, 1684–1695. [Google Scholar]
- Ranganathan, A.C.; Adam, A.P.; Aguirre-Ghiso, J.A. Opposing roles of mitogenic and stress signaling pathways in the induction of cancer dormancy. Cell Cycle 2006, 5, 1799–1807. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Demidov, O.N.; Saito, S.; Kauraniemi, P.; Phillips, C.; Amundson, S.A.; Ambrosino, C.; Sauter, G.; Nebreda, A.R.; Anderson, C.W.; et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat. Genet. 2002, 31, 210–215. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Phillips, C.; Nannenga, B.; Timofeev, O.; Donehower, L.A.; Anderson, C.W.; Appella, E.; Fornace, A.J., Jr. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat. Genet. 2004, 36, 343–350. [Google Scholar] [CrossRef]
- Demidov, O.N.; Kek, C.; Shreeram, S.; Timofeev, O.; Fornace, A.J.; Appella, E.; Bulavin, D.V. The role of the MKK6/p38 MAPK pathway in Wip1-dependent regulation of ErbB2-driven mammary gland tumorigenesis. Oncogene 2007, 26, 2502–2506. [Google Scholar] [CrossRef]
- Lavoie, J.N.; L’Allemain, G.; Brunet, A.; Muller, R.; Pouyssegur, J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar]
- Taylor, J.; Hickson, J.; Lotan, T.; Yamada, D.S.; Rinker-Schaeffer, C. Using metastasis suppressor proteins to dissect interactions among cancer cells and their microenvironment. Cancer Metastasis Rev. 2008, 27, 67–73. [Google Scholar] [CrossRef]
- Horak, C.E.; Lee, J.H.; Marshall, J.C.; Shreeve, S.M.; Steeg, P.S. The role of metastasis suppressor genes in metastatic dormancy. APMIS 2008, 116, 586–601. [Google Scholar] [CrossRef]
- Hickson, J.A.; Huo, D.; vander Griend, D.J.; Lin, A.; Rinker-Schaeffer, C.W.; Yamada, S.D. The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res. 2006, 66, 2264–2270. [Google Scholar] [CrossRef]
- Yamada, S.D.; Hickson, J.A.; Hrobowski, Y.; vander Griend, D.J.; Benson, D.; Montag, A.; Karrison, T.; Huo, D.; Rutgers, J.; Adams, S.; et al. Mitogen-activated protein kinase kinase 4 (MKK4) acts as a metastasis suppressor gene in human ovarian carcinoma. Cancer Res. 2002, 62, 6717–6723. [Google Scholar]
- Kovacevic, Z.; Chikhani, S.; Lui, G.Y.; Sivagurunathan, S.; Richardson, D.R. The iron-regulated metastasis suppressor NDRG1 targets NEDD4L, PTEN, and SMAD4 and inhibits the PI3K and ras signaling pathways. Antioxid. Redox Signal. 2013, 18, 874–887. [Google Scholar] [CrossRef]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of MicroRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef]
- Kim, R.S.; Avivar-Valderas, A.; Estrada, Y.; Bragado, P.; Sosa, M.S.; Aguirre-Ghiso, J.A.; Segall, J.E. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS One 2012, 7, e35569. [Google Scholar] [CrossRef]
- Satchi-Fainaro, R.; Ferber, S.; Segal, E.; Ma, L.; Dixit, N.; Ijaz, A.; Hlatky, L.; Abdollahi, A.; Almog, N. Prospective identification of glioblastoma cells generating dormant tumors. PLoS One 2012, 7, e44395. [Google Scholar] [CrossRef]
- Badgwell, D.B.; Lu, Z.; Le, K.; Gao, F.; Yang, M.; Suh, G.K.; Bao, J.J.; Das, P.; Andreeff, M.; Chen, W.; et al. The tumor-suppressor gene ARHI (DIRAS3) suppresses ovarian cancer cell migration through inhibition of the Stat3 and FAK/Rho signaling pathways. Oncogene 2012, 31, 68–79. [Google Scholar]
- Chaterjee, M.; van Golen, K.L. Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Res. 2011, 2011. [Google Scholar] [CrossRef]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Invest. 2008, 118, 3917–3929. [Google Scholar]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar]
- Lock, R.; Debnath, J. Extracellular matrix regulation of autophagy. Curr. Opin. Cell Biol. 2008, 20, 583–588. [Google Scholar] [CrossRef]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef]
- Amaravadi, R.K. Autophagy-induced tumor dormancy in ovarian cancer. J. Clin. Invest. 2008, 118, 3837–3840. [Google Scholar] [Green Version]
- Sankala, H.M.; Hait, N.C.; Paugh, S.W.; Shida, D.; Lepine, S.; Elmore, L.W.; Dent, P.; Milstien, S.; Spiegel, S. Involvement of sphingosine kinase 2 in p53-independent induction of p21 by the chemotherapeutic drug doxorubicin. Cancer Res. 2007, 67, 10466–10474. [Google Scholar] [CrossRef]
- Korotchkina, L.G.; Demidenko, Z.N.; Gudkov, A.V.; Blagosklonny, M.V. Cellular quiescence caused by the Mdm2 inhibitor nutlin-3A. Cell Cycle 2009, 8, 3777–3781. [Google Scholar] [CrossRef]
- Steelman, L.S.; Navolanic, P.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Martelli, A.M.; Cocco, L.; Stivala, F.; Libra, M.; Nicoletti, F.; et al. Involvement of Akt and mTOR in chemotherapeutic- and hormonal-based drug resistance and response to radiation in breast cancer cells. Cell Cycle 2011, 10, 3003–3015. [Google Scholar] [CrossRef]
- Buck, M.B.; Pfizenmaier, K.; Knabbe, C. Antiestrogens induce growth inhibition by sequential activation of p38 mitogen-activated protein kinase and transforming growth factor-beta pathways in human breast cancer cells. Mol. Endocrinol. 2004, 18, 1643–1657. [Google Scholar] [CrossRef]
- Alison, M.R.; Islam, S.; Wright, N.A. Stem cells in cancer: Instigators and propagators? J. Cell Sci. 2010, 123, 2357–2368. [Google Scholar] [CrossRef]
- Moore, N.; Houghton, J.; Lyle, S. Slow-cycling therapy-resistant cancer cells. Stem Cells Dev. 2012, 21, 1822–1830. [Google Scholar] [CrossRef]
- Kusumbe, A.P.; Bapat, S.A. Cancer stem cells and aneuploid populations within developing tumors are the major determinants of tumor dormancy. Cancer Res. 2009, 69, 9245–9253. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Invest. 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef]
- Alison, M.R.; Islam, S. Attributes of adult stem cells. J. Pathol. 2009, 217, 144–160. [Google Scholar] [CrossRef]
- Tan, B.T.; Park, C.Y.; Ailles, L.E.; Weissman, I.L. The cancer stem cell hypothesis: A work in progress. Lab. Invest. 2006, 86, 1203–1207. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar]
- Hiscox, S.; Jiang, W.G.; Obermeier, K.; Taylor, K.; Morgan, L.; Burmi, R.; Barrow, D.; Nicholson, R.I. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int. J. Cancer 2006, 118, 290–301. [Google Scholar] [CrossRef]
- Yang, A.D.; Fan, F.; Camp, E.R.; van Buren, G.; Liu, W.; Somcio, R.; Gray, M.J.; Cheng, H.; Hoff, P.M.; Ellis, L.M. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin. Cancer Res. 2006, 12, 4147–4153. [Google Scholar] [CrossRef]
- Foltys, D.; Linkermann, A.; Heumann, A.; Hoppe-Lotichius, M.; Heise, M.; Schad, A.; Schneider, J.; Bender, K.; Schmid, M.; Mauer, D.; et al. Organ recipients suffering from undifferentiated neuroendocrine small-cell carcinoma of donor origin: A case report. Transplant. Proc. 2009, 41, 2639–2642. [Google Scholar] [CrossRef]
- Jiang, Y.; Villeneuve, P.J.; Wielgosz, A.; Schaubel, D.E.; Fenton, S.S.; Mao, Y. The incidence of cancer in a population-based cohort of Canadian heart transplant recipients. Am. J. Transplant. 2010, 10, 637–645. [Google Scholar] [CrossRef]
- Baccarani, U.; Piselli, P.; Serraino, D.; Adani, G.L.; Lorenzin, D.; Gambato, M.; Buda, A.; Zanus, G.; Vitale, A.; de Paoli, A.; et al. Comparison of de novo tumours after liver transplantation with incidence rates from Italian cancer registries. Dig. Liver Dis. 2010, 42, 55–60. [Google Scholar] [CrossRef]
- Brewer, J.D.; Colegio, O.R.; Phillips, P.K.; Roenigk, R.K.; Jacobs, M.A.; van de Beek, D.; Dierkhising, R.A.; Kremers, W.K.; McGregor, C.G.; Otley, C.C. Incidence of and risk factors for skin cancer after heart transplant. Arch. Dermatol. 2009, 145, 1391–1396. [Google Scholar] [CrossRef]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Street, S.E.; Cretney, E.; Smyth, M.J. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood 2001, 97, 192–197. [Google Scholar] [CrossRef]
- Street, S.E.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J. Exp. Med. 2002, 196, 129–134. [Google Scholar] [CrossRef]
- Dighe, A.S.; Richards, E.; Old, L.J.; Schreiber, R.D. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity 1994, 1, 447–456. [Google Scholar] [CrossRef]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; Cretney, E.; Trapani, J.A.; Taniguchi, M.; Kawano, T.; Pelikan, S.B.; Crowe, N.Y.; Godfrey, D.I. Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med. 2000, 191, 661–668. [Google Scholar] [CrossRef]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 2000, 192, 755–760. [Google Scholar] [CrossRef]
- Quezada, S.A.; Peggs, K.S.; Simpson, T.R.; Allison, J.P. Shifting the equilibrium in cancer immunoediting: From tumor tolerance to eradication. Immunol. Rev. 2011, 241, 104–118. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef]
- Quesnel, B. Tumor dormancy and immunoescape. APMIS 2008, 116, 685–694. [Google Scholar] [CrossRef]
- Muller-Hermelink, N.; Braumuller, H.; Pichler, B.; Wieder, T.; Mailhammer, R.; Schaak, K.; Ghoreschi, K.; Yazdi, A.; Haubner, R.; Sander, C.A.; et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 2008, 13, 507–518. [Google Scholar] [CrossRef]
- Zhang, B.; Karrison, T.; Rowley, D.A.; Schreiber, H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J. Clin. Invest. 2008, 118, 1398–1404. [Google Scholar] [CrossRef]
- Ziegler, A.; Heidenreich, R.; Braumuller, H.; Wolburg, H.; Weidemann, S.; Mocikat, R.; Rocken, M. EpCAM, a human tumor-associated antigen promotes Th2 development and tumor immune evasion. Blood 2009, 113, 3494–3502. [Google Scholar] [CrossRef]
- Eyles, J.; Puaux, A.L.; Wang, X.; Toh, B.; Prakash, C.; Hong, M.; Tan, T.G.; Zheng, L.; Ong, L.C.; Jin, Y.; et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Invest. 2010, 120, 2030–2039. [Google Scholar] [CrossRef]
- Radoja, S.; Rao, T.D.; Hillman, D.; Frey, A.B. Mice bearing late-stage tumors have normal functional systemic T cell responses in vitro and in vivo. J. Immunol. 2000, 164, 2619–2628. [Google Scholar]
- Willimsky, G.; Blankenstein, T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature 2005, 437, 141–146. [Google Scholar] [CrossRef]
- Blohm, U.; Roth, E.; Brommer, K.; Dumrese, T.; Rosenthal, F.M.; Pircher, H. Lack of effector cell function and altered tetramer binding of tumor-infiltrating lymphocytes. J. Immunol. 2002, 169, 5522–5530. [Google Scholar]
- Kurt, R.A.; Park, J.A.; Panelli, M.C.; Schluter, S.F.; Marchalonis, J.J.; Carolus, B.; Akporiaye, E.T. T lymphocytes infiltrating sites of tumor rejection and progression display identical V beta usage but different cytotoxic activities. J. Immunol. 1995, 154, 3969–3974. [Google Scholar]
- Kurt, R.A.; Park, J.A.; Schluter, S.F.; Marchalonis, J.J.; Akporiaye, E.T. TCR v(beta) usage and clonality of T cells isolated from progressing and rejected tumor sites before and after in vitro culture. Int. Immunol. 2000, 12, 639–646. [Google Scholar] [CrossRef]
- Yao, L.; Sgadari, C.; Furuke, K.; Bloom, E.T.; Teruya-Feldstein, J.; Tosato, G. Contribution of natural killer cells to inhibition of angiogenesis by interleukin-12. Blood 1999, 93, 1612–1621. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339, 58–61. [Google Scholar] [CrossRef]
- Png, K.J.; Halberg, N.; Yoshida, M.; Tavazoie, S.F. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature 2012, 481, 190–194. [Google Scholar]
- Straume, O.; Shimamura, T.; Lampa, M.J.; Carretero, J.; Oyan, A.M.; Jia, D.; Borgman, C.L.; Soucheray, M.; Downing, S.R.; Short, S.M.; et al. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 8699–8704. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar]
- Paradis, V.; Youssef, N.; Dargere, D.; Ba, N.; Bonvoust, F.; Deschatrette, J.; Bedossa, P. Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum. Pathol. 2001, 32, 327–332. [Google Scholar] [CrossRef]
- Melk, A.; Kittikowit, W.; Sandhu, I.; Halloran, K.M.; Grimm, P.; Schmidt, B.M.; Halloran, P.F. Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int. 2003, 63, 2134–2143. [Google Scholar] [CrossRef]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Van Zant, G.; Liang, Y. Concise review: Hematopoietic stem cell aging, life span, and transplantation. Stem Cells Transl. Med. 2012, 1, 651–657. [Google Scholar] [CrossRef]
- Ewertz, M.; Jensen, M.B.; Gunnarsdottir, K.A.; Hojris, I.; Jakobsen, E.H.; Nielsen, D.; Stenbygaard, L.E.; Tange, U.B.; Cold, S. Effect of obesity on prognosis after early-stage breast cancer. J. Clin. Oncol. 2011, 29, 25–31. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Cleary, M.P.; Grossmann, M.E. Minireview: Obesity and breast cancer: The estrogen connection. Endocrinology 2009, 150, 2537–2542. [Google Scholar] [CrossRef]
- Panigrahy, D.; Edin, M.L.; Lee, C.R.; Huang, S.; Bielenberg, D.R.; Butterfield, C.E.; Barnes, C.M.; Mammoto, A.; Mammoto, T.; Luria, A.; et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Invest. 2012, 122, 178–191. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Gross, G.J. Review article: Epoxyeicosatrienoic acids: Novel mediators of cardioprotection. J. Cardiovasc. Pharmacol. Ther. 2010, 15, 112–119. [Google Scholar] [CrossRef]
- DuPage, M.; Mazumdar, C.; Schmidt, L.M.; Cheung, A.F.; Jacks, T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012, 482, 405–409. [Google Scholar] [CrossRef]
- Almog, N.; Ma, L.; Raychowdhury, R.; Schwager, C.; Erber, R.; Short, S.; Hlatky, L.; Vajkoczy, P.; Huber, P.E.; Folkman, J.; et al. Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res. 2009, 69, 836–844. [Google Scholar] [CrossRef]
- Almog, N.; Ma, L.; Schwager, C.; Brinkmann, B.G.; Beheshti, A.; Vajkoczy, P.; Folkman, J.; Hlatky, L.; Abdollahi, A. Consensus micro RNAs governing the switch of dormant tumors to the fast-growing angiogenic phenotype. PLoS One 2012, 7, e44001. [Google Scholar] [CrossRef]
- Alunni-Fabbroni, M.; Sandri, M.T. Circulating tumour cells in clinical practice: Methods of detection and possible characterization. Methods 2010, 50, 289–297. [Google Scholar] [CrossRef]
- Graves, H.; Czerniecki, B.J. Circulating tumor cells in breast cancer patients: An evolving role in patient prognosis and disease progression. Pathol. Res. Int. 2011, 2011. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Osisami, M.; Keller, E.T. Mechanisms of Metastatic Tumor Dormancy. J. Clin. Med. 2013, 2, 136-150. https://doi.org/10.3390/jcm2030136
Osisami M, Keller ET. Mechanisms of Metastatic Tumor Dormancy. Journal of Clinical Medicine. 2013; 2(3):136-150. https://doi.org/10.3390/jcm2030136
Chicago/Turabian StyleOsisami, Mary, and Evan T. Keller. 2013. "Mechanisms of Metastatic Tumor Dormancy" Journal of Clinical Medicine 2, no. 3: 136-150. https://doi.org/10.3390/jcm2030136
APA StyleOsisami, M., & Keller, E. T. (2013). Mechanisms of Metastatic Tumor Dormancy. Journal of Clinical Medicine, 2(3), 136-150. https://doi.org/10.3390/jcm2030136