
Prenatal Diagnosis of ACTG2-Related Megacystis–Microcolon–Intestinal Hypoperistalsis Syndrome—Case Report and Systematic Review

Abstract
1. Introduction
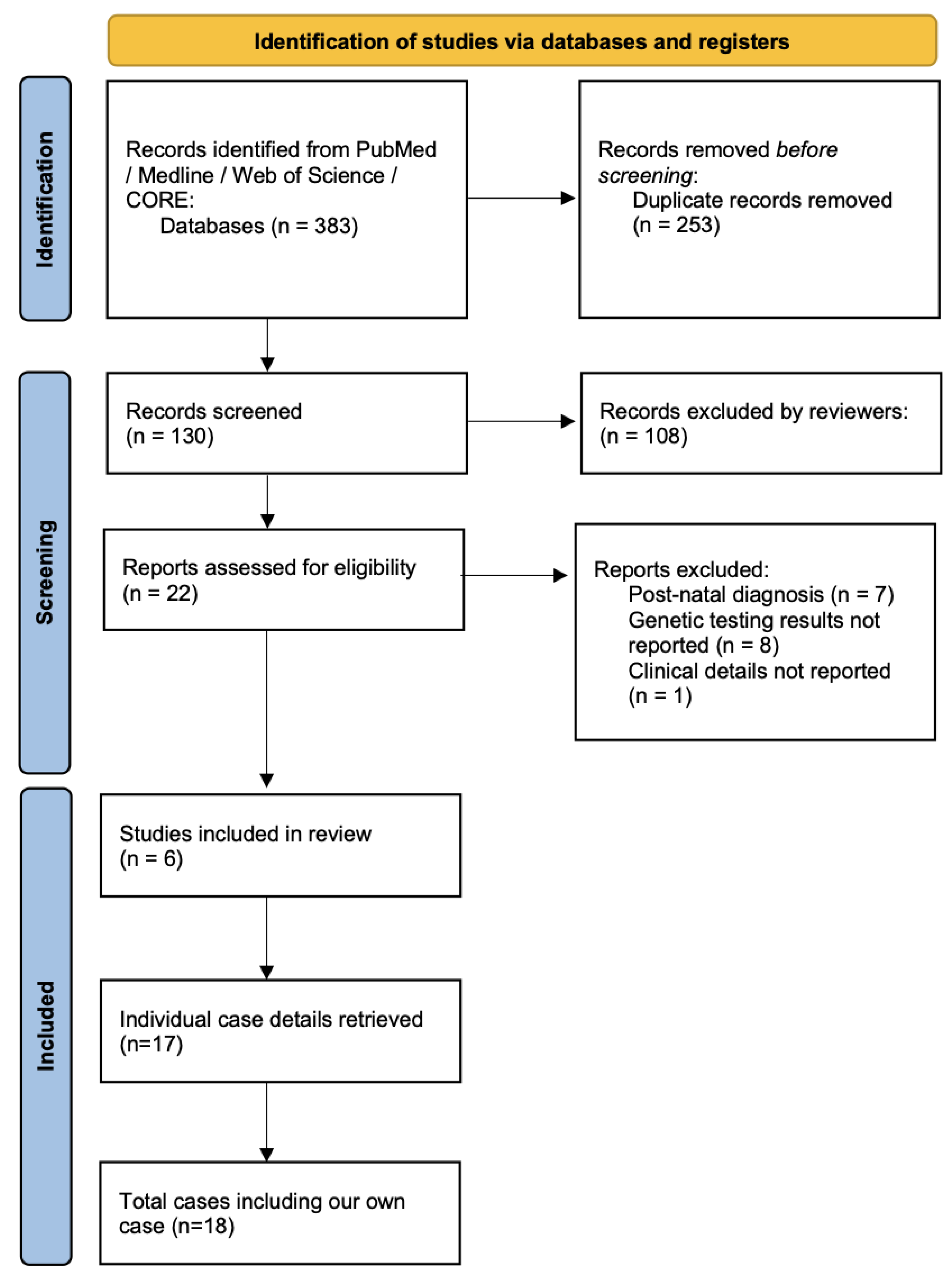
2. Materials and Methods
3. Results
3.1. Case Report
3.2. Systematic Review
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MMIHS | Megacystis–microcolon–intestinal hypoperistalsis syndrome |
| US | Ultrasound |
| WES | Whole-exome sequencing |
| TOP | Termination of pregnancy |
| GA | Gestational age |
| IUFD | Intrauterine fetal death |
| AD | Autosomal dominant |
| AR | Autosomal recessive |
References
- Ambartsumyan, L. Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome Overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019. Available online: http://www.ncbi.nlm.nih.gov/books/NBK540960/ (accessed on 10 February 2025).
- Prathapan, K.M.; King, D.E.; Raghu, V.K.; Ackerman, K.; Presel, T.; Yaworski, J.A.; Ganoza, A.; Bond, G.; Sevilla, W.M.A.; Rudolph, J.A.; et al. Megacystis Microcolon Intestinal Hypoperistalsis Syndrome: A Case Series With Long-term Follow-up and Prolonged Survival. J. Pediatr. Gastroenterol. Nutr. 2021, 72, e81–e85. [Google Scholar] [CrossRef] [PubMed]
- Thorson, W.; Diaz-Horta, O.; Foster, J.; Spiliopoulos, M.; Quintero, R.; Farooq, A.; Blanton, S.; Tekin, M. De novo ACTG2 mutations cause congenital distended bladder, microcolon, and intestinal hypoperistalsis. Hum. Genet. 2014, 133, 737–742. [Google Scholar] [CrossRef]
- Tuzovic, L.; Anyane-Yeboa, K.; Mills, A.; Glassberg, K.; Miller, R. Megacystis-microcolon-intestinal hypoperistalsis syndrome: Case report and review of prenatal ultrasonographic findings. Fetal Diagn. Ther. 2014, 36, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Billon, C.; Molin, A.; Poirsier, C.; Clemenson, A.; Dauge, C.; Grelet, M.; Sigaudy, S.; Patrier, S.; Goldenberg, A.; Layet, V.; et al. Fetal megacystis-microcolon: Genetic mutational spectrum and identification of PDCL3 as a novel candidate gene. Clin. Genet. 2020, 98, 261–273. [Google Scholar] [CrossRef]
- Halim, D.; Wilson, M.P.; Oliver, D.; Brosens, E.; Verheij, J.B.G.M.; Han, Y.; Nanda, V.; Lyu, Q.; Doukas, M.; Stoop, H.; et al. Loss of LMOD1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome in humans and mice. Proc. Natl. Acad. Sci. USA 2017, 114, E2739–E2747. [Google Scholar] [CrossRef] [PubMed]
- McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD) Online Mendelian Inheritance in Man, OMIM. Available online: https://omim.org/ (accessed on 5 March 2025).
- Krabek, R.; Smed, V.M.; Oestergaard, E.; Sundberg, K. Variant in ACTG2 Causing Megacystis Microcolon Hypoperistalsis Syndrome and Severe Familial Postpartum Bleeding. Fetal Diagn. Ther. 2022, 49, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, J.; Wang, H.; Feng, Q.; Luo, F.; Xie, J. Compound heterozygous variants in MYH11 underlie autosomal recessive megacystis-microcolon-intestinal hypoperistalsis syndrome in a Chinese family. J. Hum. Genet. 2019, 64, 1067–1073. [Google Scholar] [CrossRef]
- Yu, Q.-X.; Liu, N.; Zhen, L.; Wen, Y.-J.; Li, D.-Z. Prenatal Diagnosis of ACTG2 Visceral Myopathy Presented with Fetal Megacystis Identified in the Second Trimester. Prenat. Diagn. 2025, 45, 138–140. [Google Scholar] [CrossRef]
- Milunsky, A.; Lazier, J.; Baldwin, C.; Young, C.; Primack, D.; Milunsky, J.M. Prenatal diagnosis of chronic intestinal pseudo-obstruction and paternal somatic mosaicism for the ACTG2 pathogenic variant. Prenat. Diagn. 2017, 37, 1254–1256. [Google Scholar] [CrossRef]
- Markota, B.; Gross, A.M.; Specht, C.; Schertler, C.; Rhomberg, M.; Bemetz, U.; Scheier, M. Prenatal diagnosis of megacystis-microcolon-intestinal hypoperistalsis syndrome. Case Rep. Perinat. Med. 2019, 9, 215–218. [Google Scholar] [CrossRef]
- Fontanella, F.; Maggio, L.; Verheij, J.B.G.M.; Duin, L.K.; Adama Van Scheltema, P.N.; Cohen-Overbeek, T.E.; Pajkrt, E.; Bekker, M.; Willekes, C.; Bax, C.J.; et al. Fetal megacystis: A lot more than LUTO. Ultrasound Obstet. Gynecol. 2019, 53, 779–787. [Google Scholar] [CrossRef] [PubMed]
- White, S.M.; Chamberlain, P.; Hitchcock, R.; Sullivan, P.B.; Boyd, P.A. Megacystis-microcolon-intestinal hypoperistalsis syndrome: The difficulties with antenatal diagnosis. Case report and review of the literature. Prenat. Diagn. 2000, 20, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Ignasiak-Budzyńska, K.; Danko, M.; Książyk, J. Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome (MMIHS): Series of 4 Cases Caused by Mutation of ACTG2 (Actin Gamma 2, Smooth Muscle) Gene. Case Rep. Gastrointest. Med. 2021, 2021, 6612983. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, S.; Solinho, M.; Lourenço, C.; Brito, C. Megacystis-microcolon-intestinal Hypoperistalsis Syndrome: Four Consecutive Pregnancies with Megacystis. J. Med. Ultrasound 2024, 32, 255–258. [Google Scholar] [CrossRef]
- Tanudisastro, H.A.; Holman, K.; Ho, G.; Farnsworth, E.; Fisk, K.; Gayagay, T.; Hackett, E.; Jenkins, G.; Krishnaraj, R.; Lai, T.; et al. Australia and New Zealand renal gene panel testing in routine clinical practice of 542 families. NPJ Genom. Med. 2021, 6, 20. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Authors | Year | GA at Testing | Trigger for Testing | Ultrasound Features | Type of Genetic Testing | MMIH Gene Result | Outcome |
|---|---|---|---|---|---|---|---|
| Milunsky [11] | 2017 | 22 | US findings | Megacystis, hydronephrosis, thinning of renal cortex | Targeted | ACTG2 | TOP GA 22 |
| Wang [9] | 2019 | 13 | US findings | Megacystis, oligohydramnios | Trio WES | MYH11 | TOP GA 17 |
| Wang [9] | 2019 | 14 | US findings | Megacystis | Trio WES | MYH11 | TOP GA 16 |
| Markota [12] | 2020 | 25 | US findings | Megacystis, prominent small bowel | Targeted | ACTG2 | Livebirth at GA 34 (preterm labour) |
| Billon [5] | 2020 | 21 | US findings | Megacystis | Targeted | ACTG2 | TOP GA 24 |
| Billon [5] | 2020 | 19 | US findings | Megacystis | Targeted | ACTG2 | TOP GA 25 |
| Billon [5] | 2020 | 20 | US findings | Megacystis, echogenic bowel, omphalocele | Targeted | ACTG2 | TOP GA 29 |
| Billon [5] | 2020 | 16 | US findings | Megacystis | Trio WES | MYH11 | TOP GA 16 |
| Billon [5] | 2020 | Unknown | US findings + family history | Megacystis | Trio WES | MYH11 | IUFD GA 28 |
| Billon [5] | 2020 | 10 | US findings + family history | Megacystis | Trio WES | MYH11 | TOP GA 10 |
| Billon [5] | 2020 | 15 | US findings + family history | Megacystis | Trio WES | MYL9 | TOP GA 27 |
| Billon [5] | 2020 | Second trimester | US findings | Megacystis, bilateral pelvicalyceal dilatation, single umbilical artery | Trio WES | PDCL3 | TOP GA 30 |
| Billon [5] | 2020 | 12 | US findings + family history | Megacystis, bilateral diaphragmatic hernia | Trio WES | PDCL3 | IUFD GA 12 |
| Krabek [8] | 2023 | 27 | US findings + family history | Megacystis, mild pyelectasis | Trio WES | ACTG2 | Livebirth at term |
| Yu [10] | 2024 | 23 | US findings | Megacystis, mild pyelectasis | Trio WES | ACTG2 | TOP GA 23 |
| Yu [10] | 2024 | 17 | US findings | Megacystis | Trio WES | ACTG2 | TOP GA 23 |
| Yu [10] | 2024 | 22 | US findings | Megacystis | Trio WES | ACTG2 | TOP GA 22 |
| Ravi | 2025 | 21 | US findings | Megacystis, bilateral renal pelvis dilatation, polyhydramnios | Trio WES | ACTG2 | TOP GA 35 |
| Gene/Locus | Location | Inheritance |
|---|---|---|
| ACTG2 | 2p13.1 | AD |
| MYLK | 3q21.1 | AR |
| MYL9 | 20q11.23 | AR |
| MYH11 | 16p13.11 | AR |
| LMOD1 | 1q32.1 | AR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravi, N.; Kumar, S.; Ramachandran, A. Prenatal Diagnosis of ACTG2-Related Megacystis–Microcolon–Intestinal Hypoperistalsis Syndrome—Case Report and Systematic Review. J. Clin. Med. 2025, 14, 3204. https://doi.org/10.3390/jcm14093204
Ravi N, Kumar S, Ramachandran A. Prenatal Diagnosis of ACTG2-Related Megacystis–Microcolon–Intestinal Hypoperistalsis Syndrome—Case Report and Systematic Review. Journal of Clinical Medicine. 2025; 14(9):3204. https://doi.org/10.3390/jcm14093204
Chicago/Turabian StyleRavi, Neha, Sailesh Kumar, and Aparna Ramachandran. 2025. "Prenatal Diagnosis of ACTG2-Related Megacystis–Microcolon–Intestinal Hypoperistalsis Syndrome—Case Report and Systematic Review" Journal of Clinical Medicine 14, no. 9: 3204. https://doi.org/10.3390/jcm14093204
APA StyleRavi, N., Kumar, S., & Ramachandran, A. (2025). Prenatal Diagnosis of ACTG2-Related Megacystis–Microcolon–Intestinal Hypoperistalsis Syndrome—Case Report and Systematic Review. Journal of Clinical Medicine, 14(9), 3204. https://doi.org/10.3390/jcm14093204