
Autoimmune Pancreatitis: A Review

Abstract
1. Introduction
2. Epidemiology
3. Pathogenesis
4. Clinical Presentation
5. Evaluation and Diagnosis
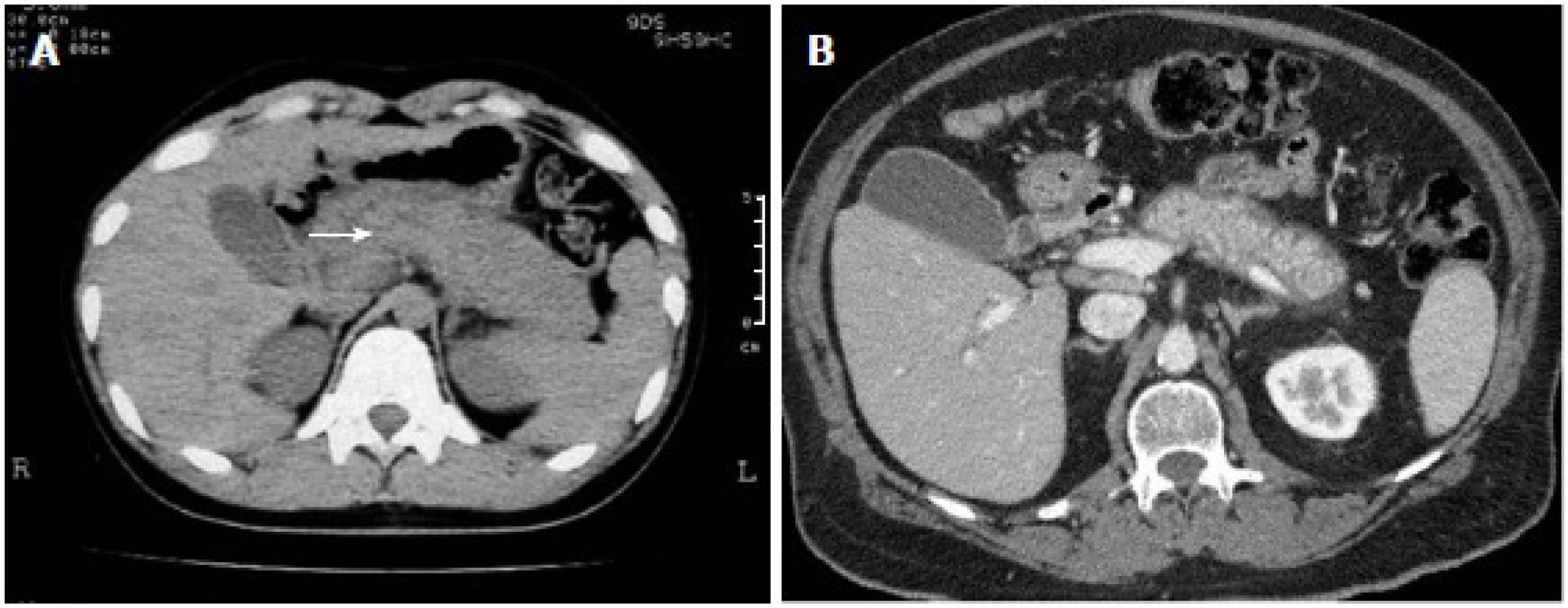
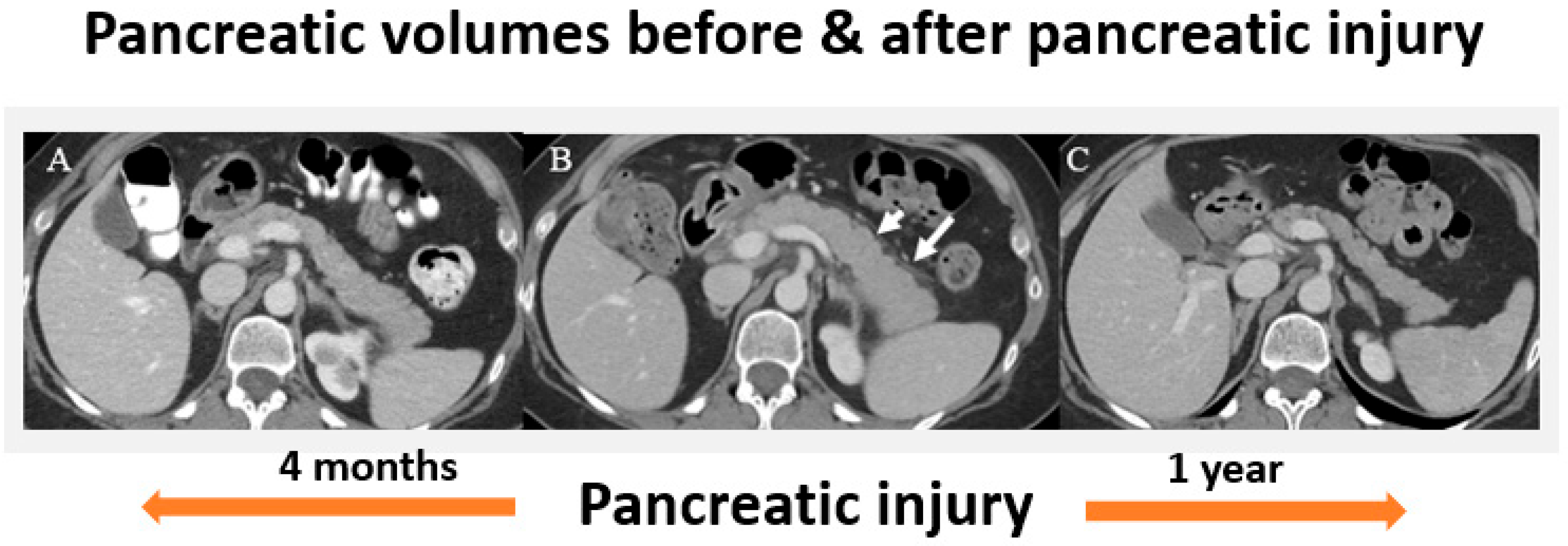
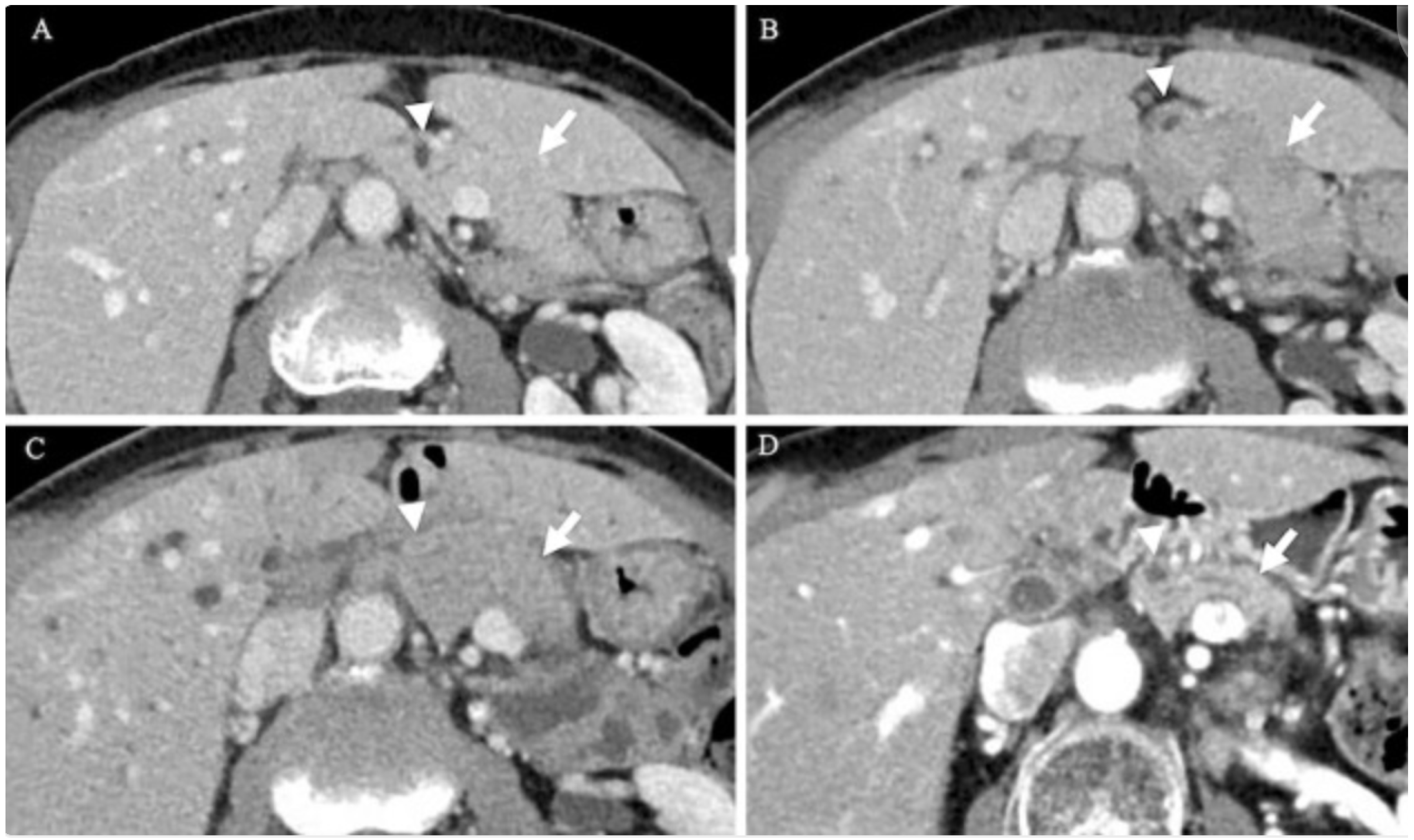
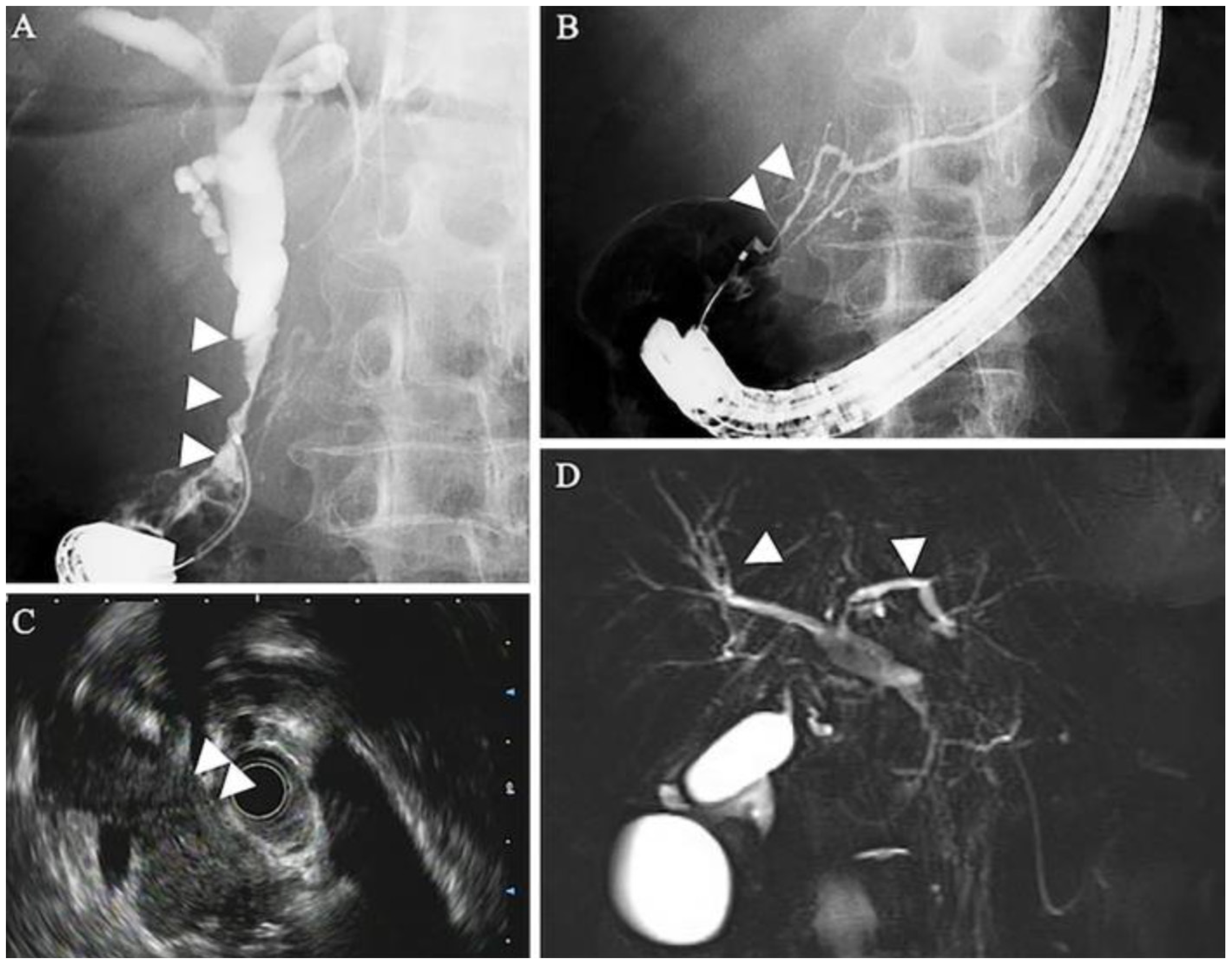
5.1. Radiographic Findings
5.2. Serologic Findings
5.3. Histologic Findings
6. Treatment and Management

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AIP-1 | AIP-2 | AIP-3 | |
|---|---|---|---|
| Demographic | Older males (>60 years) | Younger patients (~50 years) | Patients receiving ICI therapy (age variable) |
| Clinical Presentation | Painless obstructive jaundice, mild abdominal discomfort, weight loss | Recurrent acute pancreatitis, abdominal pain, jaundice | Asymptomatic or with pancreatitis-like symptoms (abdominal pain, nausea, vomiting) or incidental changes on imaging |
| Histology | IgG4+ plasma cell infiltration | Granulocytic epithelial lesions (GEL) | CD8 predominant inflammation, often mixed inflammatory infiltrates |
| Serum IgG4 Levels | Elevated (>135 mg/dL) | Normal | Variable, usually normal |
| Systemic Involvement | Common (salivary glands, kidneys, lungs, bile ducts, etc.) | Rare | Can be associated with other ICI-related autoimmune conditions |
| Key Imaging Findings | Diffuse “sausage-shaped” pancreas, capsule-like rim enhancement | Focal/diffuse involvement, strictures without upstream dilation | Normal appearing pancreas, rarely features of interstitial pancreatitis |
| Response to Steroids | Good response | Good response | Limited data |
| Risk of Relapse After Treatment | High, requires maintenance therapy | Low | Limited data |
| AIP-1 | AIP-2 | AIP-3 | |
|---|---|---|---|
| Diagnosis and Evaluation |
|
|
|
| First-Line Treatment |
|
|
|
| Second-Line Treatment |
|
|
|
| Long-Term Management and Prognosis |
|
|
|
| Other Considerations |
|
|
|
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Blaho, M.; Dítě, P.; Kunovský, L.; Kianička, B. Autoimmune pancreatitis—An ongoing challenge. Adv. Med. Sci. 2020, 65, 403–408. [Google Scholar] [CrossRef]
- Vujasinovic, M.; Löhr, J.M. Autoimmune pancreatitis: Definition, clinical presentation, and classification. In Clinical Pancreatology for Practising Gastroenterologists and Surgeons, 1st ed.; Domínguez-Muñoz, J.E., Ed.; Wiley: Hoboken, NJ, USA, 2021; pp. 347–355. [Google Scholar] [CrossRef]
- Takahashi, M.; Fujinaga, Y.; Notohara, K.; Koyama, T.; Inoue, D.; Irie, H.; Gabata, T.; Kadoya, M.; Kawa, S.; Okazaki, K.; et al. Diagnostic imaging guide for autoimmune pancreatitis. Jpn. J. Radiol. 2020, 38, 591–612. [Google Scholar] [CrossRef]
- Nikolic, S.; Maisonneuve, P.; Dahlman, I.; Löhr, J.-M.; Vujasinovic, M. Exocrine and endocrine insufficiency in autoimmune pancreatitis: A matter of treatment or time? J. Clin. Med. 2022, 11, 3724. [Google Scholar] [CrossRef]
- Qureshi, A.; Ghobrial, Y.; De Castro, J.; Siami-Namini, K.; Newman, K.A. Autoimmune pancreatitis—What we know and what do we have to know? Autoimmun. Rev. 2021, 20, 102912. [Google Scholar] [CrossRef]
- Sandrasegaran, K.; Menias, C.O. Imaging in autoimmune pancreatitis and immunoglobulin g4–related disease of the abdomen. Gastroenterol. Clin. N. Am. 2018, 47, 603–619. [Google Scholar] [CrossRef]
- Khandelwal, A.; Inoue, D.; Takahashi, N. Autoimmune pancreatitis: An update. Abdom. Radiol. 2020, 45, 1359–1370. [Google Scholar] [CrossRef]
- Nan, N.; Wang, D. Type 2 autoimmune pancreatitis associated with ulcerative colitis. Front. Immunol. 2023, 14, 1288390. [Google Scholar] [CrossRef]
- De Pretis, N.; Frulloni, L. Autoimmune pancreatitis type 2. Curr. Opin. Gastroenterol. 2020, 36, 417–420. [Google Scholar] [CrossRef]
- Sayed Ahmed, A.; Abreo, M.; Thomas, A.; Chari, S.T. Type 3 autoimmune pancreatitis (immune checkpoint inhibitor-induced pancreatitis). Curr. Opin. Gastroenterol. 2022, 38, 516–520. [Google Scholar] [CrossRef]
- Tan, S.; Day, D.; Nicholls, S.J.; Segelov, E. Immune checkpoint inhibitor therapy in oncology. JACC CardioOncol. 2022, 4, 579–597. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38. [Google Scholar] [CrossRef]
- Gallo, C.; Dispinzieri, G.; Zucchini, N.; Invernizzi, P.; Massironi, S. Autoimmune pancreatitis: Cornerstones and future perspectives. World J. Gastroenterol. 2024, 30, 817–832. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, H.; Zhou, L.; Li, W.; Yang, L.; Li, W.; Li, K.; Liu, X. Immunotherapy-associated pancreatic adverse events: Current understanding of their mechanism, diagnosis, and management. Front. Oncol. 2021, 11, 627612. [Google Scholar] [CrossRef]
- Thomas, A.S.; Abreo, M.; Ahmed, A.S.; Rao Manikonda, S.P.; Eyada, M.; Issac, A.; Abraham, F.; Jacob, J.S.; Wang, Y.; Yedururi, S.; et al. Immune checkpoint inhibitor-induced pancreatic injury: Clinical and radiological profile and response to steroids. Gastro Hep Adv. 2024, 3, 361–367. [Google Scholar] [CrossRef]
- Basyal, B.; Kc, P. Autoimmune pancreatitis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK560769/ (accessed on 1 February 2025).
- Schneider, A.; Michaely, H.; Weiss, C.; Hirth, M.; Rückert, F.; Wilhelm, T.J.; Schönberg, S.; Marx, A.; Singer, M.V.; Löhr, J.M.; et al. Prevalence and incidence of autoimmune pancreatitis in the population living in the southwest of Germany. Digestion 2017, 96, 187–198. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Hamada, S.; Tsuji, I.; Takeyama, Y.; Shimosegawa, T.; Okazaki, K. Nationwide epidemiological survey of autoimmune pancreatitis in Japan in 2016. J. Gastroenterol. 2020, 55, 462–470. [Google Scholar] [CrossRef]
- Kanno, A.; Masamune, A.; Okazaki, K.; Kamisawa, T.; Kawa, S.; Nishimori, I.; Tsuji, I.; Shimosegawa, T. Nationwide epidemiological survey of autoimmune pancreatitis in Japan in 2011. Pancreas 2015, 44, 535–539. [Google Scholar] [CrossRef]
- Li, Y.; Song, H.; Meng, X.; Li, R.; Leung, P.S.C.; Gershwin, M.E.; Zhang, S.; Sun, S.; Song, J. Autoimmune pancreatitis type 2 (Idiopathic duct-centric pancreatitis): A comprehensive review. J. Autoimmun. 2023, 140, 103121. [Google Scholar] [CrossRef]
- George, J.; Bajaj, D.; Sankaramangalam, K.; Yoo, J.W.; Joshi, N.S.; Gettinger, S.; Price, C.; Farrell, J.J. Incidence of pancreatitis with the use of immune checkpoint inhibitors (ICI) in advanced cancers: A systematic review and meta-analysis. Pancreatology 2019, 19, 587–594. [Google Scholar] [CrossRef]
- Abu-Sbeih, H.; Tang, T.; Lu, Y.; Thirumurthi, S.; Altan, M.; Jazaeri, A.A.; Dadu, R.; Coronel, E.; Wang, Y. Clinical characteristics and outcomes of immune checkpoint inhibitor-induced pancreatic injury. J. Immunother. Cancer 2019, 7, 31. [Google Scholar] [CrossRef]
- Wang, Y. (Ed.) Managing Immunotherapy Related Organ Toxicities: A Practical Guide; Springer International Publishing: Berlin/Heidelberg, Germany, 2022. [Google Scholar] [CrossRef]
- Shirwaikar Thomas, A.; Chari, S.T. Immune checkpoint inhibitor-induced (Type 3) autoimmune pancreatitis. Curr. Gastroenterol. Rep. 2023, 25, 255–259. [Google Scholar] [CrossRef]
- Thomas, A.S.; Abreo, M.; Sayed, S.A.; Sireesha Yedururi, Y.W.; Chari, S.T. Autoimmune pancreatitis secondary to immune checkpoint inhibitor therapy (Type 3 AIP): Insights into a new disease from serial pancreatic imaging. Gastroenterology 2023, 164, 154–155. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, P.; Xiang, Y.; Tao, L.; Huang, T.; Feng, Z. IgG4-related autoimmune pancreatitis and sclerosing cholangitis: A case report and literature review. Medicine 2024, 103, e37922. [Google Scholar] [CrossRef]
- Kamisawa, T.; Zen, Y.; Pillai, S.; Stone, J.H. IgG4-related disease. Lancet 2015, 385, 1460–1471. [Google Scholar] [CrossRef]
- Kokubo, K.; Onodera, A.; Kiuchi, M.; Tsuji, K.; Hirahara, K.; Nakayama, T. Conventional and pathogenic Th2 cells in inflammation, tissue repair, and fibrosis. Front. Immunol. 2022, 13, 945063. [Google Scholar] [CrossRef]
- Nista, E.C.; De Lucia, S.S.; Manilla, V.; Schepis, T.; Pellegrino, A.; Ojetti, V.; Pignataro, G.; Zileri Dal Verme, L.; Franceschi, F.; Gasbarrini, A.; et al. Autoimmune pancreatitis: From pathogenesis to treatment. Int. J. Mol. Sci. 2022, 23, 12667. [Google Scholar] [CrossRef]
- Minaga, K.; Watanabe, T.; Hara, A.; Yoshikawa, T.; Kamata, K.; Kudo, M. Plasmacytoid dendritic cells as a new therapeutic target for autoimmune pancreatitis and IgG4-related disease. Front. Immunol. 2021, 12, 713779. [Google Scholar] [CrossRef]
- Psarras, A.; Emery, P.; Vital, E.M. Type I interferon–mediated autoimmune diseases: Pathogenesis, diagnosis and targeted therapy. Rheumatology 2017, 56, kew431. [Google Scholar] [CrossRef]
- Ku, Y.; Hong, S.-M.; Fujikura, K.; Kim, S.J.; Akita, M.; Abe-Suzuki, S.; Shiomi, H.; Masuda, A.; Itoh, T.; Azuma, T.; et al. IL-8 expression in granulocytic epithelial lesions of idiopathic duct-centric pancreatitis (type 2 autoimmune pancreatitis). Am. J. Surg. Pathol. 2017, 41, 1129–1138. [Google Scholar] [CrossRef]
- Loos, M.; Lauffer, F.; Schlitter, A.M.; Kleeff, J.; Friess, H.; Klöppel, G.; Esposito, I. Potential role of Th17 cells in the pathogenesis of type 2 autoimmune pancreatitis. Virchows Arch. 2015, 467, 641–648. [Google Scholar] [CrossRef]
- Osaki, M.; Sakaguchi, S. Soluble CTLA-4 regulates immune homeostasis and promotes resolution of inflammation by suppressing type 1 but allowing type 2 immunity. Immunity 2025, 58, 889–908.e13. [Google Scholar] [CrossRef]
- Zhang, M.; Ni, J.; Xu, W.-D.; Wen, P.-F.; Qiu, L.-J.; Wang, X.-S.; Pan, H.-F.; Ye, D.-Q. Association of CTLA-4 variants with susceptibility to inflammatory bowel disease: A meta-analysis. Hum. Immunol. 2014, 75, 227–233. [Google Scholar] [CrossRef]
- Klocke, K.; Sakaguchi, S.; Holmdahl, R.; Wing, K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc. Natl. Acad. Sci. USA 2016, 113, E2383–E2392. [Google Scholar] [CrossRef]
- Lanzillotta, M.; Vujasinovic, M.; Löhr, J.; Della Torre, E. Update on autoimmune pancreatitis and igg4-related disease. United Eur. Gastroenterol. J. 2024, 13, 107–115. [Google Scholar] [CrossRef]
- Xu, B.; Cai, Z.-P.; Liu, W. IgG4-related cholangiopathy, pancreatopathy and lymphadenopathy. Am. J. Med. Sci. 2023, 366, e60–e61. [Google Scholar] [CrossRef]
- Wu, S.; Wang, H. IgG4-related digestive diseases: Diagnosis and treatment. Front. Immunol. 2023, 14, 1278332. [Google Scholar] [CrossRef]
- Li, M.-Z.; Guo, T.; Feng, Y.-L.; Zhang, S.-Y.; Bai, X.-Y.; Wu, X.; Xu, K.; Yang, A.-M. Diabetes mellitus in patients with type 1 autoimmune pancreatitis at diagnosis and after corticosteroid therapy. Hepatobiliary Pancreat. Dis. Int. 2024, 23, 393–398. [Google Scholar] [CrossRef]
- Noguchi, K.; Nakai, Y.; Mizuno, S.; Isayama, H.; Hirano, K.; Kanai, S.; Nakamura, T.; Uchino, R.; Takahara, N.; Kogure, H.; et al. Insulin secretion improvement during steroid therapy for autoimmune pancreatitis according to the onset of diabetes mellitus. J. Gastroenterol. 2020, 55, 198–204. [Google Scholar] [CrossRef]
- Matsushiro, M.; Haraguchi, T.; Yamazaki, Y.; Hamamoto, Y.; Seino, Y. Effects of steroid therapy on pancreatic endocrine function in IgG4-related aip: Evaluation by arginine stimulation test. JCEM Case Rep. 2025, 3, luaf048. [Google Scholar] [CrossRef]
- Nishimori, I.; Tamakoshi, A.; Kawa, S.; Tanaka, S.; Takeuchi, K.; Kamisawa, T.; Saisho, H.; Hirano, K.; Okamura, K.; Yanagawa, N.; et al. Research Committee on Intractable Pancreatic Diseases, the Ministry of Health and Welfare of Japan. Influence of steroid therapy on the course of diabetes mellitus in patients with autoimmune pancreatitis: Findings from a nationwide survey in Japan. Pancreas 2006, 32, 244–248. [Google Scholar] [CrossRef]
- Hart, P.A.; Levy, M.J.; Smyrk, T.C.; Takahashi, N.; Abu Dayyeh, B.K.; Clain, J.E.; Gleeson, F.C.; Pearson, R.K.; Petersen, B.T.; Topazian, M.D.; et al. Clinical profiles and outcomes in idiopathic duct-centric chronic pancreatitis (type 2 autoimmune pancreatitis): The Mayo Clinic experience. Gut 2016, 65, 1702–1709. [Google Scholar] [CrossRef]
- Massironi, S.; Fanetti, I.; Viganò, C.; Pirola, L.; Fichera, M.; Cristoferi, L.; Capurso, G.; Invernizzi, P.; Danese, S. Systematic review—Pancreatic involvement in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2022, 55, 1478–1491. [Google Scholar] [CrossRef]
- Lorenzo, D.; Maire, F.; Stefanescu, C.; Gornet, J.-M.; Seksik, P.; Serrero, M.; Bournet, B.; Marteau, P.; Amiot, A.; Laharie, D.; et al. Features of autoimmune pancreatitis associated with inflammatory bowel diseases. Clin. Gastroenterol. Hepatol. 2018, 16, 59–67. [Google Scholar] [CrossRef]
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S.; Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef]
- Shimosegawa, T. Clinical diagnostic criteria for autoimmune pancreatitis. In The Pancreas, 1st ed.; Beger, H.G., Büchler, M.W., Hruban, R.H., Mayerle, J., Neoptolemos, J.P., Shimosegawa, T., Warshaw, A.L., Whitcomb, D.C., Zhao, Y., Groß, C., Eds.; Wiley: Hoboken, NJ, USA, 2023; pp. 561–567. [Google Scholar] [CrossRef]
- Maruyama, M.; Watanabe, T.; Kanai, K.; Oguchi, T.; Muraki, T.; Hamano, H.; Arakura, N.; Kawa, S. International consensus diagnostic criteria for autoimmune pancreatitis and its japanese amendment have improved diagnostic ability over existing criteria. Gastroenterol. Res. Pract. 2013, 2013, 456965. [Google Scholar] [CrossRef]
- Rehnitz, C.; Klauss, M.; Singer, R.; Ehehalt, R.; Werner, J.; Büchler, M.W.; Kauczor, H.-U.; Grenacher, L. Morphologic patterns of autoimmune pancreatitis in CT and MRI. Pancreatology 2011, 11, 240–251. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.H.; Kim, S.Y.; Byun, J.H.; Kim, H.J.; Kim, M.-H.; Lee, M.-G.; Lee, S.S. Comparison of diagnostic performance between CT and MRI in differentiating non-diffuse-type autoimmune pancreatitis from pancreatic ductal adenocarcinoma. Eur. Radiol. 2018, 28, 5267–5274. [Google Scholar] [CrossRef]
- O’Reilly, D.A.; Malde, D.J.; Duncan, T.; Rao, M.; Filobbos, R. Review of the diagnosis, classification and management of autoimmune pancreatitis. World J. Gastrointest. Pathophysiol. 2014, 5, 71–81. [Google Scholar] [CrossRef]
- Das, J.P.; Postow, M.A.; Friedman, C.F.; Do, R.K.; Halpenny, D.F. Imaging findings of immune checkpoint inhibitor associated pancreatitis. Eur. J. Radiol. 2020, 131, 109250. [Google Scholar] [CrossRef]
- Tanabe, K.; Yokoyama, K.; Kanno, A.; Ikeda, E.; Ando, K.; Nagai, H.; Koyanagi, T.; Sakaguchi, M.; Nakaya, T.; Tamada, K.; et al. Immune checkpoint inhibitor-induced pancreatitis with pancreatic enlargement mimicking autoimmune pancreatitis: A case report and review of the literature. Intern. Med. 2024, 63, 791–798. [Google Scholar] [CrossRef]
- Yokode, M.; Shiokawa, M.; Kodama, Y. Review of diagnostic biomarkers in autoimmune pancreatitis: Where are we now? Diagnostics 2021, 11, 770. [Google Scholar] [CrossRef]
- Chen, L.; Orr, C.E.; Wang, T. Prevalence of histological features resembling autoimmune pancreatitis in neoplastic pancreas resections. Histopathology 2020, 77, 673–677. [Google Scholar] [CrossRef]
- Deshpande, V.; Gupta, R.; Sainani, N.; Sahani, D.V.; Virk, R.; Ferrone, C.; Khosroshahi, A.; Stone, J.H.; Lauwers, G.Y. Subclassification of autoimmune pancreatitis: A histologic classification with clinical significance. Am. J. Surg. Pathol. 2011, 35, 26–35. [Google Scholar] [CrossRef]
- Okazaki, K.; Chari, S.T.; Frulloni, L.; Lerch, M.M.; Kamisawa, T.; Kawa, S.; Kim, M.-H.; Lévy, P.; Masamune, A.; Webster, G.; et al. International consensus for the treatment of autoimmune pancreatitis. Pancreatology 2017, 17, 1–6. [Google Scholar] [CrossRef]
- Löhr, J.-M.; Beuers, U.; Vujasinovic, M.; Alvaro, D.; Frøkjær, J.B.; Buttgereit, F.; Capurso, G.; Culver, E.L.; de-Madaria, E.; Della-Torre, E.; et al. European Guideline on IgG4-related digestive disease—UEG and SGF evidence-based recommendations. United Eur. Gastroenterol. J. 2020, 8, 637–666. [Google Scholar] [CrossRef]
- Hart, P.A.; Kamisawa, T.; Brugge, W.R.; Chung, J.B.; Culver, E.L.; Czakó, L.; Frulloni, L.; Go, V.L.W.; Gress, T.M.; Kim, M.-H.; et al. Long-term outcomes of autoimmune pancreatitis: A multicentre, international analysis. Gut 2013, 62, 1771–1776. [Google Scholar] [CrossRef]
- Matsubayashi, H.; Yoneyama, M.; Nanri, K.; Sugimoto, S.; Shinjo, K.; Kakushima, N.; Tanaka, M.; Ito, S.; Takao, M.; Ono, H. Determination of steroid response by abdominal ultrasound in cases with autoimmune pancreatitis. Dig. Liver Dis. 2013, 45, 1034–1040. [Google Scholar] [CrossRef]
- Matsubayashi, H.; Ishiwatari, H.; Imai, K.; Kishida, Y.; Ito, S.; Hotta, K.; Yabuuchi, Y.; Yoshida, M.; Kakushima, N.; Takizawa, K.; et al. Steroid therapy and steroid response in autoimmune pancreatitis. Int. J. Mol. Sci. 2019, 21, 257. [Google Scholar] [CrossRef]
- Tacelli, M.; Celsa, C.; Magro, B.; Barresi, L.; Guastella, S.; Capurso, G.; Frulloni, L.; Cabibbo, G.; Cammà, C. Risk factors for rate of relapse and effects of steroid maintenance therapy in patients with autoimmune pancreatitis: Systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2019, 17, 1061–1072.e8. [Google Scholar] [CrossRef]
- Akiyama, M.; Takeuchi, T. IgG4-related disease: Beyond glucocorticoids. Drugs Aging 2018, 35, 275–287. [Google Scholar] [CrossRef]
- Brito-Zerón, P.; Kostov, B.; Bosch, X.; Acar-Denizli, N.; Ramos-Casals, M.; Stone, J.H. Therapeutic approach to IgG4-related disease: A systematic review. Medicine 2016, 95, e4002. [Google Scholar] [CrossRef]
- Chiabrando, F.; Lanzillotta, M.; Palumbo, D.; Pedica, F.; Caruso, M.; Capurso, G.; Della-Torre, E. Treating type 2 autoimmune pancreatitis with colchicine: A case series. Ann. Intern. Med. 2021, 174, 1775–1776. [Google Scholar] [CrossRef]
- Satish, D.; Lin, I.-H.; Flory, J.; Gerdes, H.; Postow, M.A.; Faleck, D.M. Exocrine pancreatic insufficiency induced by immune checkpoint inhibitors. Oncologist 2023, 28, 1085–1093. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vemulapalli, V.; Natha, C.; Shirwaikar Thomas, A. Autoimmune Pancreatitis: A Review. J. Clin. Med. 2025, 14, 3076. https://doi.org/10.3390/jcm14093076
Vemulapalli V, Natha C, Shirwaikar Thomas A. Autoimmune Pancreatitis: A Review. Journal of Clinical Medicine. 2025; 14(9):3076. https://doi.org/10.3390/jcm14093076
Chicago/Turabian StyleVemulapalli, Varun, Cristina Natha, and Anusha Shirwaikar Thomas. 2025. "Autoimmune Pancreatitis: A Review" Journal of Clinical Medicine 14, no. 9: 3076. https://doi.org/10.3390/jcm14093076
APA StyleVemulapalli, V., Natha, C., & Shirwaikar Thomas, A. (2025). Autoimmune Pancreatitis: A Review. Journal of Clinical Medicine, 14(9), 3076. https://doi.org/10.3390/jcm14093076