

Clinical Ophthalmic Outcomes and Impact of Single Large-Scale Mitochondrial DNA Deletions


Abstract
1. Introduction
Patients
2. Materials and Methods
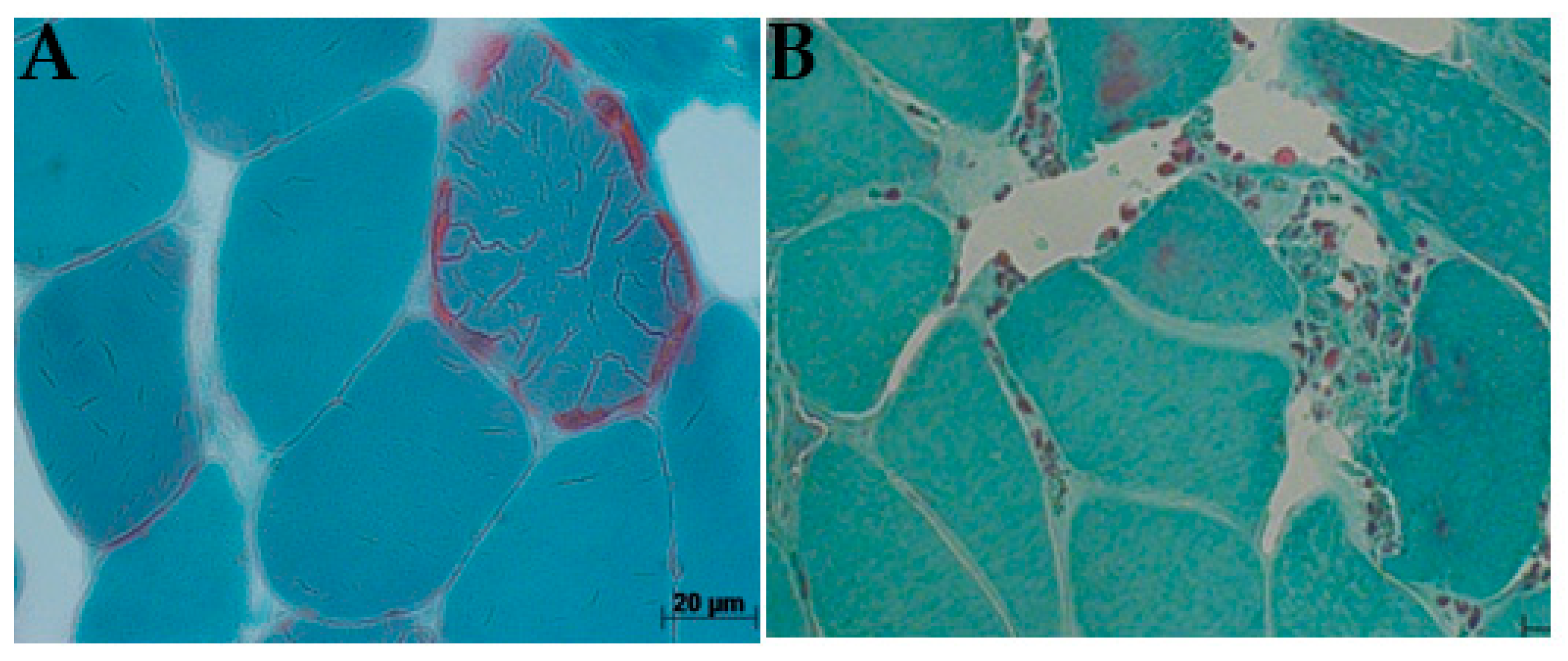
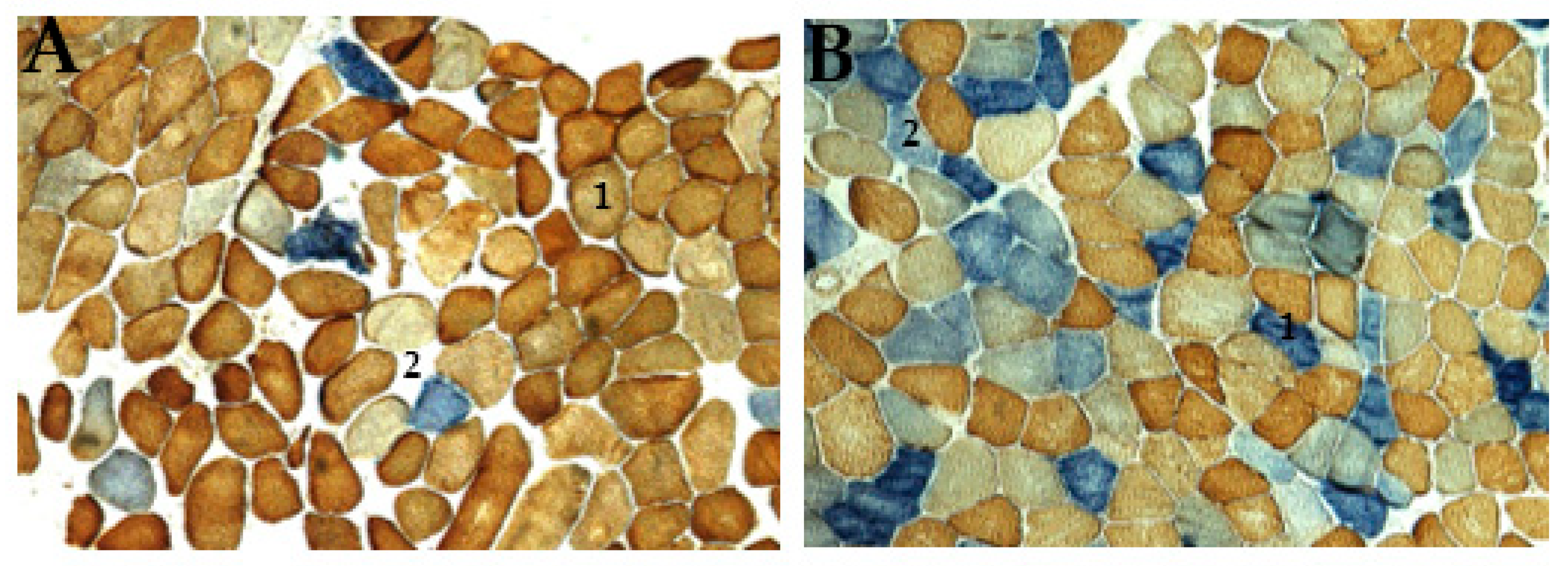
2.1. Histological Analysis
COX/SDH Stain
2.2. Molecular Genetic Analysis
Screening of the Common Deletion (CD)
2.3. Statistical Analysis
3. Results
3.1. Clinical Characteristics
3.2. Analytical Correlation Between Patients Age and mtDNA Deletion
3.3. Comparative Analysis of Age at Disease Onset and Clinical Symptoms
3.4. Genetics Evaluations Reveal the Presence of the mtDNA 4977 bp Common Deletion (CD)
3.5. Histological Evaluation Reveals mtDNA Disorder
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| mtDNA | mitochondrial DNA |
| CPEO | chronic progressive external ophthalmoplegia |
| KSS | Kearns–Sayre Syndrome |
| kb | kilobase |
| P | patients |
| G | gender |
| F | female |
| M | male |
| A | age at biopsy |
| O | onset of biopsy |
| DS | deletion size |
| kb | kilobase |
| VI | vision impairment |
| DM2 | diabetes mellitus type 2 |
| Mm | mitochondrial myopathy |
| UTI | urinary tract infection |
| IDA | iron deficiency anemia |
| IHD | ischemic heart disease |
| LFI | lactose and fructose intolerance |
| CK | chronic keratopathy |
| DE | depressive episode |
| CD | common deletion |
References
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499. [Google Scholar] [PubMed]
- Jokinen, R.; Marttinen, P.; Sandell, H.K.; Manninen, T.; Teerenhovi, H.; Wai, T.; Teoli, D.; Loredo-Osti, J.C.; Shoubridge, E.A.; Battersby, B.J. Gimap3 regulates tissue-specific mitochondrial DNA segregation. PLoS Genet. 2010, 6, e1001161. [Google Scholar]
- Gonzalez-Freire, M.; de Cabo, R.; Bernier, M.; Sollott, S.J.; Fabbri, E.; Navas, P.; Ferrucci, L. Reconsidering the role of mitochondria in aging. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1334–1342. [Google Scholar]
- Sciacco, M.; Bonilla, E.; Schon, E.A.; DiMauro, S.; Moraes, C.T. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum. Mol. Genet. 1994, 3, 13–19. [Google Scholar] [CrossRef]
- Salvatore, D.; Guido, D. Mitochondrial DNA and disease. Ann. Med. 2005, 37, 222–232. [Google Scholar]
- Chinnery, P.F.; Turnbull, D.M. Clinical features, investigation, and management of patients with defects of mitochondrial DNA (Editorial). J. Neurol. Neurosurg. Psychiatry 1997, 63, 559–563. [Google Scholar]
- Procaccio, V.; Neckelmann, N.; Paquis-Flucklinger, V.; Bannwarth, S.; Jimenez, R.; Davila, A.; Poole, J.C.; Wallace, D.C. Detection of low levels of the mitochondrial tRNA (Leu(UUR)) 3243ANG mutation in blood-derived from patients with diabetes. Mol. Diagn. Ther. 2006, 10, 381–389. [Google Scholar] [PubMed]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondfrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef]
- Moraes, C.T.; DiMauro, S.; Zeviani, M.; Lombes, A.; Shanske, S.; Miranda, A.F.; Nakase, H.; Bonilla, E.; Werneck, L.C.; Servidei, S.; et al. Mitochondrial DNA Deletions in Progressive External Ophthalmoplegia and Kearns–Sayre Syndrome. N. Engl. J. Med. 1989, 320, 1293–1299. [Google Scholar] [CrossRef]
- Meißner, C.; von Wurm, N.; Oehmichen, M. Detection of the age-dependent 4977 bp deletion of mitochondrial DNA. Int. J. Leg. Med 1997, 110, 288–289. [Google Scholar]
- Debrosse, S.; Parikh, S. Neurologic Disorders Due to Mitochondrial DNA Mutations. Semin. Pediatr. Neurol. 2012, 19, 194–202. [Google Scholar] [PubMed]
- Orsucci, D.; Caldarazzo Ienco, E.; Mancuso, M.; Siciliano, G. POLG1-related and other “mitochondrial Parkinsonisms”: An over-view. J. Mol. Neurosci. 2011, 44, 17–24. [Google Scholar]
- Laforêt, P.; Lombès, A.; Eymard, B.; Danan, C.; Chevallay, M.; Rouche, A.; Frachon, P.; Fardeau, M. Chronic progressive external ophthalmoplegia with ragged-red fibers: Clinical, morphological and genetic investigations in 43 patients. Neuromuscul. Disord. 1995, 5, 399–413. [Google Scholar]
- Kearns, T.P.; Sayre, G.P. Retinitis pigmentosa, external ophthalmoplegia, and complete heart block. Arch. Ophthal 1958, 60, 280–289. [Google Scholar]
- Goldstein, A.; Falk, M.J. Mitochondrial DNA Deletion Syndromes. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Rutten, J., Oberstein, S.A.L., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2003; pp. 1993–2020. [Google Scholar]
- Coulter-Mackie, M.B.; Applegarth, D.A.; Toone, J.R.; Gagnier, L. A Protocol for Detection of Mitochondrial DNA Deletions: Characterization of a Novel Deletion. Clin. Biochem. 1998, 31, 627–632. [Google Scholar]
- Yamashita, S.; Nishino, I.; Nonaka, I.; Goto, Y.I. Genotype and phenotype analyses in 136 patients with single large-scale mitochondrial DNA deletions. J. Hum. Genet. 2008, 53, 598–606. [Google Scholar]
- Filosto, M.; Tomelleri, G.; Tonin, P.; Scarpelli, M.; Vattemi, G.; Rizzuto, N.; Padovani, A.; Simonati, A. Neuropathology of mitochondrial diseases. Biosci. Rep. 2007, 27, 23–30. [Google Scholar] [CrossRef]
- Himani, S.; Archna, S.; Chandresh, S.; Sunesh, K.J.; Singh, N. Mutations in the mitochondrial DNA D-loop region are frequent in cervical cancer. Cancer Cell Int. 2005, 5, 34. [Google Scholar]
- Zeviani, M.; Servidei, S.; Gellera, C.; Bertini, E.; DiMauro, S.; DiDonato, S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 1989, 339, 309–311. [Google Scholar] [PubMed]
- Deschauer, M.; Kiefer, R.; Blakely, E.L. A novel Twinkle gene mutation in autosomal dominant progressive external ophthalmoplegia. Neuromuscul. Disord. 2003, 13, 568–572. [Google Scholar] [CrossRef]
- Lin, P.S.; Cheng, Y.M.; Wei, Y.H. Large-scale mitochondrial DNA deletions in skeletal muscle of patients with end-stage renal disease. Free Radic. Biol. Med. 2000, 29, 454–463. [Google Scholar]
- Jaksch, M.; Horvath, R.; Horn, N.; Auer, D.P.; Macmillan, C.; Peters, J.; Gerbitz, D.; Kraegeloh-Mann, I.; Muntau, A.; Karcagi, V.; et al. Homozygosity (E140K) in SCO2 causes delayed infantile onset of cardiomyopathy and neuropathy. Neurology 2001, 57, 1440–1446. [Google Scholar] [PubMed]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA depletion syndromes: Review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 2013, 10, 186–198. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Lott, M.T.; Voljavec, A.S.; Soueidan, S.A.; Costigan, D.A.; Wallace, D.C. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: A slip-replication model and metabolic therapy. Proc. Natl. Acad. Sci. USA 1989, 86, 7952–7956. [Google Scholar]
- Hanisch, F.; Kornhuber, M.; Alston, C.L.; Taylor, R.W.; Deschauer, M.; Zierz, S. SANDO syndrome in a cohort of 107 patients with CPEO and mitochondrial DNA deletions. J. Neurol. Neurosurg. Psychiatry 2014, 86, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Porteous, W.K.; James, A.M.; Sheard, P.W.; Porteous, C.M.; Packer, M.A.; Hyslop, S.J.; Melton, J.V.; Pang, C.Y.; Wei, Y.H.; Murphy, M.P. Bioenergetic consequences of accumulating the common 4977-bp mitochondrial DNA deletion. Eur. J. Biochem. 1998, 257, 192–201. [Google Scholar]
- Holt, I.J.; Harding, A.E.; Cooper, J.M.; Schapira, A.H.; Toscano, A.; Clark, J.B.; Morgan-Hughes, J.A. Mitochondrial myopathies: Clinical and biochemical features of 30 patients with major deletions of muscle mitochondrial DNA. Ann. Neurol. 1989, 26, 699–708. [Google Scholar]
- Nickander, K.K.; Schmelzer, J.D.; Low, P.A. Assessment of the “common” 4.8-kb mitochondrial DNA deletion and identification of several closely related deletions in the dorsal root ganglion of aging and streptozotocin rats. J. Peripher. Nerv. Syst. 2002, 7, 96–103. [Google Scholar]
- Tabaku, M.; Legius, E.; Robberecht, W.; Sciot, R.; Fryns, J.P.; Cassiman, J.J.; Matthijs, G. A novel 7.4 kb mitochondrial deletion in a patient with congenital progressive external ophthalmoplegia, muscle weakness, and mental retardation. Genet. Couns. 1999, 10, 285–293. [Google Scholar] [PubMed]
- Gattermann, N.; Berneburg, M.; Heinisch, J.; Aul, C.; Schneider, W. Detection of the aging-associated 5-Kb common deletion of mitochondrial DNA in blood and bone marrow of hematologically normal adults. Absence of the deletion in clonal bone marrow disorders. Leukemia 1995, 9, 1704–1710. [Google Scholar] [PubMed]
- Diba, K.; Miyawaki, H. Regulation of hippocampal firing by network oscillations during sleep. Curr. Biol. 2016, 26, 893–902. [Google Scholar]
- Dai, P.; Yang, W.; Jiang, S.; Gu, R.; Yuan, H.; Han, D.; Guo, W.; Cao, J. Correlation of cochlear blood supply with mitochondrial DNA common deletion in presbyacusis. Acta Otolaryngol. 2004, 124, 130–136. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, M.A.; Federico, A.; Minetti, C.; Moggio, M. Redefining phenotypes associated with mitochondrial DNA single deletion. J. Neurol. 2015, 262, 1301–1309. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P | G | A (yrs) | O (yrs) | Clinical Symptoms | DS (kb) |
|---|---|---|---|---|---|
| 1 | F | 47 | 20 | CPEO, ptosis, LFI, CK | 5.0 |
| 2 | F | 24 | 16 | CPEO | 5.5 |
| 3 | F | 42 | 31 | CPEO, KSS | 5.0 |
| 4 | F | 19 | 19 | CPEO | 4.5 |
| 5 | F | 29 | 21 | CPEO, ptosis, Mm | 5.0 |
| 6 | F | 32 | 17 | CPEO, ptosis, myopathy, KSS | 5.5 |
| 7 | F | 43 | 30 | CPEO, ptosis, perinatal hypoxia | 2.5 |
| 8 | F | 30 | 11 | CPEO, ptosis, UTI, IDA | 5.0 |
| 9 | F | 34 | 34 | CPEO | 6.0 |
| 10 | F | 21 | 21 | CPEO, VI | 5.5 |
| 11 | F | 37 | 37 | CPEO | 9.0 |
| 12 | F | 34 | 34 | CPEO | 5.5 |
| 13 | F | 27 | 21 | CPEO, subacute dysphagia, DM2 ptosis, myopathy, | 3.0 |
| 14 | F | 54 | 50 | CPEO, ptosis, Mm, glaucoma, UTI | 5.0 |
| 15 | F | 61 | 34 | CPEO | 5.0 |
| 16 | F | 74 | 22 | CPEO, ptosis, DM2 absolute arrhythmia, IHD | 4.0 |
| 17 | F | 49 | 40 | CPEO, ptosis, DM2 absolute arrhythmia, IHD | 2.5 |
| 18 | F | 14 | 10 | CPEO, ptosis | 7.5 |
| 19 | F | 55 | 25 | CPEO, ptosis, cataract, DE | 5.5 |
| 20 | F | 53 | 53 | CPEO, Mm | 5.5 |
| 21 | F | 49 | 35 | CPEO, ptosis, migraine | 5.0 |
| 22 | F | 50 | 15 | CPEO, ptosis | 2.0 |
| 23 | F | 27 | 17 | CPEO, Ptosis, VI, DM2 | 5.0 |
| 24 | F | 40 | 38 | CPEO, ptosis, Mm | 2.0 |
| 25 | F | 36 | 27 | CPEO | 2.5 |
| 26 | M | 15 | 14 | CPEO | 5.0 |
| 27 | M | 65 | 33 | CPEO, Mm | 2.0 |
| 28 | M | 38 | 31 | CPEO, mitochondrial myopathy | 7.5 |
| 29 | M | 25 | 24 | CPEO, myopathy, intracerebellar hemorrhage | 6.0 |
| 30 | M | 33 | 26 | CPEO | 5.0 |
| 31 | M | 42 | 22 | CPEO, ptosis | 5.0 |
| 32 | M | 32 | 32 | CPEO, mitochondrial myopathy | 3.5 |
| 33 | M | 66 | 48 | CPEO, ptosis, DM, IHD, hyperlipidemia | 5.0 |
| 34 | M | 71 | 16 | CPEO, ptosis, DM, Mm | 4.5 |
| 35 | M | 49 | 41 | CPEO, ptosis, arrhythmia, hyperlipidemia, arterial hypertonia | 5.5 |
| 36 | M | 73 | 44 | CPEO, ptosis, diabetes mellitus type 2, bradycardia, arterial hypertonia | 4.0 |
| Age Group (Years) | No. of Patients | Mean Age (±SD) | Range of Deletion (kb) | Frequency of CD |
|---|---|---|---|---|
| ≤18 | 8 | 14.50 (±5.0) | 2.0–7.5 | 2 (18.18%) |
| >18 | 28 | 31.90 (±9.5) | 2.0–9.0 | 9 (81.82%) |
| Patients | No. of Patients | With CD | p-Value | Without CD | p-Value |
|---|---|---|---|---|---|
| Total | 36 | 11 (30.56%) | 25 (69.44%) | ||
| Ptosis | 20 | 8 (40.00%) | 0.013 * | 12 (60.00%) | 0.032 * |
| Mm | 9 | 3 (33.33%) | 0.286 | 6 (66.67%) | 0.019 * |
| VI | 4 | 2 (50.00%) | 0.337 | 2 (50.00%) | 0.668 |
| DM | 6 | 3 (50.00%) | 0.354 | 3 (50.00%) | 0.415 |
| KSS | 2 | 2 (5.56%) | 0.038 * | No value |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erhunmwunse, M.O.; Joshi, P.R. Clinical Ophthalmic Outcomes and Impact of Single Large-Scale Mitochondrial DNA Deletions. J. Clin. Med. 2025, 14, 2537. https://doi.org/10.3390/jcm14082537
Erhunmwunse MO, Joshi PR. Clinical Ophthalmic Outcomes and Impact of Single Large-Scale Mitochondrial DNA Deletions. Journal of Clinical Medicine. 2025; 14(8):2537. https://doi.org/10.3390/jcm14082537
Chicago/Turabian StyleErhunmwunse, Michael Otakhor, and Pushpa Raj Joshi. 2025. "Clinical Ophthalmic Outcomes and Impact of Single Large-Scale Mitochondrial DNA Deletions" Journal of Clinical Medicine 14, no. 8: 2537. https://doi.org/10.3390/jcm14082537
APA StyleErhunmwunse, M. O., & Joshi, P. R. (2025). Clinical Ophthalmic Outcomes and Impact of Single Large-Scale Mitochondrial DNA Deletions. Journal of Clinical Medicine, 14(8), 2537. https://doi.org/10.3390/jcm14082537