
Diagnostic Role of Immunofluorescence Analysis in Primary Ciliary Dyskinesia-Suspected Individuals

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Public and Patient Involvement Statement
2.3. X-Ray Imaging
2.4. Nasal Nitric Oxide (nNO)
2.5. High-Speed Video Microscopy (HSVM)
2.6. Immunofluorescence Analysis
2.7. Statistical Analysis
3. Results
3.1. Clinical Features
3.2. HSVM Analysis
3.3. Genetic Results
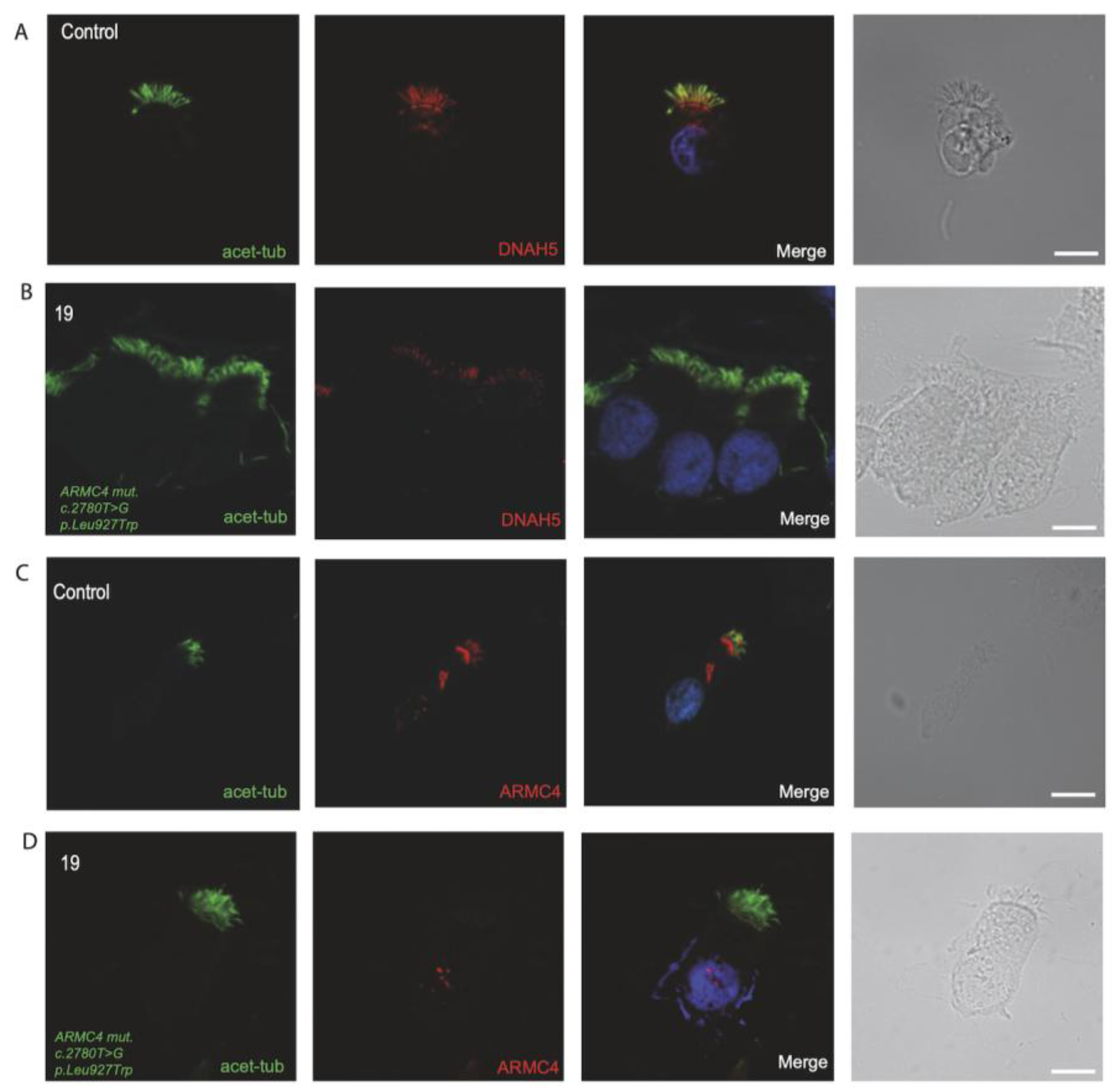
3.4. Immunofluorescence Analysis for Genetically Analyzed Individuals
3.5. Diagnostic IF Analysis for Non-Genetically Analyzed Individuals
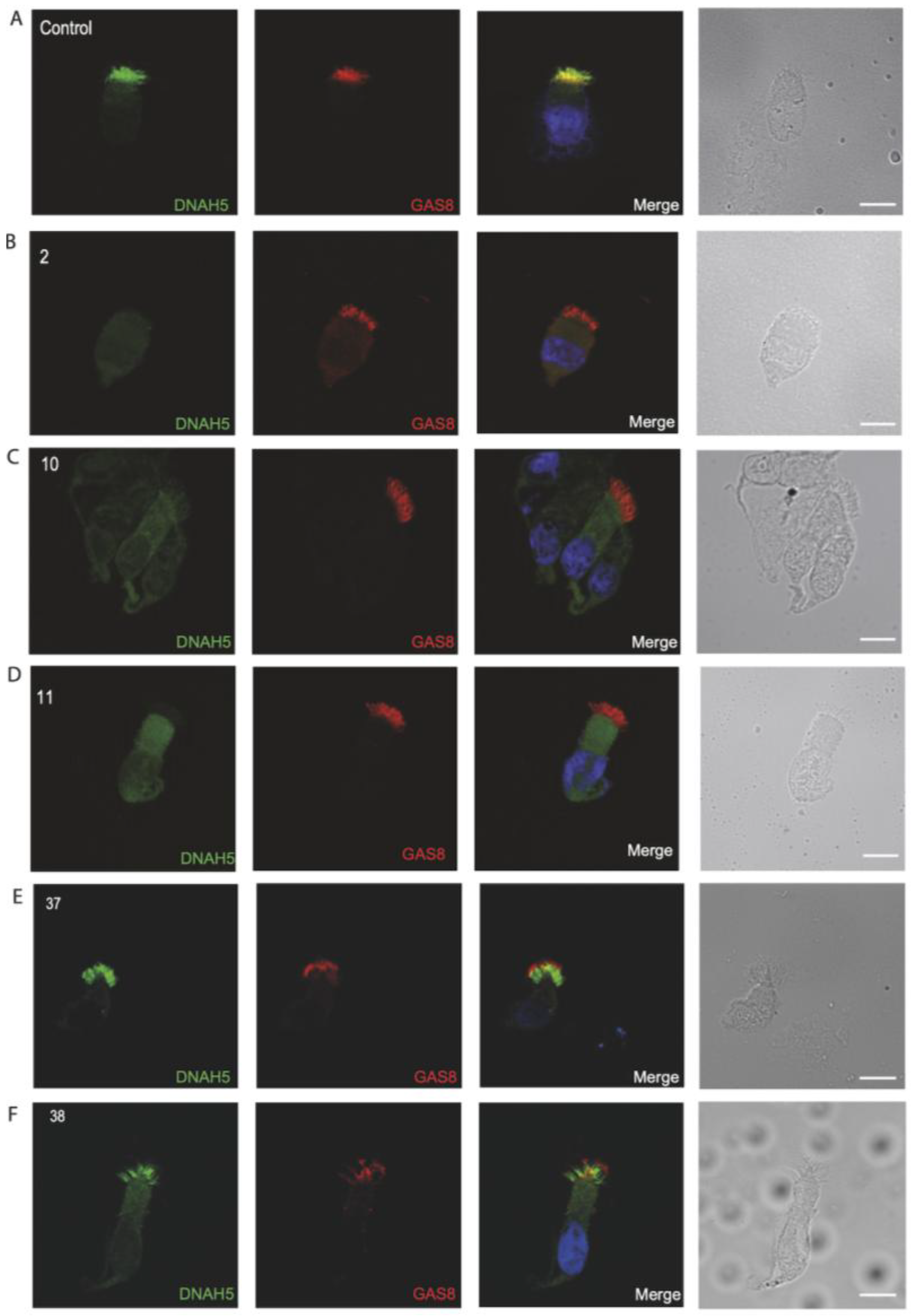
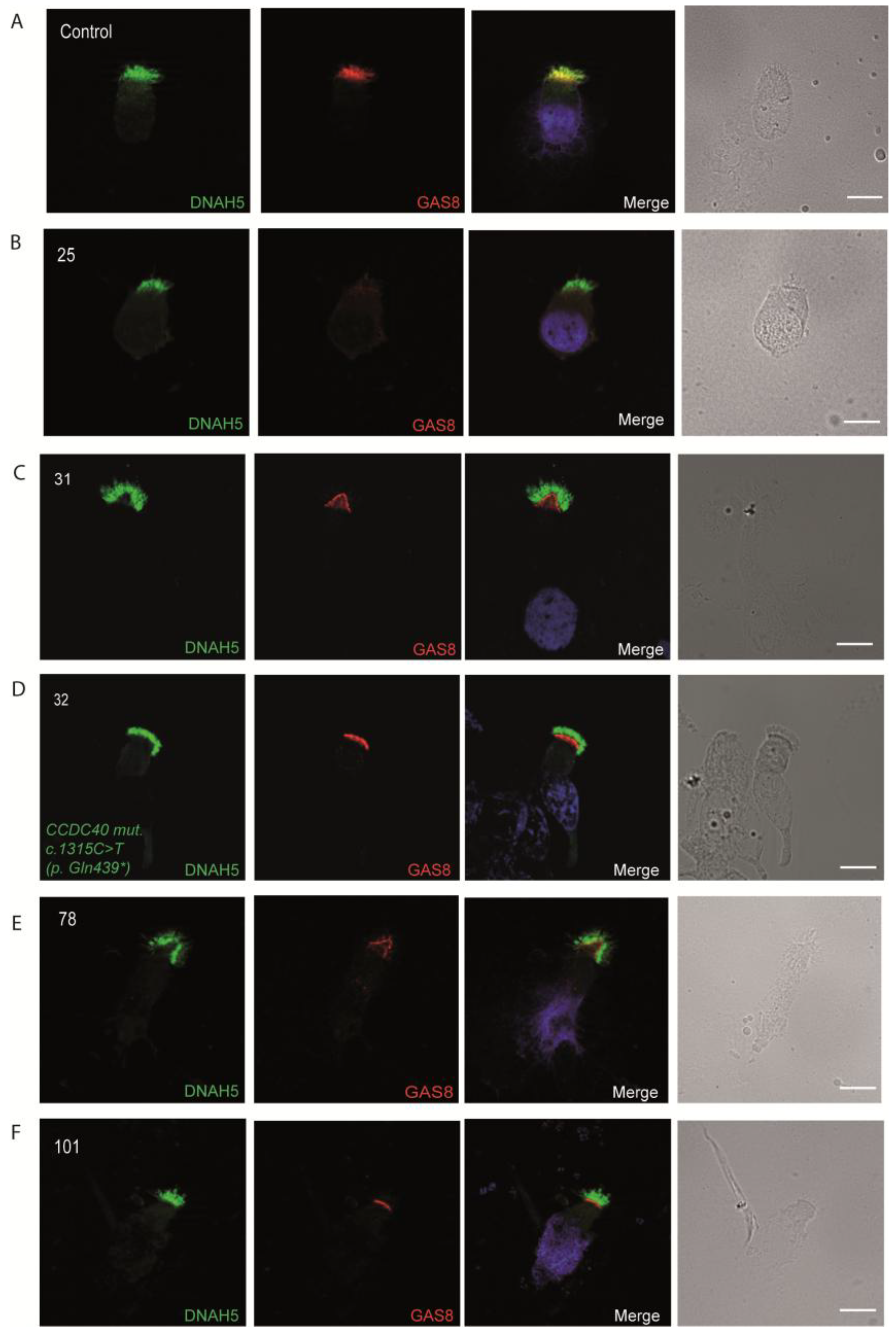
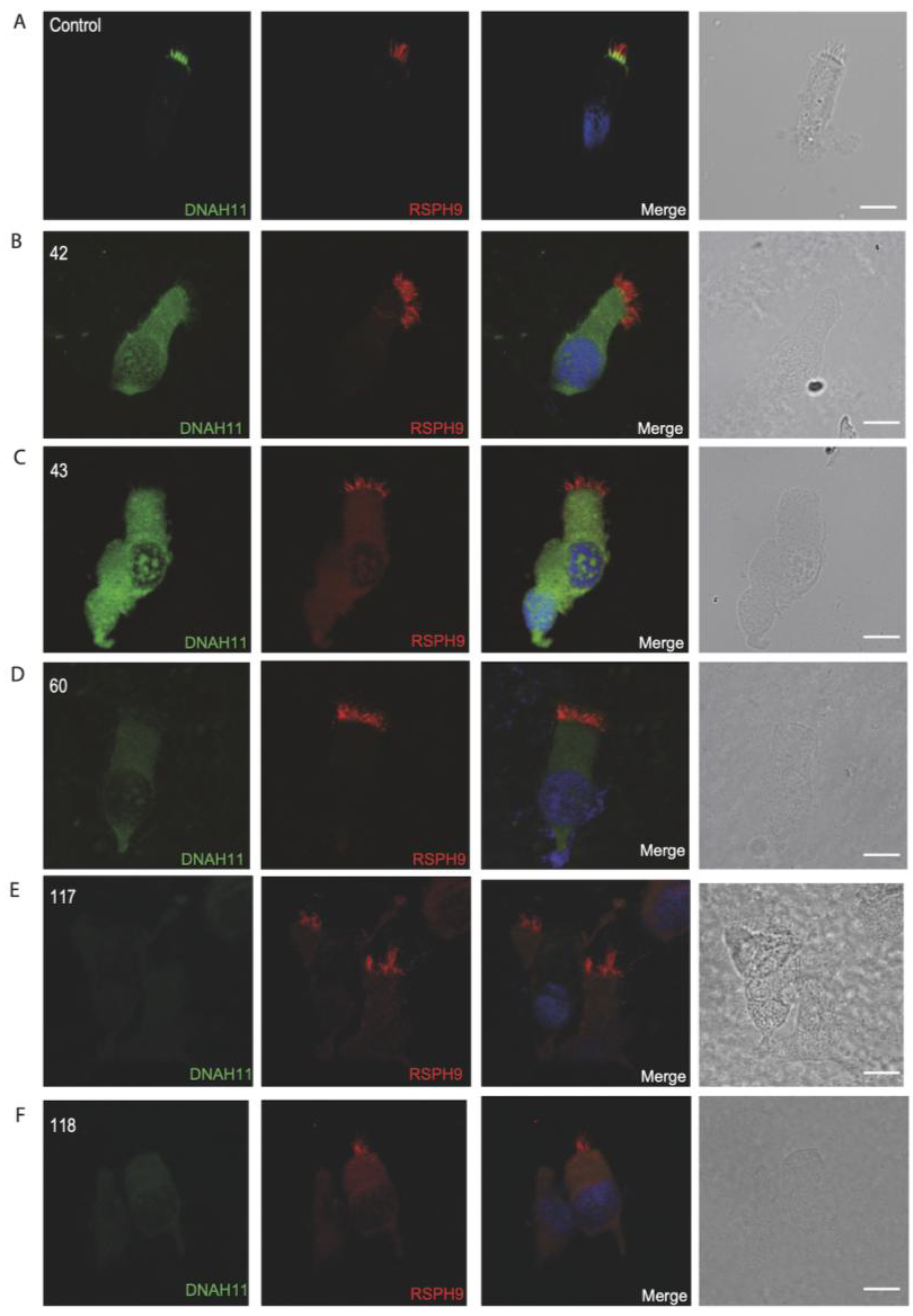
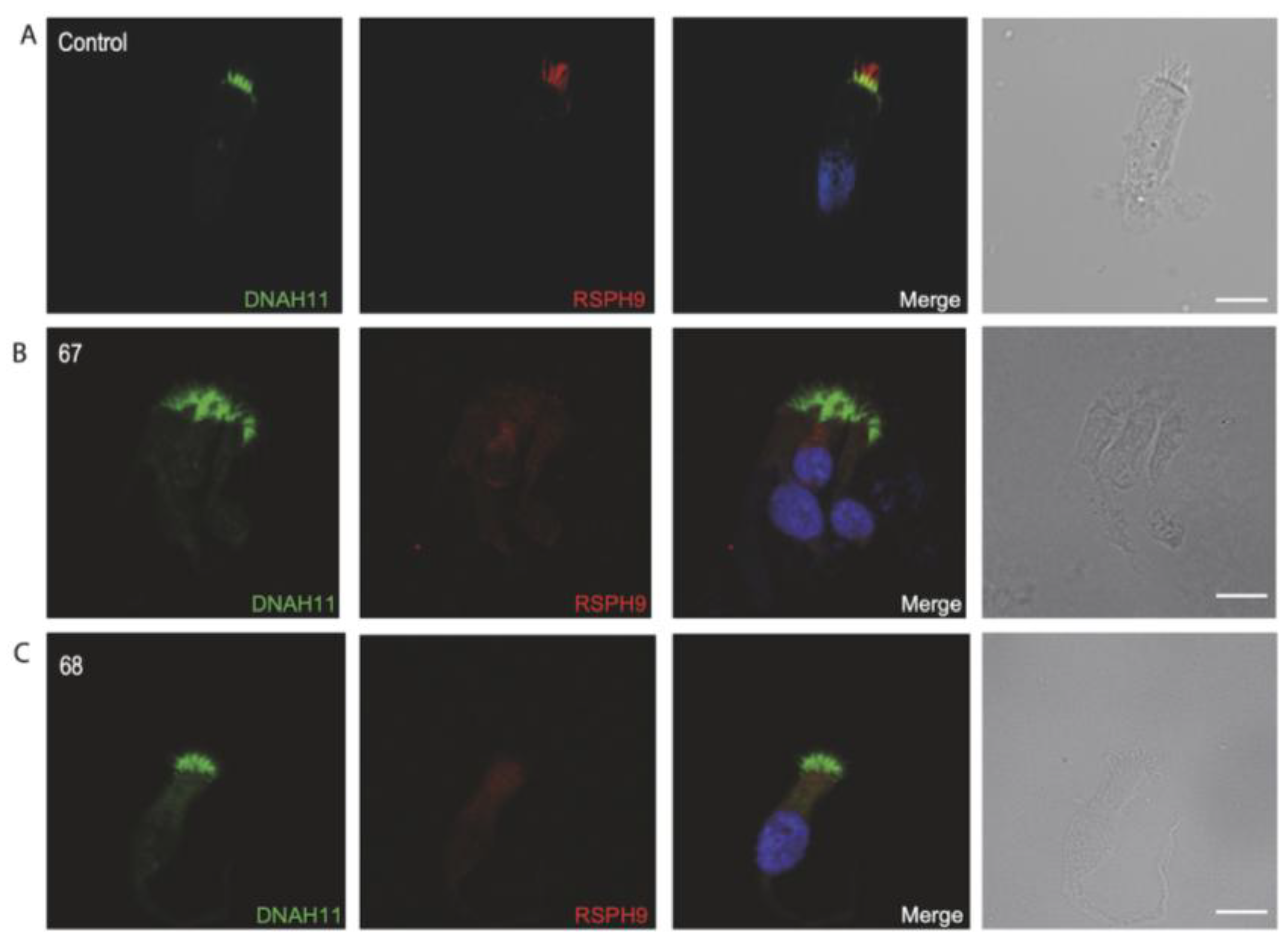
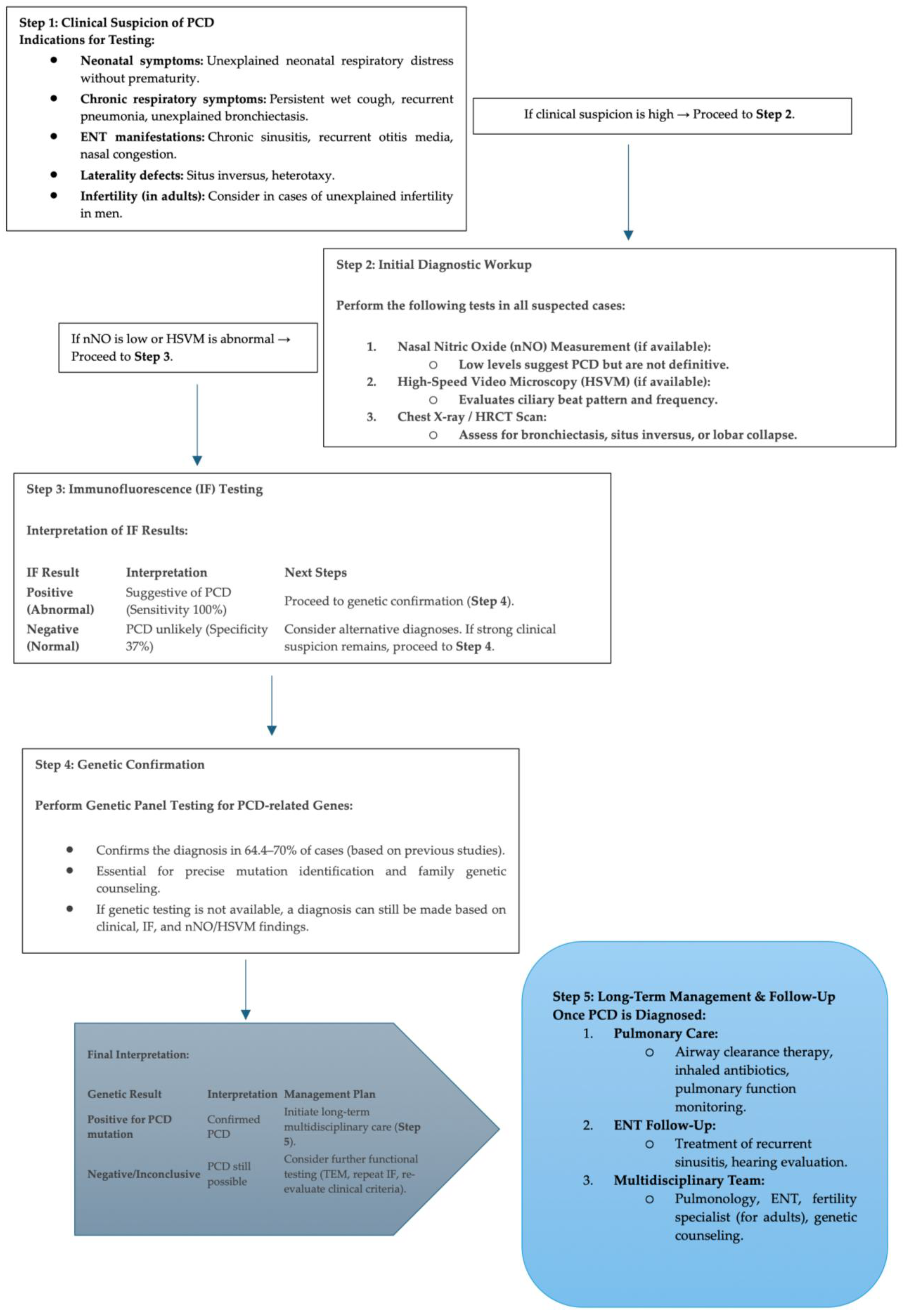
3.6. Proposed Diagnostic Algorithm for PCD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mirra, V.; Werner, C.; Santamaria, F. Primary Ciliary Dyskinesia: An Update on Clinical Aspects, Genetics, Diagnosis, and Future Treatment Strategies. Front. Pediatr. 2017, 5, 135. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Ferkol, T.W. Advances in the Genetics of Primary Ciliary Dyskinesia. Chest 2018, 154, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Reilly, M.L.; Benmerah, A. Ciliary kinesins beyond IFT: Cilium length, disassembly, cargo transport and signalling. Biol. Cell 2019, 111, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Shi, X.; Zhou, Y.; Duan, J.; He, L.; Song, X.; Huang, Z.; Chen, R.; Li, J.; Jia, N. Genetic spectrum and genotype–phenotype correlations in DNAH5-mutated primary ciliary dyskinesia: A systematic review. Orphanet J. Rare Dis. 2025, 20, 97. [Google Scholar] [CrossRef]
- Olbrich, H.; Schmidts, M.; Werner, C.; Onoufriadis, A.; Loges, N.T.; Raidt, J.; Banki, N.F.; Shoemark, A.; Burgoyne, T.; Al Turki, S.; et al. Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012, 91, 672–684. [Google Scholar] [CrossRef]
- Loges, N.T.; Antony, D.; Maver, A.; Deardorff, M.A.; Gulec, E.Y.; Gezdirici, A.; Nothe-Menchen, T.; Hoben, I.M.; Jelten, L.; Frank, D.; et al. Recessive DNAH9 Loss-of-Function Mutations Cause Laterality Defects and Subtle Respiratory Ciliary-Beating Defects. Am. J. Hum. Genet. 2018, 103, 995–1008. [Google Scholar] [CrossRef]
- Zhao, L.; Hou, Y.; Picariello, T.; Craige, B.; Witman, G.B. Proteome of the central apparatus of a ciliary axoneme. J. Cell Biol. 2019, 218, 2051–2070. [Google Scholar] [CrossRef]
- Merveille, A.C.; Davis, E.E.; Becker-Heck, A.; Legendre, M.; Amirav, I.; Bataille, G.; Belmont, J.; Beydon, N.; Billen, F.; Clement, A.; et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011, 43, 72–78. [Google Scholar] [CrossRef]
- Becker-Heck, A.; Zohn, I.E.; Okabe, N.; Pollock, A.; Lenhart, K.B.; Sullivan-Brown, J.; McSheene, J.; Loges, N.T.; Olbrich, H.; Haeffner, K.; et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011, 43, 79–84. [Google Scholar] [CrossRef]
- Loges, N.T.; Olbrich, H.; Becker-Heck, A.; Häffner, K.; Heer, A.; Reinhard, C.; Schmidts, M.; Kispert, A.; Zariwal, M.A.; Leigh, M.W.; et al. Deletions and Point Mutations of Cause Primary Ciliary Dyskinesia Due to Dynein Arm Defects. Am. J. Hum. Genet. 2009, 85, 883–889. [Google Scholar] [CrossRef]
- Mitchison, H.M.; Schmidts, M.; Loges, N.T.; Freshour, J.; Dritsoula, A.; Hirst, R.A.; O’Callaghan, C.; Blau, H.; Al Dabbagh, M.; Olbrich, H.; et al. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012, 44, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.V.; Shook, B.; Cantino, M.; Schwartz, D.; Jahnke, C.; et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Druley, T.E.; Zariwala, M.A.; Pate, A.C.; Levinson, B.T.; Van Arendonk, L.G.; Thornton, K.C.; Giacalone, J.C.; Albee, A.J.; Wilson, K.S.; et al. Whole-Exome Capture and Sequencing Identifies Mutation as a Cause of Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 2012, 91, 685–693. [Google Scholar] [CrossRef]
- Paff, T.; Loges, N.T.; Aprea, I.; Wu, K.; Bakey, Z.; Haarman, E.G.; Daniels, J.M.A.; Sistermans, E.A.; Bogunovic, N.; Dougherty, G.W.; et al. Mutations in PIH1D3 Cause X-Linked Primary Ciliary Dyskinesia with Outer and Inner Dynein Arm Defects. Am. J. Hum. Genet. 2017, 100, 160–168. [Google Scholar] [CrossRef]
- Zariwala, M.A.; Gee, H.Y.; Kurkowiak, M.; Al-Mutairi, D.A.; Leigh, M.W.; Hurd, T.W.; Hjeij, R.; Dell, S.D.; Chaki, M.; Dougherty, G.W.; et al. Is Mutated in Primary Ciliary Dyskinesia and Interacts with LRRC6. Am. J. Hum. Genet. 2013, 93, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Kott, E.; Duquesnoy, P.; Copin, B.; Legendre, M.; Dastot-Le Moal, F.; Montantin, G.; Jeanson, L.; Tamalet, A.; Papon, J.F.; Siffroi, J.P.; et al. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 958–964. [Google Scholar] [CrossRef]
- Hjeij, R.; Aprea, I.; Poeta, M.; Nothe-Menchen, T.; Bracht, D.; Raidt, J.; Honecker, B.I.; Dougherty, G.W.; Olbrich, H.; Schwartz, O.; et al. Pathogenic variants in CLXN encoding the outer dynein arm docking-associated calcium-binding protein calaxin cause primary ciliary dyskinesia. Genet. Med. 2023, 25, 100798. [Google Scholar] [CrossRef]
- Hjeij, R.; Lindstrand, A.; Francis, R.; Zariwala, M.A.; Liu, X.; Li, Y.; Damerla, R.; Dougherty, G.W.; Abouhamed, M.; Olbrich, H.; et al. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet. 2013, 93, 357–367. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Paff, T.; Antony, D.; Shoemark, A.; Micha, D.; Kuyt, B.; Schmidts, M.; Petridi, S.; Dankert-Roelse, J.E.; Haarman, E.G.; et al. Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 92, 88–98. [Google Scholar] [CrossRef]
- Hjeij, R.; Onoufriadis, A.; Watson, C.M.; Slagle, C.E.; Klena, N.T.; Dougherty, G.W.; Kurkowiak, M.; Loges, N.T.; Diggle, C.P.; Morante, N.F.; et al. CCDC151 mutations cause primary ciliary dyskinesia by disruption of the outer dynein arm docking complex formation. Am. J. Hum. Genet. 2014, 95, 257–274. [Google Scholar] [CrossRef]
- Wallmeier, J.; Shiratori, H.; Dougherty, G.W.; Edelbusch, C.; Hjeij, R.; Loges, N.T.; Menchen, T.; Olbrich, H.; Pennekamp, P.; Raidt, J.; et al. TTC25 Deficiency Results in Defects of the Outer Dynein Arm Docking Machinery and Primary Ciliary Dyskinesia with Left-Right Body Asymmetry Randomization. Am. J. Hum. Genet. 2016, 99, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Schultz, R.; Elenius, V.; Fassad, M.R.; Freke, G.; Rogers, A.; Shoemark, A.; Koistinen, T.; Mohamed, M.A.; Lim, J.S.Y.; Mitchison, H.M.; et al. CFAP300 mutation causing primary ciliary dyskinesia in Finland. Front. Genet. 2022, 13, 985227. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Li, H.; Wu, P.; Yang, Q.; Wan, X.; Wu, Y. LRRC56 deletion causes primary ciliary dyskinesia in mice characterized by dynein arms defects. Biol. Open 2025, 14, bio061846. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.S.; Barbato, A.; Collins, S.A.; Goutaki, M.; Behan, L.; Caudri, D.; Dell, S.; Eber, E.; Escudier, E.; Hirst, R.A.; et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 2017, 49, 1601090. [Google Scholar] [CrossRef]
- Shoemark, A.; Frost, E.; Dixon, M.; Ollosson, S.; Kilpin, K.; Patel, M.; Scully, J.; Rogers, A.V.; Mitchison, H.M.; Bush, A.; et al. Accuracy of Immunofluorescence in the Diagnosis of Primary Ciliary Dyskinesia. Am. J. Respir. Crit. Care Med. 2017, 196, 94–101. [Google Scholar] [CrossRef]
- Shoemark, A. Applications of emerging transmission electron microscopy technology in PCD research and diagnosis. Ultrastruct. Pathol. 2017, 41, 408–414. [Google Scholar] [CrossRef]
- Raidt, J.; Wallmeier, J.; Hjeij, R.; Onnebrink, J.G.; Pennekamp, P.; Loges, N.T.; Olbrich, H.; Haffner, K.; Dougherty, G.W.; Omran, H.; et al. Ciliary beat pattern and frequency in genetic variants of primary ciliary dyskinesia. Eur. Respir. J. 2014, 44, 1579–1588. [Google Scholar] [CrossRef]
- Farley, H.; Rubbo, B.; Bukowy-Bieryllo, Z.; Fassad, M.; Goutaki, M.; Harman, K.; Hogg, C.; Kuehni, C.E.; Lopes, S.; Nielsen, K.G.; et al. Proceedings of the 3rd BEAT-PCD Conference and 4th PCD Training School. BMC Proc. 2018, 12, 64. [Google Scholar] [CrossRef]
- Wallmeier, J.; Nielsen, K.G.; Kuehni, C.E.; Lucas, J.S.; Leigh, M.W.; Zariwala, M.A.; Omran, H. Motile ciliopathies. Nat. Rev. Dis. Primers 2020, 6, 77. [Google Scholar] [CrossRef]
- Chen, W.; Guo, Z.; Li, M.; Sheng, W.; Huang, G. Next-Generation Sequencing-Based Copy Number Variation Analysis in Chinese Patients with Primary Ciliary Dyskinesia Revealed Novel DNAH5 Copy Number Variations. Phenomics 2024, 4, 24–33. [Google Scholar] [CrossRef]
- Emiralioglu, N.; Taskiran, E.Z.; Kosukcu, C.; Bilgic, E.; Atilla, P.; Kaya, B.; Gunaydin, O.; Yuzbasioglu, A.; Tugcu, G.D.; Ademhan, D.; et al. Genotype and phenotype evaluation of patients with primary ciliary dyskinesia: First results from Turkey. Pediatr. Pulmonol. 2020, 55, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, G.W.; Mizuno, K.; Nothe-Menchen, T.; Ikawa, Y.; Boldt, K.; Ta-Shma, A.; Aprea, I.; Minegishi, K.; Pang, Y.P.; Pennekamp, P.; et al. CFAP45 deficiency causes situs abnormalities and asthenospermia by disrupting an axonemal adenine nucleotide homeostasis module. Nat. Commun. 2020, 11, 5520. [Google Scholar] [CrossRef] [PubMed]
- Al-Mutairi, D.A.; Alsabah, B.H.; Alkhaledi, B.A.; Pennekamp, P.; Omran, H. Identification of a novel founder variant in DNAI2 cause primary ciliary dyskinesia in five consanguineous families derived from a single tribe descendant of Arabian Peninsula. Front. Genet. 2022, 13, 1017280. [Google Scholar] [CrossRef]
- Guo, T.; Lu, C.; Yang, D.; Lei, C.; Liu, Y.; Xu, Y.; Yang, B.; Wang, R.; Luo, H. Case Report: DNAAF4 Variants Cause Primary Ciliary Dyskinesia and Infertility in Two Han Chinese Families. Front. Genet. 2022, 13, 934920. [Google Scholar] [CrossRef]
- Goutaki, M.; Meier, A.B.; Halbeisen, F.S.; Lucas, J.S.; Dell, S.D.; Maurer, E.; Casaulta, C.; Jurca, M.; Spycher, B.D.; Kuehni, C.E. Clinical manifestations in primary ciliary dyskinesia: Systematic review and meta-analysis. Eur. Respir. J. 2016, 48, 1081–1095. [Google Scholar] [CrossRef]
- Hannah, W.B.; Seifert, B.A.; Truty, R.; Zariwala, M.A.; Ameel, K.; Zhao, Y.; Nykamp, K.; Gaston, B. The global prevalence and ethnic heterogeneity of primary ciliary dyskinesia gene variants: A genetic database analysis. Lancet Respir. Med. 2022, 10, 459–468. [Google Scholar] [CrossRef]
- Zhao, X.; Bian, C.; Liu, K.; Xu, W.; Liu, Y.; Tian, X.; Bai, J.; Xu, K.-F.; Zhang, X. Clinical characteristics and genetic spectrum of 26 individuals of Chinese origin with primary ciliary dyskinesia. Orphanet J. Rare Dis. 2021, 16, 293. [Google Scholar] [CrossRef]
- Poplawska, K.; Griffiths, A.; Temme, R.; Adamko, D.J.; Nykamp, K.; Shapiro, A.J. Deletions in DNAL1 Cause Primary Ciliary Dyskinesia Across North American Indigenous Populations. J. Pediatr. 2023, 261, 113362. [Google Scholar] [CrossRef]
- Rodriguez, N.M.; Schreck, L.D.; Pedersen, E.S.L.; Cizeau, I.; Müller, L.; Kruljac, C.; Lucas, J.S.; Goutaki, M.; Kuehni, C.E. Diagnostic testing in people with primary ciliary dyskinesia: An international participatory study. PLoS Glob. Public Health 2023, 3, e0001522. [Google Scholar] [CrossRef]
- Pedersen, E.S.L.; Goutaki, M.; Schreck, L.D.; Rindlisbacher, B.; Dixon, L.; Lucas, J.S.; Kuehni, C.E. Questionnaire-assessed genotypes and associations with symptoms in primary ciliary dyskinesia. ERJ Open Res. 2024, 10, 00288-2024. [Google Scholar] [CrossRef]
- Baz-Redon, N.; Rovira-Amigo, S.; Fernandez-Cancio, M.; Castillo-Corullon, S.; Cols, M.; Caballero-Rabasco, M.A.; Asensio, O.; Martin de Vicente, C.; Martinez-Colls, M.D.M.; Torrent-Vernetta, A.; et al. Immunofluorescence Analysis as a Diagnostic Tool in a Spanish Cohort of Patients with Suspected Primary Ciliary Dyskinesia. J. Clin. Med. 2020, 9, 3603. [Google Scholar] [CrossRef] [PubMed]
- Ta-Shma, A.; Hjeij, R.; Perles, Z.; Dougherty, G.W.; Abu Zahira, I.; Letteboer, S.J.F.; Antony, D.; Darwish, A.; Mans, D.A.; Spittler, S.; et al. Homozygous loss-of-function mutations in MNS1 cause laterality defects and likely male infertility. PLoS Genet. 2018, 14, e1007602. [Google Scholar] [CrossRef]
- Yue, Y.; Huang, Q.; Zhu, P.; Zhao, P.; Tan, X.; Liu, S.; Li, S.; Han, X.; Cheng, L.; Li, B.; et al. Identification of Pathogenic Mutations and Investigation of the NOTCH Pathway Activation in Kartagener Syndrome. Front. Genet. 2019, 10, 749. [Google Scholar] [CrossRef]
- Olbrich, H.; Cremers, C.; Loges, N.T.; Werner, C.; Nielsen, K.G.; Marthin, J.K.; Philipsen, M.; Wallmeier, J.; Pennekamp, P.; Menchen, T.; et al. Loss-of-Function GAS8 Mutations Cause Primary Ciliary Dyskinesia and Disrupt the Nexin-Dynein Regulatory Complex. Am. J. Hum. Genet. 2015, 97, 546–554. [Google Scholar] [CrossRef]
- Jeanson, L.; Thomas, L.; Copin, B.; Coste, A.; Sermet-Gaudelus, I.; Dastot-Le Moal, F.; Duquesnoy, P.; Montantin, G.; Collot, N.; Tissier, S.; et al. Mutations in GAS8, a Gene Encoding a Nexin-Dynein Regulatory Complex Subunit, Cause Primary Ciliary Dyskinesia with Axonemal Disorganization. Hum. Mutat. 2016, 37, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Zlotina, A.; Barashkova, S.; Zhuk, S.; Skitchenko, R.; Usoltsev, D.; Sokolnikova, P.; Artomov, M.; Alekseenko, S.; Simanova, T.; Goloborodko, M.; et al. Characterization of pathogenic genetic variants in Russian patients with primary ciliary dyskinesia using gene panel sequencing and transcript analysis. Orphanet J. Rare Dis. 2024, 19, 310. [Google Scholar] [CrossRef]
- Nussbaumer, M.; Kieninger, E.; Tschanz, S.A.; Savas, S.T.; Casaulta, C.; Goutaki, M.; Blanchon, S.; Jung, A.; Regamey, N.; Kuehni, C.E.; et al. Diagnosis of primary ciliary dyskinesia: Discrepancy according to different algorithms. ERJ Open Res. 2021, 7, 00353-2021. [Google Scholar] [CrossRef]
- Guo, S.; Tang, D.; Chen, Y.; Yu, H.; Gu, M.; Geng, H.; Fang, J.; Wu, B.; Ruan, L.; Li, K.; et al. Association of novel DNAH11 variants with asthenoteratozoospermia lead to male infertility. Hum. Genom. 2024, 18, 97. [Google Scholar] [CrossRef]
- Maddirevula, S.; Awartani, K.; Coskun, S.; AlNaim, L.F.; Ibrahim, N.; Abdulwahab, F.; Hashem, M.; Alhassan, S.; Alkuraya, F.S. A genomics approach to females with infertility and recurrent pregnancy loss. Hum. Genet. 2020, 139, 605–613. [Google Scholar] [CrossRef]
- Schreck, L.D.; Pedersen, E.S.L.; Dexter, K.; Manion, M.; Bellu, S.; Cizeau, I.; Dexter, K.; Dixon, L.; Fernández, T.L.; Grieder, S.; et al. Infertility and pregnancy outcomes among adults with primary ciliary dyskinesia. Hum. Reprod. Open 2024, 2024, hoae039. [Google Scholar] [CrossRef]
- Frommer, A.; Hjeij, R.; Loges, N.T.; Edelbusch, C.; Jahnke, C.; Raidt, J.; Werner, C.; Wallmeier, J.; Grosse-Onnebrink, J.; Olbrich, H.; et al. Immunofluorescence Analysis and Diagnosis of Primary Ciliary Dyskinesia with Radial Spoke Defects. Am. J. Respir. Cell Mol. Biol. 2015, 53, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Mabrouk, I.; Al-Harthi, N.; Mani, R.; Montantin, G.; Tissier, S.; Lagha, R.; Ben Abdallah, F.; Hassan, M.M.; Alhomrani, M.; Gaber, A.; et al. Combining RSPH9 founder mutation screening and next-generation sequencing analysis is efficient for primary ciliary dyskinesia diagnosis in Saudi patients. J. Hum. Genet. 2022, 67, 381–386. [Google Scholar] [CrossRef]
- Shoemark, A.; Boon, M.; Brochhausen, C.; Bukowy-Bieryllo, Z.; De Santi, M.M.; Goggin, P.; Griffin, P.; Hegele, R.G.; Hirst, R.A.; Leigh, M.W.; et al. International consensus guideline for reporting transmission electron microscopy results in the diagnosis of primary ciliary dyskinesia (BEAT PCD TEM Criteria). Eur. Respir. J. 2020, 55, 1900725. [Google Scholar] [CrossRef] [PubMed]
- Fassad, M.R.; Patel, M.P.; Shoemark, A.; Cullup, T.; Hayward, J.; Dixon, M.; Rogers, A.V.; Ollosson, S.; Jackson, C.; Goggin, P.; et al. Clinical utility of NGS diagnosis and disease stratification in a multiethnic primary ciliary dyskinesia cohort. J. Med. Genet. 2020, 57, 322–330. [Google Scholar] [CrossRef]
- Horani, A.; Ferkol, T.W.; Dutcher, S.K.; Brody, S.L. Genetics and biology of primary ciliary dyskinesia. Paediatr. Respir. Rev. 2016, 18, 18–24. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Individual ID | Age | Sex | Symptoms | Bronchiectasis | Situs Inversus | Consanguinity | Genetic Variant | HSVM | DNAH5 | GAS8 | DNAH11 | RSPH9 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 19 y | F | Recurrent Lung Infection, Hearing Defect | yes | yes | yes | NA | negative | normal | normal | normal | |
| 6 | 11 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect | no | no | no | Minimal residue movement | normal | normal | normal | normal | |
| 9 | 19 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis | no | yes | yes | DNAAF1 hom: c.1385A>C; p.Gln462Pro | Minimal residue movement | negative | normal | normal | normal |
| 10 | 15 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect | yes | no | yes | Minimal residue movement | negative | normal | normal | normal | |
| 11 | 24 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Lobectomy | yes | no | yes | Immotile cilia | negative | normal | normal | normal | |
| 12 | 24 y | M | Recurrent Lung Infection | yes | no | no | Minimal residue movement | normal | normal | normal | normal | |
| 14 | 15 y | F | Recurrent Lung Infection, Hearing Defect | no | yes | yes | Almost immotile cilia | negative | normal | normal | normal | |
| 15 | 12 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis | no | no | yes | Minimal residue movement | negative | normal | normal | normal | |
| 17 | 13 y | F | Recurrent Lung Infection, Low Nasal NO | yes | no | no | Immotile cilia | negative | normal | normal | normal | |
| 18 | 17 y | F | Recurrent Lung Infection | no | yes | yes | Almost immotile cilia | normal | normal | normal | normal | |
| 19 | 22 y | F | Recurrent Lung Infection, Hearing Defect | no | yes | yes | ARMC4 hom: c.2780T>; p.Leu927Trp | Minimal residue movement and reduced amplitude | proximal | normal | normal | normal |
| 20 | 20 y | F | Recurrent Lung Infection | no | no | yes | Almost immotile cilia | normal | normal | normal | normal | |
| 21 | 22 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect | yes | no | yes | Minimal residue movement | normal | normal | normal | normal | |
| 24 | 20 y | F | Recurrent Lung Infection, Low Nasal NO | yes | no | yes | Almost immotile cilia | negative | normal | normal | normal | |
| 25 | 10 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis | yes | yes | yes | Minimal residue movement | normal | negative | normal | normal | |
| 28 | 13 y | M | Recurrent Lung Infection, Low Nasal NO | yes | no | yes | Reduced amplitude | normal | negative | normal | normal | |
| 29 | 20 y | M | Recurrent Lung Infection, Low Nasal NO, Hearing Defect | yes | yes | no | Minimal residue movement | normal | negative | normal | normal | |
| 30 | 15 y | M | Recurrent Lung Infection, Low Nasal NO | yes | no | no | DNAH5 hom: c.2710G>; p.Glu904Ter | Almost immotile cilia | negative | normal | normal | normal |
| 31 | 22 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis | yes | no | yes | Reduced amplitude | normal | negative | normal | normal | |
| 32 | 8 y | M | Recurrent Lung Infection | no | no | yes | CCDC40 hom: c.1315C>T; p. Gln439Ter | Reduced amplitude | normal | negative | normal | normal |
| 33 | 21 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis | yes | yes | yes | DNAH5 hom: c.7615T>; p.Trp2539Arg | Minimal residue movement | negative | normal | normal | normal |
| 36 | 15 y | F | Recurrent Lung Infection, Low Nasal NO | yes | no | no | Hyperkinetic cilia | normal | normal | normal | normal | |
| 37 | 22 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis | no | no | yes | Almost immotile cilia | proximal | normal | normal | normal | |
| 38 | 16 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis | yes | yes | yes | Almost immotile cilia | proximal | normal | normal | normal | |
| 42 | 14 y | F | Low Nasal NO | no | yes | yes | Stiff pattern | normal | normal | negative | normal | |
| 43 | 24 y | F | Low Nasal NO, Recurrent Lung Infection, Hearing Defect | no | no | no | NA | normal | normal | negative | normal | |
| 44 | 16 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect | yes | no | yes | Minimal residue movement | negative | normal | normal | normal | |
| 46 | 16 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect | yes | no | yes | Minimal residue movement | proximal | normal | normal | normal | |
| 48 | 14 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis | yes | no | yes | Reduced amplitude | normal | negative | normal | normal | |
| 51 | 13 y | M | Recurrent Lung Infection, Sinusitis, Hearing Defect | yes | no | yes | Almost immotile cilia | normal | normal | normal | normal | |
| 53 | 20 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Lobectomy | yes | no | no | NA | normal | normal | normal | normal | |
| 60 | 24 y | F | Recurrent lung infection, Low Nasal NO, Infertility | no | no | no | Stiff pattern | normal | normal | negative | normal | |
| 63 | 5 y | M | Recurrent Lung Infection, Hearing Loss, Low Nasal NO | no | no | yes | Stiff pattern | normal | normal | normal | normal | |
| 66 | 21 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis | yes | no | yes | Minimal residue movement | proximal | normal | normal | normal | |
| 67 | 20 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect | yes | no | yes | Stiff pattern | normal | normal | normal | negative | |
| 68 | 17 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect, Lobectomy | yes | no | yes | Stiff pattern | normal | normal | normal | negative | |
| 72 | 14 y | M | Recurrent Lung Infection, Low Nasal NO | yes | yes | no | Minimal residue movement | normal | normal | normal | normal | |
| 73 | 21 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect | yes | no | yes | Minimal residue movement | normal | normal | normal | normal | |
| 76 | 16 y | M | Recurrent Lung Infection, Sinusitis, Hearing Defect | yes | no | yes | Minimal residue movement | negative | normal | normal | normal | |
| 77 | 19 y | M | Recurrent Lung Infection, Low Nasal NO | yes | no | yes | NA | normal | normal | normal | normal | |
| 78 | 12 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis | no | yes | yes | Minimal residue movement | normal | negative | normal | normal | |
| 83 | 15 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect | no | yes | yes | NA | normal | normal | normal | normal | |
| 96 | 22 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect | yes | no | yes | Minimal residue movement | negative | normal | normal | normal | |
| 100 | 12 y | F | Recurrent Lung Infection, Low Nasal NO | no | yes | yes | Almost immotile cilia | normal | normal | normal | normal | |
| 101 | 23 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Lobectomy | yes | no | yes | Reduced amplitude | normal | negative | normal | normal | |
| 102 | 24 y | F | Recurrent Lung Infection, Hearing Defect | yes | yes | yes | NA | normal | normal | normal | normal | |
| 107 | 17 y | F | Recurrent Lung Infection, Hearing Defect, Sinusitis | yes | yes | yes | DNAAF1 hom: c.1349dupC; p.Pro451fs Ter6 | Minimal residue movement | negative | normal | normal | normal |
| 109 | 27 y | F | Recurrent Lung Infection, Hearing Defect, Sinusitis | no | yes | yes | DNAAF1 hom: c.1228_1232delCCAGA; p.Pro410fs Ter8 | Almost immotile cilia | negative | normal | normal | normal |
| 110 | 22 y | F | Recurrent Lung Infection, Sinusitis | yes | yes | yes | Almost immotile cilia | negative | normal | normal | normal | |
| 117 | 14 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis | yes | no | yes | Stiff pattern | normal | normal | negative | normal | |
| 118 | 17 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Lobectomy | yes | no | yes | Stiff pattern | normal | normal | negative | normal | |
| 119 | 18 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis, Hearing Defect | yes | yes | no | DNAH5 hom: c.5747G>A; p.Trp1916Ter | Minimal residue movement | negative | normal | normal | normal |
| #PH 1 | 7 y | M | Recurrent Lung Infection, Low Nasal NO, Sinusitis | NA | NA | NA | Minimal residue movement | normal | normal | normal | normal | |
| #PH 2 | 12 y | F | Recurrent Lung Infection, Low Nasal NO, Sinusitis | yes | yes | yes | Minimal residue movement | normal | normal | normal | normal |
| Clinical Statistics Gender | N Median | Valid Percent | DNAH5 Abnormality n (% Within Group) | GAS8 Abnormality n (% Within Group) | DNAH11 Abnormality n (% Within Group) | RSPH9 Abnormality n (% Within Group) | p Value | |
|---|---|---|---|---|---|---|---|---|
| Female | 23 | 1.4444 | 55.6 44.4 | 14 (63.6%) 8 (36.4%) | 2 (25.0%) 6 (75.0%) | 3 (60.0%) 2 (40.0%) | 1 (50.0%) 1 (50.0%) | 0.432 |
| Male | 31 | |||||||
| Age, median (min-max) | 17 (5–32) | |||||||
| Consanguinity, n | 41 | 79.2 | 86.4% | 87.5% | 60% | 100% | 0.409 | |
| Sinusitis, n | 33 | 61.1 | 72.7% | 62.5% | 20% | 100% | 0.121 | |
| Bronchiectasis, n | 36 | 67.9 | 72.7% | 75% | 20% | 100% | 0.144 | |
| Recurrent lung infection, n | 53 | 98.1 | 100% | 100% | 80% | 100% | - | |
| Hearing defect, n | 22 | 40.7 | 50% | 12.5% | 40% | 100% | 0.105 | |
| Situs inversus totalis, n | 20 | 37.7 | 45.5% | 37.5% | 20% | 0% | 0.529 | |
| Lobectomy, n | 5 | 9.3 | 4.5% | 12.5% | 20% | 50% | 0.438 | |
| Infertility, n | 1 | 1.9 | 0% | 0% | 20% | 0% | - | |
| Lab Statistics | n | Median (Min–Max) | Valid Percent | |
|---|---|---|---|---|
| Nasal NO (ppm) | 43 | 9 (5–16) | ||
| High-speed video microscopy | minimal residual | 22 | 39.6 | |
| hyperkinetic | 1 | 2.1 | ||
| immotile | 4 | 8.3 | ||
| almost immotile | 10 | 22.9 | ||
| reduced | 6 | 12.5 | ||
| stiff | 7 | 14.6 | ||
| Immunofluorescence labeling | Normal | 17 | 9.2 | |
| DNAH5 abnormal | 22 * | 11.8 | ||
| GAS8 abnormal | 8 | 4.3 | ||
| DNAH11 abnormal | 5 | 2.7 | ||
| RSPH9 abnormal | 2 | 1 | ||
| ARMC4 abnormal | 1 * | 0.5 | ||
| Genetic PCD (+) | Genetic PCD (−) | Total | |
|---|---|---|---|
| IF-Positive (TP) | 8 | 17 (FP) | 25 |
| IF-Negative (FN) | 0 | 29 (TN) | 29 |
| Total | 8 | 46 | 54 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karakoç, E.; Hjeij, R.; Kaya, Z.B.; Emiralioğlu, N.; Ademhan Tural, D.; Atilla, P.; Özçelik, U.; Omran, H. Diagnostic Role of Immunofluorescence Analysis in Primary Ciliary Dyskinesia-Suspected Individuals. J. Clin. Med. 2025, 14, 1941. https://doi.org/10.3390/jcm14061941
Karakoç E, Hjeij R, Kaya ZB, Emiralioğlu N, Ademhan Tural D, Atilla P, Özçelik U, Omran H. Diagnostic Role of Immunofluorescence Analysis in Primary Ciliary Dyskinesia-Suspected Individuals. Journal of Clinical Medicine. 2025; 14(6):1941. https://doi.org/10.3390/jcm14061941
Chicago/Turabian StyleKarakoç, Elif, Rim Hjeij, Zeynep Bengisu Kaya, Nagehan Emiralioğlu, Dilber Ademhan Tural, Pergin Atilla, Uğur Özçelik, and Heymut Omran. 2025. "Diagnostic Role of Immunofluorescence Analysis in Primary Ciliary Dyskinesia-Suspected Individuals" Journal of Clinical Medicine 14, no. 6: 1941. https://doi.org/10.3390/jcm14061941
APA StyleKarakoç, E., Hjeij, R., Kaya, Z. B., Emiralioğlu, N., Ademhan Tural, D., Atilla, P., Özçelik, U., & Omran, H. (2025). Diagnostic Role of Immunofluorescence Analysis in Primary Ciliary Dyskinesia-Suspected Individuals. Journal of Clinical Medicine, 14(6), 1941. https://doi.org/10.3390/jcm14061941