
Hematologic and Immunologic Overlap Between COVID-19 and Idiopathic Pulmonary Fibrosis


Abstract
1. Introduction
2. Materials and Methods
3. Hematologic Alterations in COVID-19
3.1. Lymphopenia, Neutrophilia, and Neutrophil-to-Lymphocyte Ratio (NLR)
3.2. Platelet Count and Function
3.3. Coagulation Abnormalities and Hypercoagulability
3.4. Cytokine Storm and Inflammatory Mediators
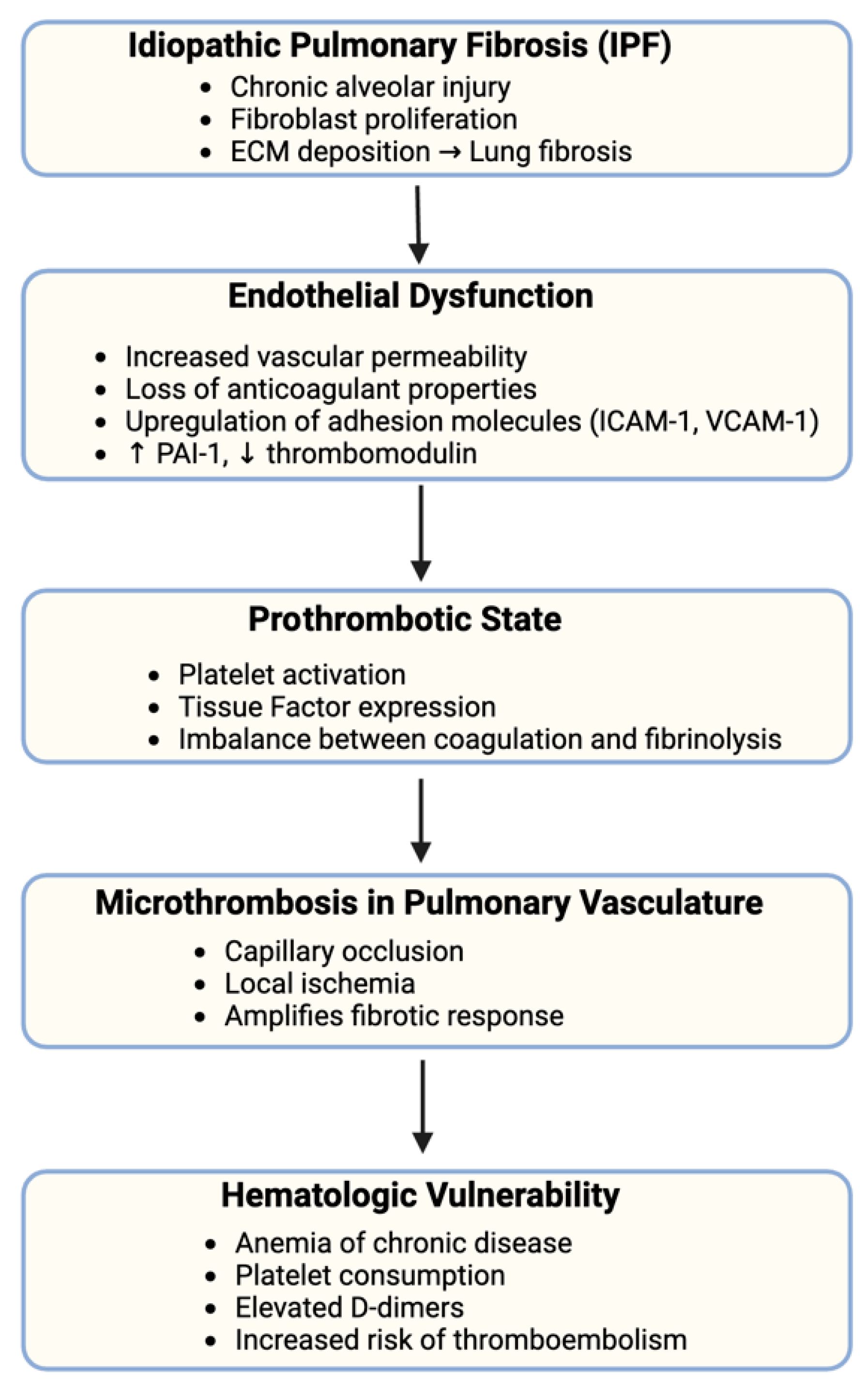
4. IPF and Hematologic Vulnerability
4.1. Endothelial Dysfunction and Microthrombosis in IPF
4.2. Chronic Inflammation and Immune Activation in IPF
4.3. Myeloid Cell Activity and Coagulation Pathways in IPF
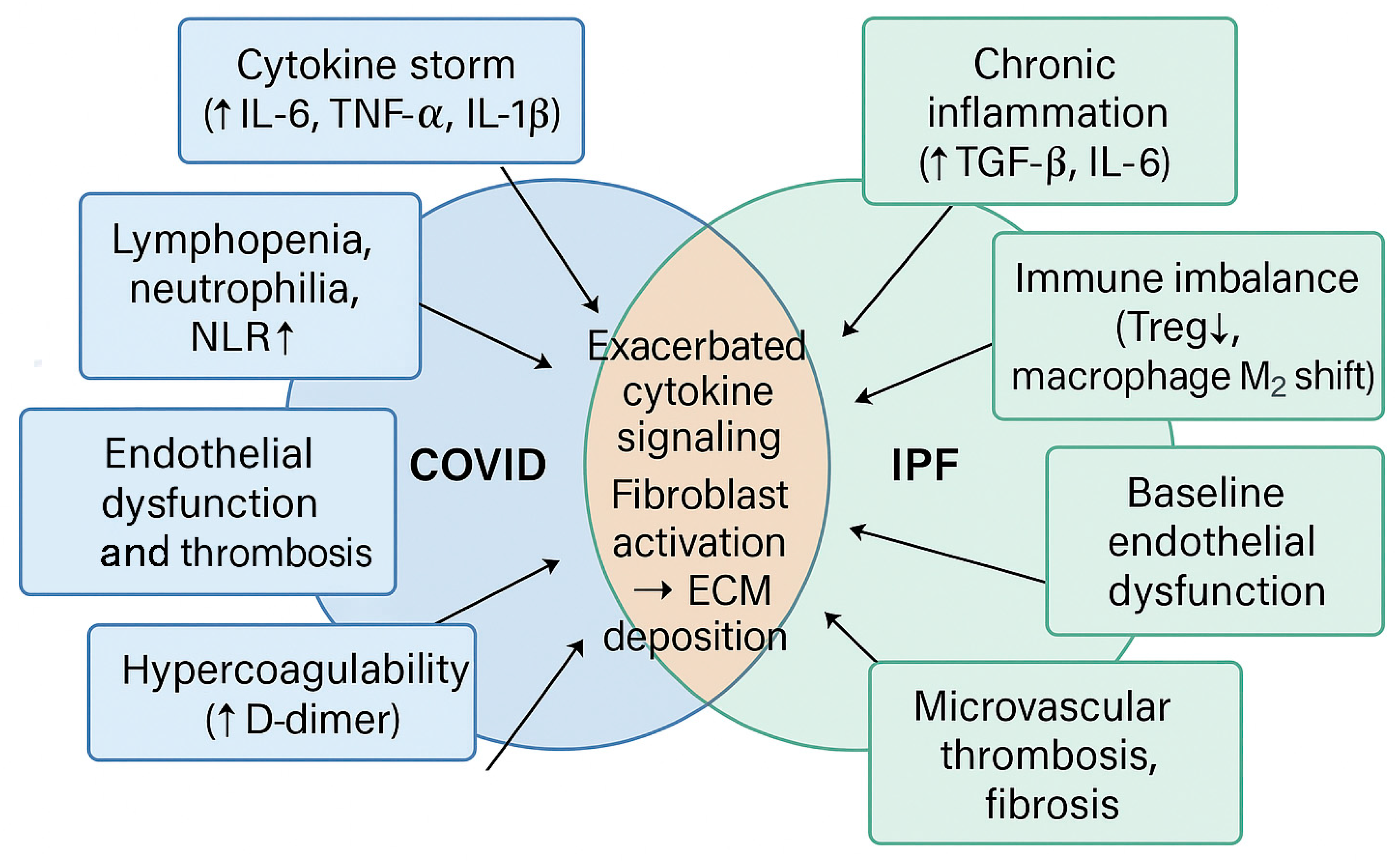
5. Intersection Between COVID-19 and IPF: Shared Hematologic Mechanisms and Pathological Synergies
5.1. Worsening of IPF Through COVID-19-Induced Pulmonary Thrombosis
5.2. Systemic Inflammation and Its Impact on Fibrosis Progression
5.3. Immune Imbalance and Exaggerated Responses to Viral Infection
5.4. Cytokine Expression: TGF-β, IL-6, and TNF-α
5.5. Impact of COVID-19 Vaccination on Hematologic and Fibrotic Pathways
6. Clinical Implications
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Lai, C.-C.; Shih, T.-P.; Ko, W.-C.; Tang, H.-J.; Hsueh, P.-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef] [PubMed]
- Zaim, S.; Chong, J.H.; Sankaranarayanan, V.; Harky, A. COVID-19 and Multiorgan Response. Curr. Probl. Cardiol. 2020, 45, 100618. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.G.; Qin, L.; Puah, S.H. COVID-19 acute respiratory distress syndrome (ARDS): Clinical features and differences from typical pre-COVID-19 ARDS. Med. J. Aust. 2020, 213, 54–56.e1. [Google Scholar] [CrossRef] [PubMed]
- van Eijk, L.E.; Binkhorst, M.; Bourgonje, A.R.; Offringa, A.K.; Mulder, D.J.; Bos, E.M.; Kolundzic, N.; Abdulle, A.E.; van der Voort, P.H.; Olde Rikkert, M.G.; et al. COVID-19: Immunopathology, pathophysiological mechanisms, and treatment options. J. Pathol. 2021, 254, 307–331. [Google Scholar] [CrossRef] [PubMed]
- El-Kassas, M.; Alboraie, M.; Elbadry, M.; El Sheemy, R.; Abdellah, M.; Afify, S.; Madkour, A.; Zaghloul, M.; Awad, A.; Wifi, M.-N.; et al. Non-pulmonary involvement in COVID-19: A systemic disease rather than a pure respiratory infection. World J. Clin. Cases 2023, 11, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, D.; Bongo, S.; Copponi, A.; Rossi, V.; Di Giorgio, R.; Bernardini, S.; Ippoliti, L.; Morello, M. A Review of the Hematological Picture of Severe COVID-19 Infection. Cureus 2025, 17, e78797. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, F.; Solidoro, P.; Gavelli, F.; Apostolo, D.; Bellan, M. Idiopathic Pulmonary Fibrosis and Post-COVID-19 Lung Fibrosis: Links and Risks. Microorganisms 2023, 11, 895. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.-L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef] [PubMed]
- Banu, N.; Panikar, S.S.; Leal, L.R.; Leal, A.R. Protective role of ACE2 and its downregulation in SARS-CoV-2 infection leading to Macrophage Activation Syndrome: Therapeutic implications. Life Sci. 2020, 256, 117905. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed]
- Koudstaal, T.; Wijsenbeek, M.S. Idiopathic pulmonary fibrosis. Presse Médicale 2023, 52, 104166. [Google Scholar] [CrossRef] [PubMed]
- Hyldgaard, C.; Møller, J.; Bendstrup, E. Changes in management of idiopathic pulmonary fibrosis: Impact on disease severity and mortality. Eur. Clin. Respir. J. 2020, 7, 1807682. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, S.F.; Lakhani, D.A.; Sohail, A.H.; Maurer, J.; Sofka, S.; Sarwari, A.; Hadi, Y.B. Patients with idiopathic pulmonary fibrosis have poor clinical outcomes with COVID-19 disease: A propensity matched multicentre research network analysis. BMJ Open Resp. Res. 2021, 8, e000969. [Google Scholar] [CrossRef] [PubMed]
- Suri, C.; Pande, B.; Sahithi, L.S.; Sahu, T.; Verma, H.K. Interplay between Lung Diseases and Viral Infections: A Comprehensive Review. Microorganisms 2024, 12, 2030. [Google Scholar] [CrossRef] [PubMed]
- Patange, A.P.; Desai, J.V.; Pujari, B.; Marwah, A.; Dey, A. Dynamic Assessment of Hematological Parameters as Predictive Biomarkers for Disease Severity and Prognosis in COVID-19 Patients: A Longitudinal Study. Cureus 2024, 16, e63593. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.I.; Nordwall, J.A.; Shaw-Saliba, K.; Aberg, J.A.; Gardner, E.M.; Goodman, A.L.; Kumarasamy, N.; Vasudeva, S.; Vock, D.M.; North, C.M.; et al. Blood absolute lymphocyte count and trajectory are important in understanding severe COVID-19. BMC Infect. Dis. 2025, 25, 67. [Google Scholar] [CrossRef] [PubMed]
- Kalfaoglu, B.; Almeida-Santos, J.; Tye, C.A.; Satou, Y.; Ono, M. T-cell dysregulation in COVID-19. Biochem. Biophys. Res. Commun. 2021, 538, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Elahi, S. Hematopoietic responses to SARS-CoV-2 infection. Cell Mol. Life Sci. 2022, 79, 187. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Liu, Y.; Chen, B.; Yang, H.; Hu, H.; Liu, Y.; Zhao, Y. Prognostic value of lymphocyte count in severe COVID-19 patients with corticosteroid treatment. Signal Transduct. Target. Ther. 2021, 6, 106. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.; Ricci, E.; Migliorati, G.; Gentili, M.; Riccardi, C. How Glucocorticoids Affect the Neutrophil Life. Int. J. Mol. Sci. 2018, 19, 4090. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.J.A.; Ribeiro, L.R.; Lima, K.V.B.; Lima, L.N.G.C. Adaptive immunity to SARS-CoV-2 infection: A systematic review. Front. Immunol. 2022, 13, 1001198. [Google Scholar] [CrossRef] [PubMed]
- Rha, M.-S.; Shin, E.-C. Activation or exhaustion of CD8+ T cells in patients with COVID-19. Cell Mol. Immunol. 2021, 18, 2325–2333. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Schulte, S.; Wildner, N.H.; Wittner, M.; Brehm, T.T.; Ramharter, M.; Woost, R.; Lohse, A.W.; Jacobs, T.; Schulze Zur Wiesch, J. Analysis of Co-inhibitory Receptor Expression in COVID-19 Infection Compared to Acute Plasmodium falciparum Malaria: LAG-3 and TIM-3 Correlate with T Cell Activation and Course of Disease. Front. Immunol. 2020, 11, 1870. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.-H.; Hsu, W.-L.; Peng, G.-R.; Yu, W.-C.; Lin, W.-C.; Hu, S.; Yu, S.-H. Role of the Immune Microenvironment in SARS-CoV-2 Infection. Cell Transpl. 2021, 30, 09636897211010632. [Google Scholar] [CrossRef] [PubMed]
- Notarbartolo, S. T-Cell Immune Responses to SARS-CoV-2 Infection and Vaccination. Vaccines 2024, 12, 1126. [Google Scholar] [CrossRef] [PubMed]
- Davitt, E.; Davitt, C.; Mazer, M.B.; Areti, S.S.; Hotchkiss, R.S.; Remy, K.E. COVID-19 disease and immune dysregulation. Best Pr. Res. Clin. Haematol. 2022, 35, 101401. [Google Scholar] [CrossRef] [PubMed]
- Puhach, O.; Meyer, B.; Eckerle, I. SARS-CoV-2 viral load and shedding kinetics. Nat. Rev. Microbiol. 2022, 21, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Lord, J.M. Neutrophils and COVID-19: Active Participants and Rational Therapeutic Targets. Front. Immunol. 2021, 12, 680134. [Google Scholar] [CrossRef] [PubMed]
- Rong, N.; Wei, X.; Liu, J. The Role of Neutrophil in COVID-19: Positive or Negative. J. Innate Immun. 2024, 16, 80–95. [Google Scholar] [CrossRef] [PubMed]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Q.; Yin, Y.; Zhang, Y.; Cao, Y.; Lin, X.; Huang, L.; Hoffmann, D.; Lu, M.; Qiu, Y. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front. Immunol. 2020, 11, 2063. [Google Scholar] [CrossRef] [PubMed]
- El Azhary, K.; Ghazi, B.; Kouhen, F.; El Bakkouri, J.; Chamlal, H.; El Ghanmi, A.; Badou, A. Clinical Impact of Neutrophil Variation on COVID-19 Complications. Diagnostics 2025, 15, 457. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps (NETs) as markers of disease severity in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Toori, K.U.; Qureshi, M.A.; Chaudhry, A.; Safdar, M.F. Neutrophil to lymphocyte ratio (NLR) in COVID-19: A cheap prognostic marker in a resource constraint setting. Pak. J. Med. Sci. 2021, 37, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Basbus, L.; Lapidus, M.I.; Martingano, I.; Puga, M.C.; Pollán, J. Neutrophil to lymphocyte ratio as a prognostic marker in COVID-19. Medicina 2020, 80 (Suppl. 3), 31–36. [Google Scholar] [PubMed]
- Ulloque-Badaracco, J.R.; Ivan Salas-Tello, W.; Al-kassab-Córdova, A.; Alarcón-Braga, E.A.; Benites-Zapata, V.A.; Maguiña, J.L.; Hernandez, A.V. Prognostic value of neutrophil-to-lymphocyte ratio in COVID-19 patients: A systematic review and meta-analysis. Int. J. Clin. Pr. 2021, 75, e14596. [Google Scholar] [CrossRef] [PubMed]
- Asperges, E.; Albi, G.; Zuccaro, V.; Sambo, M.; Pieri, T.C.; Calia, M.; Colaneri, M.; Maiocchi, L.; Melazzini, F.; Lasagna, A.; et al. Dynamic NLR and PLR in Predicting COVID-19 Severity: A Retrospective Cohort Study. Infect. Dis. Ther. 2023, 12, 1625–1640. [Google Scholar] [CrossRef] [PubMed]
- Todor, S.-B.; Bîrluțiu, V.; Topîrcean, D.; Mihăilă, R.-G. Role of biological markers and CT severity score in predicting mortality in patients with COVID-19: An observational retrospective study. Exp. Ther. Med. 2022, 24, 698. [Google Scholar] [CrossRef] [PubMed]
- Wool, G.D.; Miller, J.L. The Impact of COVID-19 Disease on Platelets and Coagulation. Pathobiology 2021, 88, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Q.; Wang, Y.; Wu, Y.; Xu, J.; Yu, Y.; Shang, Y. Thrombocytopenia and its association with mortality in patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhou, Q.; Xu, J. Mechanism of thrombocytopenia in COVID-19 patients. Ann. Hematol. 2020, 99, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, M.G.; Alanazi, N.; Yousef, A.; Alanazi, N.; Alotaibi, B.; Aljurf, M.; El Fakih, R. COVID-19 associated with immune thrombocytopenia: A systematic review and meta-analysis. Expert. Rev. Hematol. 2022, 15, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, N.; Colucci, M. The Prothrombotic State Associated with SARS-CoV-2 Infection: Pathophysiological Aspects. Mediterr. J. Hematol. Infect. Dis. 2021, 13, e2021045. [Google Scholar] [CrossRef] [PubMed]
- Sciaudone, A.; Corkrey, H.; Humphries, F.; Koupenova, M. Platelets and SARS-CoV-2 During COVID-19: Immunity, Thrombosis, and Beyond. Circ. Res. 2023, 132, 1272–1289. [Google Scholar] [CrossRef] [PubMed]
- Obeagu, E.I.; Obeagu, G.U.; Aja, P.M.; Okoroiwu, G.I.A.; Ubosi, N.I.; Pius, T.; Ashiru, M.; Akaba, K.; Adias, T.C. Soluble platelet selectin and platelets in COVID-19: A multifaceted connection. Ann. Med. Surg. 2024, 86, 4634–4642. [Google Scholar] [CrossRef] [PubMed]
- Agrati, C.; Sacchi, A.; Tartaglia, E.; Vergori, A.; Gagliardini, R.; Scarabello, A.; Bibas, M. The Role of P-Selectin in COVID-19 Coagulopathy: An Updated Review. Int. J. Mol. Sci. 2021, 22, 7942. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Pushpakumar, S.; Zheng, Y.; Smolenkova, I.; Akinterinwa, O.E.; Luulay, B.; Tyagi, S.C. Novel mechanism of the COVID-19 associated coagulopathy (CAC) and vascular thromboembolism. NPJ Viruses 2023, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Abou-Ismail, M.Y.; Diamond, A.; Kapoor, S.; Arafah, Y.; Nayak, L. The hypercoagulable state in COVID-19: Incidence, pathophysiology, and management. Thromb. Res. 2020, 194, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; Van De Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Nemec, H.M.; Ferenczy, A.; Christie, B.D.; Ashley, D.W.; Montgomery, A. Correlation of D-dimer and Outcomes in COVID-19 Patients. Am. Surg. 2022, 88, 2115–2118. [Google Scholar] [CrossRef] [PubMed]
- Beidollahkhani, S.; Fayedeh, F.; Shoja, A.; Hassan Nejad, E.; Hoseinpour, M.; Fazlpour, F.; Payandeh, A.; Pezeshki Rad, M.; Moodi Ghalibaf, A. d-dimer as a biomarker for COVID-19-associated pulmonary thromboembolism: A narrative review from molecular pathways to the imaging findings. Egypt. J. Bronchol. 2023, 17, 44. [Google Scholar] [CrossRef]
- Wang, L.; He, W.-B.; Yu, X.-M.; Hu, D.-L.; Jiang, H. Prolonged prothrombin time at admission predicts poor clinical outcome in COVID-19 patients. World J. Clin. Cases 2020, 8, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Yan, H.; Chen, H.; He, C.; Lin, C.; He, H.; Zhang, S.; Shi, S.; Lin, K. COVID-19 and coagulation dysfunction in adults: A systematic review and meta-analysis. J. Med. Virol. 2021, 93, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Yan, Z.; Montano, M.; Sozmen, E.G.; Dixit, K.; Suryawanshi, R.K.; Matsui, Y.; Helmy, E.; Kaushal, P.; Makanani, S.K.; et al. Fibrin drives thromboinflammation and neuropathology in COVID-19. Nature 2024, 633, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Zheng, X.; Zhao, S.; Liu, H.; Chen, E.; Xie, R.; Li, R.; Chen, Y. Disseminated intravascular coagulation: Cause, molecular mechanism, diagnosis, and therapy. MedComm 2025, 6, e70058. [Google Scholar] [CrossRef] [PubMed]
- Hunt, B.J.; Levi, M. Re The source of elevated plasma D-dimer levels in COVID-19 infection. Br. J. Haematol. 2020, 190, E133–E134. [Google Scholar] [CrossRef] [PubMed]
- Hanff, T.C.; Mohareb, A.M.; Giri, J.; Cohen, J.B.; Chirinos, J.A. Thrombosis in COVID-19. Am. J. Hematol. 2020, 95, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, D.; Sperhake, J.-P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients with COVID-19: A Prospective Cohort Study. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Pezzuto, F.; Fortarezza, F.; Hofman, P.; Kern, I.; Panizo, A.; Von Der Thüsen, J.; Timofeev, S.; Gorkiewicz, G.; Lunardi, F. Pulmonary pathology and COVID-19: Lessons from autopsy. The experience of European Pulmonary Pathologists. Virchows Arch. 2020, 477, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Jayarangaiah, A.; Kariyanna, P.T.; Chen, X.; Jayarangaiah, A.; Kumar, A. COVID-19-Associated Coagulopathy: An Exacerbated Immunothrombosis Response. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620943293. [Google Scholar] [CrossRef] [PubMed]
- Montazersaheb, S.; Hosseiniyan Khatibi, S.M.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Ghasemian Sorbeni, F.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Dharra, R.; Kumar Sharma, A.; Datta, S. Emerging aspects of cytokine storm in COVID-19: The role of proinflammatory cytokines and therapeutic prospects. Cytokine 2023, 169, 156287. [Google Scholar] [CrossRef] [PubMed]
- Nazerian, Y.; Ghasemi, M.; Yassaghi, Y.; Nazerian, A.; Hashemi, S.M. Role of SARS-CoV-2-induced cytokine storm in multi-organ failure: Molecular pathways and potential therapeutic options. Int. Immunopharmacol. 2022, 113 Pt B, 109428. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.C.; Hoiland, R.L.; Stukas, S.; Wellington, C.L.; Sekhon, M.S. Confronting the controversy: Interleukin-6 and the COVID-19 cytokine storm syndrome. Eur. Respir. J. 2020, 56, 2003006. [Google Scholar] [CrossRef] [PubMed]
- Paranga, T.G.; Mitu, I.; Pavel-Tanasa, M.; Rosu, M.F.; Miftode, I.-L.; Constantinescu, D.; Obreja, M.; Plesca, C.E.; Miftode, E. Cytokine Storm in COVID-19: Exploring IL-6 Signaling and Cytokine-Microbiome Interactions as Emerging Therapeutic Approaches. Int. J. Mol. Sci. 2024, 25, 11411. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Willey, J. The interplay between inflammation and thrombosis in COVID-19: Mechanisms, therapeutic strategies, and challenges. Thromb. Update 2022, 8, 100117. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Filho, S.P.D.M.; De Souza Pinheiro, R.; Martins, F.S.; Giroldi, P.J.; E Melo, R.H.; De Oliveira, E.L.; Dos Santos, A.B.; Medeiros, D.C.O.; Lopes, J.A.; Chaves, Y.O.; et al. Kinetics of IL-6, C-reactive Protein and Fibrinogen Levels in COVID-19 Outpatients Who Evolved to Hypoxemia. Clin. Pathol. 2024, 17, 2632010X231222795. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2021, 17, 46–64. [Google Scholar] [CrossRef] [PubMed]
- Escher, R.; Breakey, N.; Lämmle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef] [PubMed]
- Zeylabi, F.; Nameh Goshay Fard, N.; Parsi, A.; Pezeshki, S.M.S. Bone marrow alterations in COVID-19 infection: The root of hematological problems. Curr. Res. Transl. Med. 2023, 71, 103407. [Google Scholar] [CrossRef] [PubMed]
- Shouman, S.; El-Kholy, N.; Hussien, A.E.; El-Derby, A.M.; Magdy, S.; Abou-Shanab, A.M.; Elmehrath, A.O.; Abdelwaly, A.; Helal, M.; El-Badri, N. SARS-CoV-2-associated lymphopenia: Possible mechanisms and the role of CD147. Cell Commun. Signal 2024, 22, 349. [Google Scholar] [CrossRef] [PubMed]
- Reusch, N.; De Domenico, E.; Bonaguro, L.; Schulte-Schrepping, J.; Baßler, K.; Schultze, J.L.; Aschenbrenner, A.C. Neutrophils in COVID-19. Front. Immunol. 2021, 12, 652470. [Google Scholar] [CrossRef] [PubMed]
- Landau, N.; Shoenfeld, Y.; Negru, L.; Segal, G. Exploring the pathways of inflammation and coagulopathy in COVID-19: A narrative tour into a viral rabbit hole. Int. Rev. Immunol. 2022, 41, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Hakim, N.N.; Chi, J.; Olazagasti, C.; Liu, J.M. Secondary hemophagocytic lymphohistiocytosis versus cytokine release syndrome in severe COVID-19 patients. Exp. Biol. Med. 2021, 246, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Opoka-Winiarska, V.; Grywalska, E.; Roliński, J. Could hemophagocytic lymphohistiocytosis be the core issue of severe COVID-19 cases? BMC Med. 2020, 18, 214. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, R.; Seino, K. Macrophage activation syndrome and COVID-19. Inflamm. Regen. 2020, 40, 19. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Yu, J.; Gao, W.; Guo, W.; Zhu, J.; Wang, T. Hemophagocytosis, hyper-inflammatory responses, and multiple organ damages in COVID-19-associated hyperferritinemia. Ann. Hematol. 2022, 101, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Boonpheng, B.; Ungprasert, P. Risk of venous thromboembolism in patients with idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Sarcoidosis Vasc. Diffus. Lung Dis. 2018, 35, 109–114. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, H.H.; Park, H.J.; Kim, S.; Kim, Y.-J.; Lee, J.S.; Kim, H.C. Venous thromboembolism in patients with idiopathic pulmonary fibrosis, based on nationwide claim data. Ther. Adv. Respir. Dis. 2023, 17, 17534666231155772. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.M.; Waldman, W.J.; Knight, D.A.; Allen, J.N.; Nadasdy, T.; Frambach, G.E.; Ross, P.; Marsh, C.B. Idiopathic Pulmonary Fibrosis Related to Endothelial Injury and Antiendothelial Cell Antibodies. Hum. Immunol. 2006, 67, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Crooks, M.G.; Hart, S.P. Coagulation and anticoagulation in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2015, 24, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed]
- May, J.; Mitchell, J.A.; Jenkins, R.G. Beyond epithelial damage: Vascular and endothelial contributions to idiopathic pulmonary fibrosis. J. Clin. Investig. 2023, 133, e172058. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, F.S.; Lanzetti, M.; Nesi, R.T.; Nagato, A.C.; Silva, C.P.E.; Kennedy-Feitosa, E.; Melo, A.C.; Cattani-Cavalieri, I.; Porto, L.C.; Valenca, S.S. Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries. Antioxidants 2023, 12, 548. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, K.; Hao, D.; Li, X.; Zhu, Y.; Yu, H.; Chen, H. Pulmonary fibrosis: Pathogenesis and therapeutic strategies. MedComm 2024, 5, e744. [Google Scholar] [CrossRef] [PubMed]
- Borek, I.; Birnhuber, A.; Voelkel, N.F.; Marsh, L.M.; Kwapiszewska, G. The vascular perspective on acute and chronic lung disease. J. Clin. Investig. 2023, 133, e170502. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, C.; Burtey, S.; Sabatier, F.; Cauchois, R.; Lano, G.; Abdili, E.; Daviet, F.; Arnaud, L.; Brunet, P.; Hraiech, S.; et al. Circulating Endothelial Cells as a Marker of Endothelial Injury in Severe COVID-19. J. Infect. Dis. 2020, 222, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Hasegawa, Y.; Tsuchiya, Y.; Shimokata, K. Expression of Cell Adhesion Molecules in the Lungs of Patients with Idiopathic Pulmonary Fibrosis. Chest 1995, 108, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Agassandian, M.; Tedrow, J.R.; Sembrat, J.; Kass, D.J.; Zhang, Y.; Goncharova, E.A.; Kaminski, N.; Mallampalli, R.K.; Vuga, L.J. VCAM-1 is a TGF-β1 inducible gene upregulated in idiopathic pulmonary fibrosis. Cell Signal 2015, 27, 2467–2473. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Hattori, N.; Senoo, T.; Takayama, Y.; Masuda, T.; Nakashima, T.; Iwamoto, H.; Fujitaka, K.; Hamada, H.; Kohno, N. Inhibition of Plasminogen Activator Inhibitor-1 Attenuates Transforming Growth Factor-β-Dependent Epithelial Mesenchymal Transition and Differentiation of Fibroblasts to Myofibroblasts. PLoS ONE 2016, 11, e0148969. [Google Scholar] [CrossRef] [PubMed]
- Frischmuth, T.; Hindberg, K.; Aukrust, P.; Ueland, T.; Brækkan, S.K.; Hansen, J.; Morelli, V.M. Elevated plasma levels of plasminogen activator inhibitor-1 are associated with risk of future incident venous thromboembolism. J. Thromb. Haemost. 2022, 20, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Bringardner, B.D.; Baran, C.P.; Eubank, T.D.; Marsh, C.B. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid. Redox Signal 2008, 10, 287–301. [Google Scholar] [CrossRef] [PubMed]
- She, Y.X.; Yu, Q.Y.; Tang, X.X. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discov. 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, A.; Soler, P.; Kambouchner, M.; Loiseau, P.; Milleron, B.; Valeyre, D.; Hance, A.J.; Tazi, A. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-β and IL-10. Eur. Respir. J. 2003, 22, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Epstein Shochet, G.; Brook, E.; Bardenstein-Wald, B.; Shitrit, D. TGF-β pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir. Res. 2020, 21, 56. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, G.; Liu, A.; Herzog, E.L. Evolving Perspectives on Innate Immune Mechanisms of IPF. Front. Mol. Biosci. 2021, 8, 676569. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Chen, Y.; Ma, L.; Hu, F.; Xie, L. Macrophage polarization and its impact on idiopathic pulmonary fibrosis. Front. Immunol. 2024, 15, 1444964. [Google Scholar] [CrossRef] [PubMed]
- Cilli, A.; Hanta, I.; Uzer, F.; Coskun, F.; Sevinc, C.; Deniz, P.P.; Parlak, M.; Altunok, E.; Tertemiz, K.C.; Ursavas, A. Characteristics and outcomes of COVID-19 patients with IPF: A multi-center retrospective study. Respir. Med. Res. 2022, 81, 100900. [Google Scholar] [CrossRef] [PubMed]
- van Geffen, C.; Deißler, A.; Quante, M.; Renz, H.; Hartl, D.; Kolahian, S. Regulatory Immune Cells in Idiopathic Pulmonary Fibrosis: Friends or Foes? Front. Immunol. 2021, 12, 663203. [Google Scholar] [CrossRef] [PubMed]
- Florez-Sampedro, L.; Song, S.; Melgert, B.N. The diversity of myeloid immune cells shaping wound repair and fibrosis in the lung. Regeneration 2018, 5, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Liu, Q.; Li, H.; Zhang, M.; You, L.; Lin, Y.; Wang, K.; Gou, Q.; Wang, Z.; Zhou, S.; et al. The role of monocytes in thrombotic diseases: A review. Front. Cardiovasc. Med. 2023, 10, 1113827. [Google Scholar] [CrossRef] [PubMed]
- Rehill, A.M.; Leon, G.; McCluskey, S.; Schoen, I.; Hernandez-Santana, Y.; Annett, S.; Klavina, P.; Robson, T.; Curtis, A.M.; Renné, T.; et al. Glycolytic reprogramming fuels myeloid cell-driven hypercoagulability. J. Thromb. Haemost. 2024, 22, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Pokhreal, D.; Crestani, B.; Helou, D.G. Macrophage Implication in IPF: Updates on Immune, Epigenetic, and Metabolic Pathways. Cells 2023, 12, 2193. [Google Scholar] [CrossRef] [PubMed]
- van der Poll, T. Tissue factor as an initiator of coagulation and inflammation in the lung. Crit. Care 2008, 12 (Suppl. 6), S3. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, E.; Scaf, B.; Munts, C.; van Hunnik, A.; Trevelyan, C.J.; Verheule, S.; Spronk, H.M.H.; Turner, N.A.; Ten Cate, H.; Schotten, U.; et al. Coagulation Factor Xa Induces Proinflammatory Responses in Cardiac Fibroblasts via Activation of Protease-Activated Receptor-1. Cells 2021, 10, 2958. [Google Scholar] [CrossRef] [PubMed]
- Posma, J.J.; Grover, S.P.; Hisada, Y.; Owens, A.P.; Antoniak, S.; Spronk, H.M.; Mackman, N. Roles of Coagulation Proteases and PARs (Protease-Activated Receptors) in Mouse Models of Inflammatory Diseases. Arter. Thromb. Vasc. Biol. 2019, 39, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Schuliga, M.; Grainge, C.; Westall, G.; Knight, D. The fibrogenic actions of the coagulant and plasminogen activation systems in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2018, 97, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Mercer, P.F.; Chambers, R.C. Coagulation and coagulation signalling in fibrosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Loskutoff, D.J.; Quigley, J.P. PAI-1, fibrosis, and the elusive provisional fibrin matrix. J. Clin. Investig. 2000, 106, 1441–1443. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhang, N.; Liu, Y.; Yang, X.; He, Y.; Li, Q.; Shen, X.; Zhu, Y.; Yang, Y. The Interaction Between Pulmonary Fibrosis and COVID-19 and the Application of Related Anti-Fibrotic Drugs. Front. Pharmacol. 2021, 12, 805535. [Google Scholar] [CrossRef] [PubMed]
- Kangro, K.; Wolberg, A.S.; Flick, M.J. Fibrinogen, Fibrin, and Fibrin Degradation Products in COVID-19. Curr. Drug Targets 2022, 23, 1593–1602. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Pan, J.Y. Anatomical and Pathological Observation and Analysis of SARS and COVID-19: Microthrombosis Is the Main Cause of Death. Biol. Proced. Online 2021, 23, 4. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, L.P.M.H.; Becker, L.M.; Teuwen, L.-A.; Boeckx, B.; Jansen, S.; Feys, S.; Verleden, S.; Liesenborghs, L.; Stalder, A.K.; Libbrecht, S.; et al. The pulmonary vasculature in lethal COVID-19 and idiopathic pulmonary fibrosis at single-cell resolution. Cardiovasc. Res. 2023, 119, 520–535. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, D.; Ernst, A.; Kalluri, R. Idiopathic Pulmonary Fibrosis Is Associated with Endothelial to Mesenchymal Transition. Am. J. Respir. Cell Mol. Biol. 2010, 43, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Falleni, M.; Tosi, D.; Savi, F.; Chiumello, D.; Bulfamante, G. Endothelial-Mesenchymal Transition in COVID-19 lung lesions. Pathol. Res. Pr. 2021, 221, 153419. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Zanza, C.; Romenskaya, T.; Manetti, A.C.; Franceschi, F.; La Russa, R.; Bertozzi, G.; Maiese, A.; Savioli, G.; Volonnino, G.; Longhitano, Y. Cytokine Storm in COVID-19: Immunopathogenesis and Therapy. Medicina 2022, 58, 144. [Google Scholar] [CrossRef] [PubMed]
- Oatis, D.; Herman, H.; Balta, C.; Ciceu, A.; Simon-Repolski, E.; Mihu, A.G.; Lepre, C.C.; Russo, M.; Trotta, M.C.; Gravina, A.G.; et al. Dynamic shifts in lung cytokine patterns in post-COVID-19 interstitial lung disease patients: A pilot study. Ther. Adv. Chronic Dis. 2024, 15, 20406223241236257. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Pan, H.; Li, R.; He, K.; Zhang, H.; Liu, L. Increased Circulating Cytokines Have a Role in COVID-19 Severity and Death with a More Pronounced Effect in Males: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2022, 13, 802228. [Google Scholar] [CrossRef] [PubMed]
- Kottmann, R.M.; Hogan, C.M.; Phipps, R.P.; Sime, P.J. Determinants of initiation and progression of idiopathic pulmonary fibrosis. Respirology 2009, 14, 917–933. [Google Scholar] [CrossRef] [PubMed]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Ruta, V.M.; Man, A.M.; Alexescu, T.G.; Motoc, N.S.; Tarmure, S.; Ungur, R.A.; Todea, D.A.; Coste, S.C.; Valean, D.; Pop, M.C. Neutrophil-To-Lymphocyte Ratio and Systemic Immune-Inflammation Index-Biomarkers in Interstitial Lung Disease. Medicina 2020, 56, 381. [Google Scholar] [CrossRef] [PubMed]
- Buonacera, A.; Stancanelli, B.; Colaci, M.; Malatino, L. Neutrophil to Lymphocyte Ratio: An Emerging Marker of the Relationships between the Immune System and Diseases. Int. J. Mol. Sci. 2022, 23, 3636. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Chen, Q.; Xu, M.; Huang, J.; Ye, H. Communication between alveolar macrophages and fibroblasts via the TNFSF12-TNFRSF12A pathway promotes pulmonary fibrosis in severe COVID-19 patients. J. Transl. Med. 2024, 22, 698. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.I.; Puritz, C.H.; Senkow, K.J.; Markov, N.S.; Diaz, E.; Jonasson, E.; Yu, Z.; Swaminathan, S.; Lu, Z.; Fenske, S.; et al. Profibrotic monocyte-derived alveolar macrophages are expanded in patients with persistent respiratory symptoms and radiographic abnormalities after COVID-19. Nat. Immunol. 2024, 25, 2097–2109. [Google Scholar] [CrossRef] [PubMed]
- Oatis, D.; Balta, C.; Herman, H.; Ciceu, A.; Simon-Repolski, E.; Mihu, A.G.; Lepre, C.C.; Russo, M.; Trotta, M.C.; D’Amico, G.; et al. The interplay between lung galectins and pro-fibrotic markers in post-COVID-19 fibrogenesis: A pilot study. Life Sci. 2025, 361, 123326. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. The Impact of TGF-β on Lung Fibrosis: From Targeting to Biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, E.; Casitas, R.; Díaz-García, E.; García-Tovar, S.; Galera, R.; Torres-Vargas, M.; Fernández-Velilla, M.; López-Fernández, C.; Añón, J.M.; Quintana-Díaz, M.; et al. TGF-β1 overexpression in severe COVID-19 survivors and its implications for early-phase fibrotic abnormalities and long-term functional impairment. Front. Immunol. 2024, 15, 1401015. [Google Scholar] [CrossRef] [PubMed]
- Arguinchona, L.M.; Zagona-Prizio, C.; Joyce, M.E.; Chan, E.D.; Maloney, J.P. Microvascular significance of TGF-β axis activation in COVID-19. Front. Cardiovasc. Med. 2022, 9, 1054690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z.; Wei, R.; Miao, X.; Sun, S.; Liang, G.; Chu, C.; Zhao, L.; Zhu, X.; Guo, Q.; et al. IL (Interleukin)-6 Contributes to Deep Vein Thrombosis and Is Negatively Regulated by miR-338-5p. Arter. Thromb. Vasc. Biol. 2020, 40, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tang, G.; Liu, Y.; Zhang, L.; Chen, B.; Han, Y.; Fu, Z.; Wang, L.; Hu, G.; Ma, Q.; et al. The role of IL-6 in coronavirus, especially in COVID-19. Front. Pharmacol. 2022, 13, 1033674. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal 2009, 11, 2245–2263. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Tabarsi, P.; Jamaati, H.; Dalil Roofchayee, N.; Dezfuli, N.K.; Hashemian, S.M.; Moniri, A.; Marjani, M.; Malekmohammad, M.; Mansouri, D.; et al. Increased Serum Levels of Soluble TNF-α Receptor Is Associated with ICU Mortality in COVID-19 Patients. Front. Immunol. 2021, 12, 592727. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, L.K.A.; Thompson-Figueroa, J.; Leclair, T.; Sullivan, M.J.; Poynter, M.E.; Irvin, C.G.; Bates, J.H.T. Tumor necrosis factor-alpha overexpression in lung disease: A single cause behind a complex phenotype. Am. J. Respir. Crit. Care Med. 2005, 171, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Epstein Shochet, G.; Brook, E.; Israeli-Shani, L.; Edelstein, E.; Shitrit, D. Fibroblast paracrine TNF-α signaling elevates integrin A5 expression in idiopathic pulmonary fibrosis (IPF). Respir. Res. 2017, 18, 122. [Google Scholar] [CrossRef] [PubMed]
- Faraj, S.S.; Jalal, P.J. IL1β, IL-6, and TNF-α cytokines cooperate to modulate a complicated medical condition among COVID-19 patients: Case-control study. Ann. Med. Surg. 2023, 85, 2291–2297. [Google Scholar] [CrossRef] [PubMed]
- Borges, L.; Pithon-Curi, T.C.; Curi, R.; Hatanaka, E. COVID-19 and Neutrophils: The Relationship between Hyperinflammation and Neutrophil Extracellular Traps. Mediat. Inflamm. 2020, 2020, 8829674. [Google Scholar] [CrossRef] [PubMed]
- Heukels, P.; Moor, C.C.; Von Der Thüsen, J.H.; Wijsenbeek, M.S.; Kool, M. Inflammation and immunity in IPF pathogenesis and treatment. Respir. Med. 2019, 147, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Meng, M.; Kumar, R.; Wu, Y.; Huang, J.; Deng, Y.; Weng, Z.; Yang, L. Lymphopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A systemic review and meta-analysis. Int. J. Infect. Dis. 2020, 96, 131–135. [Google Scholar] [CrossRef] [PubMed]
- McKenna, E.; Wubben, R.; Isaza-Correa, J.M.; Melo, A.M.; Mhaonaigh, A.U.; Conlon, N.; O’Donnell, J.S.; Ní Cheallaigh, C.; Hurley, T.; Stevenson, N.J.; et al. Neutrophils in COVID-19: Not Innocent Bystanders. Front. Immunol. 2022, 13, 864387. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, M.; Rabaan, A.A.; Alwarthan, S.; Alhajri, M.; Halwani, M.A.; Alshengeti, A.; Najim, M.A.; Alwashmi, A.S.S.; Alshehri, A.A.; Alshamrani, S.A.; et al. Regulatory T Cells (Tregs) and COVID-19: Unveiling the Mechanisms, and Therapeutic Potentialities with a Special Focus on Long COVID. Vaccines 2023, 11, 699. [Google Scholar] [CrossRef] [PubMed]
- Achaiah, A.; Rathnapala, A.; Pereira, A.; Bothwell, H.; Dwivedi, K.; Barker, R.; Iotchkova, V.; Benamore, R.; Hoyles, R.K.; Ho, L.-P. Neutrophil lymphocyte ratio as an indicator for disease progression in Idiopathic Pulmonary Fibrosis. BMJ Open Resp. Res. 2022, 9, e001202. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.J.A.; Ribeiro, L.R.; Gouveia, M.I.M.; Marcelino, B.D.R.; Santos, C.S.D.; Lima, K.V.B.; Lima, L.N.G.C. Hyperinflammatory Response in COVID-19: A Systematic Review. Viruses 2023, 15, 553. [Google Scholar] [CrossRef] [PubMed]
- Alhamad, E.H.; Cal, J.G.; Shakoor, Z.; Almogren, A.; AlBoukai, A.A. Cytokine gene polymorphisms and serum cytokine levels in patients with idiopathic pulmonary fibrosis. BMC Med. Genet. 2013, 14, 66. [Google Scholar] [CrossRef] [PubMed]
- Fukihara, J.; Kondoh, Y. COVID-19 and interstitial lung diseases: A multifaceted look at the relationship between the two diseases. Respir. Investig. 2023, 61, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.R.; Søgaard, O.S.; Tolstrup, M.; Stærke, N.B.; Lundgren, J.; Østergaard, L.; Hvas, A.-M. Inflammation and Platelet Activation After COVID-19 Vaccines—Possible Mechanisms Behind Vaccine-Induced Immune Thrombocytopenia and Thrombosis. Front. Immunol. 2021, 12, 779453. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Rubin, L.; Peng, T.; Liu, L.; Xing, X.; Lazarovici, P.; Zheng, W. Cytokine storm in COVID-19: From viral infection to immune responses, diagnosis and therapy. Int. J. Biol. Sci. 2022, 18, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Magrì, T.; Lerede, M.; Comes, A.; Richeldi, L. COVID-19 Vaccine in Patients with Exacerbation of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022, 206, 219–221. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Pathological Aspect | COVID-19 | IPF | Combined/Amplified Effects | References |
|---|---|---|---|---|
| Pulmonary Thrombosis | - Widespread microvascular thrombosis - Elevated D-dimer and fibrinogen - Endothelial injury | - Baseline microvascular remodeling - Prothrombotic endothelial phenotype | Exacerbation of hypoxia, fibrotic progression, increased risk of acute exacerbation | [80] |
| Systemic Inflammation | - Acute cytokine storm (↑ IL-6, IL-1β, TNF-α) - Intense neutrophilia, hyperinflammation, and neutrophil extracellular traps | - Chronic low-grade inflammation - Persistent release of profibrotic cytokines | Synergistic inflammatory damage, increased fibroblast activity, rapid ECM deposition | [119,139,140,141] |
| Immune Dysregulation | - Lymphopenia - Neutrophil/monocyte overactivation - Treg cell dysfunction | - Impaired immune tolerance - Chronic macrophage activation - High neutrophil-to-lymphocyte ratio | Exaggerated immune response, delayed viral clearance, enhanced tissue damage | [141,142,143,144,145] |
| Cytokine Expression (TGF-β, IL-6, TNF-α) | - Upregulation of all three - Associated with severe COVID-19 and ARDS | - Chronically elevated in lung tissue and circulation | Accelerated fibrosis, epithelial–mesenchymal transition (EMT), fibroblast proliferation | [146,147] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mara, G.; Nini, G.; Frenț, S.M.; Cotoraci, C. Hematologic and Immunologic Overlap Between COVID-19 and Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2025, 14, 5229. https://doi.org/10.3390/jcm14155229
Mara G, Nini G, Frenț SM, Cotoraci C. Hematologic and Immunologic Overlap Between COVID-19 and Idiopathic Pulmonary Fibrosis. Journal of Clinical Medicine. 2025; 14(15):5229. https://doi.org/10.3390/jcm14155229
Chicago/Turabian StyleMara, Gabriela, Gheorghe Nini, Stefan Marian Frenț, and Coralia Cotoraci. 2025. "Hematologic and Immunologic Overlap Between COVID-19 and Idiopathic Pulmonary Fibrosis" Journal of Clinical Medicine 14, no. 15: 5229. https://doi.org/10.3390/jcm14155229
APA StyleMara, G., Nini, G., Frenț, S. M., & Cotoraci, C. (2025). Hematologic and Immunologic Overlap Between COVID-19 and Idiopathic Pulmonary Fibrosis. Journal of Clinical Medicine, 14(15), 5229. https://doi.org/10.3390/jcm14155229