

Expanding the Spectrum of CSF3R-Mutated Myeloid Neoplasm Beyond Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia: A Comprehensive Analysis of 13 Cases

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection and Data Collection
2.2. Hematologic and Morphologic Analysis
2.3. Cytogenetic and FISH Analysis
2.4. Molecular Studies
2.5. Statistical Analysis
2.6. Ethical Considerations
3. Results
3.1. Clinical Findings
3.2. Peripheral Blood Findings
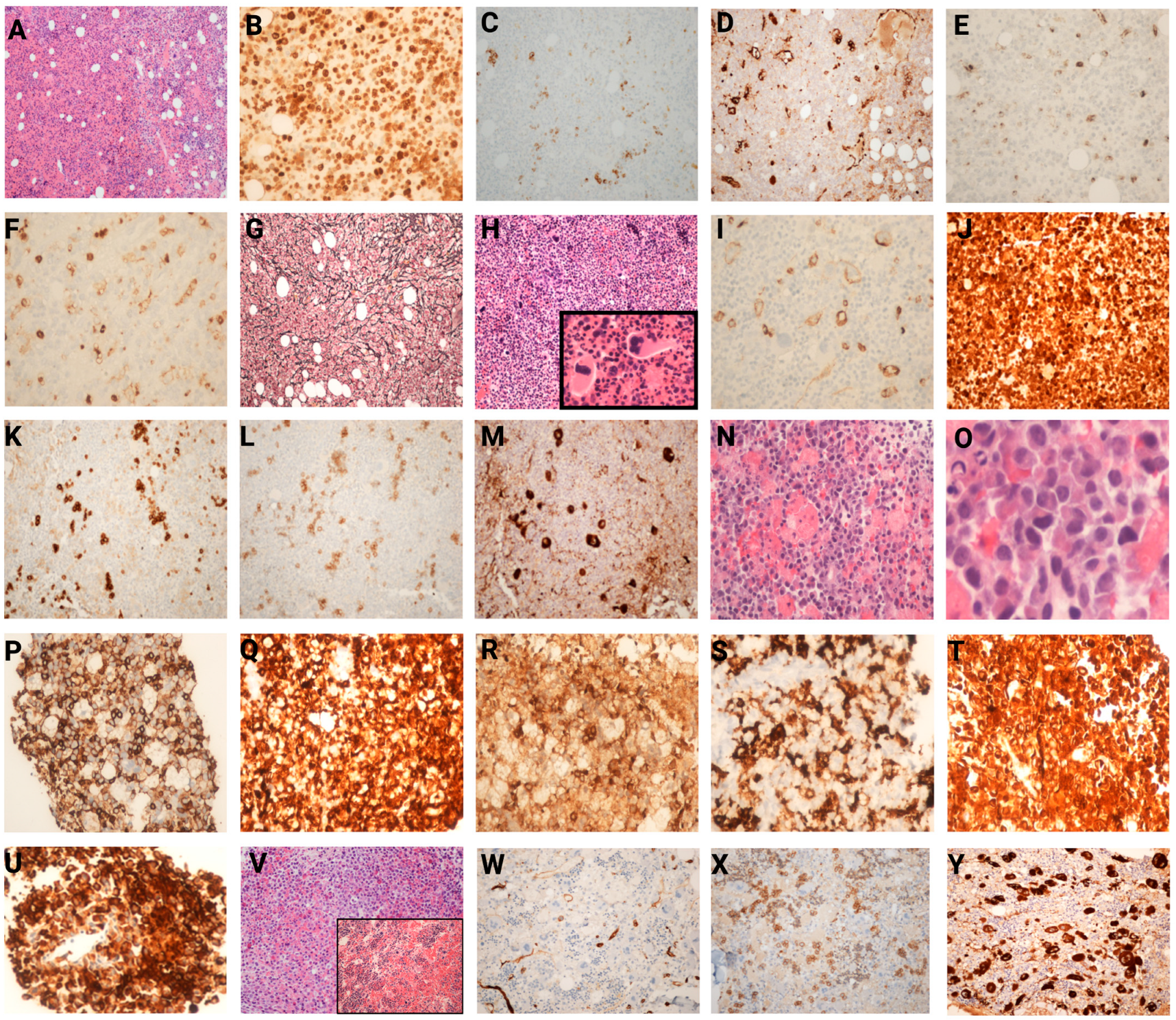
3.3. Histopathological Findings in Bone Marrow
3.4. Cytogenetic and FISH Analysis
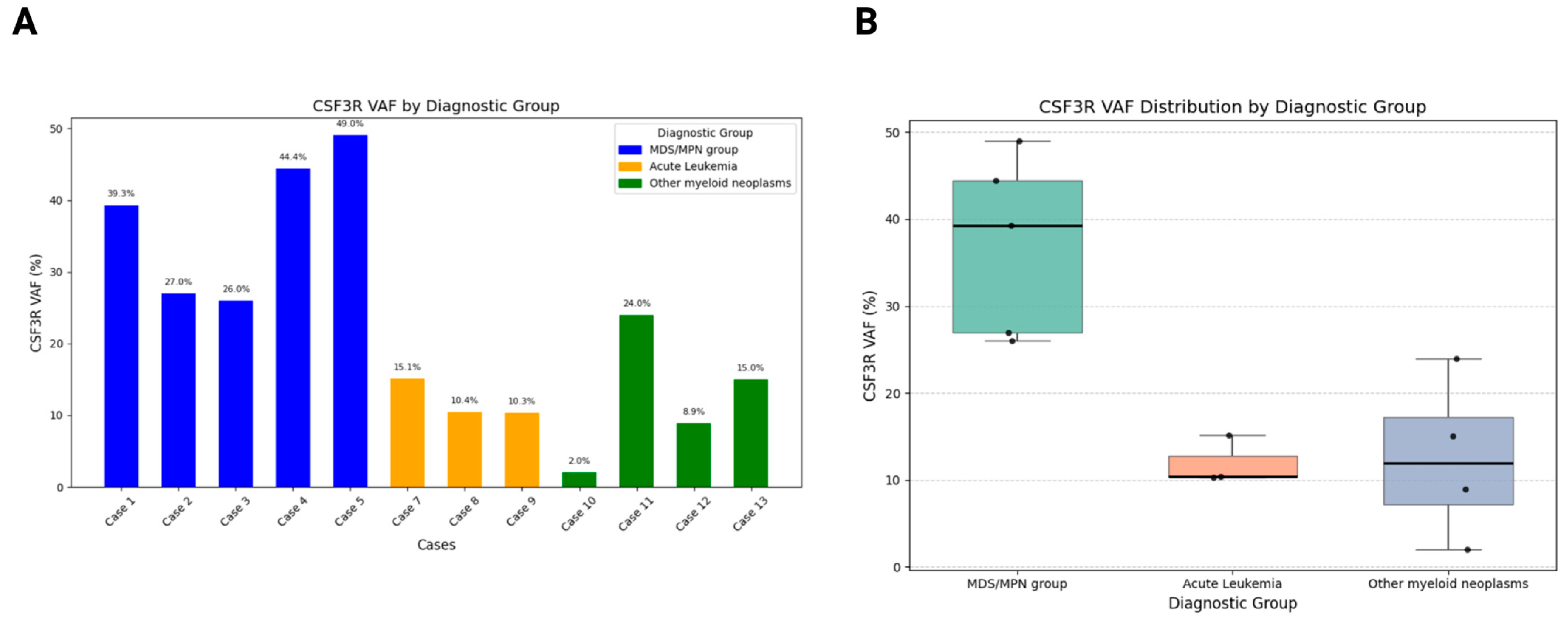
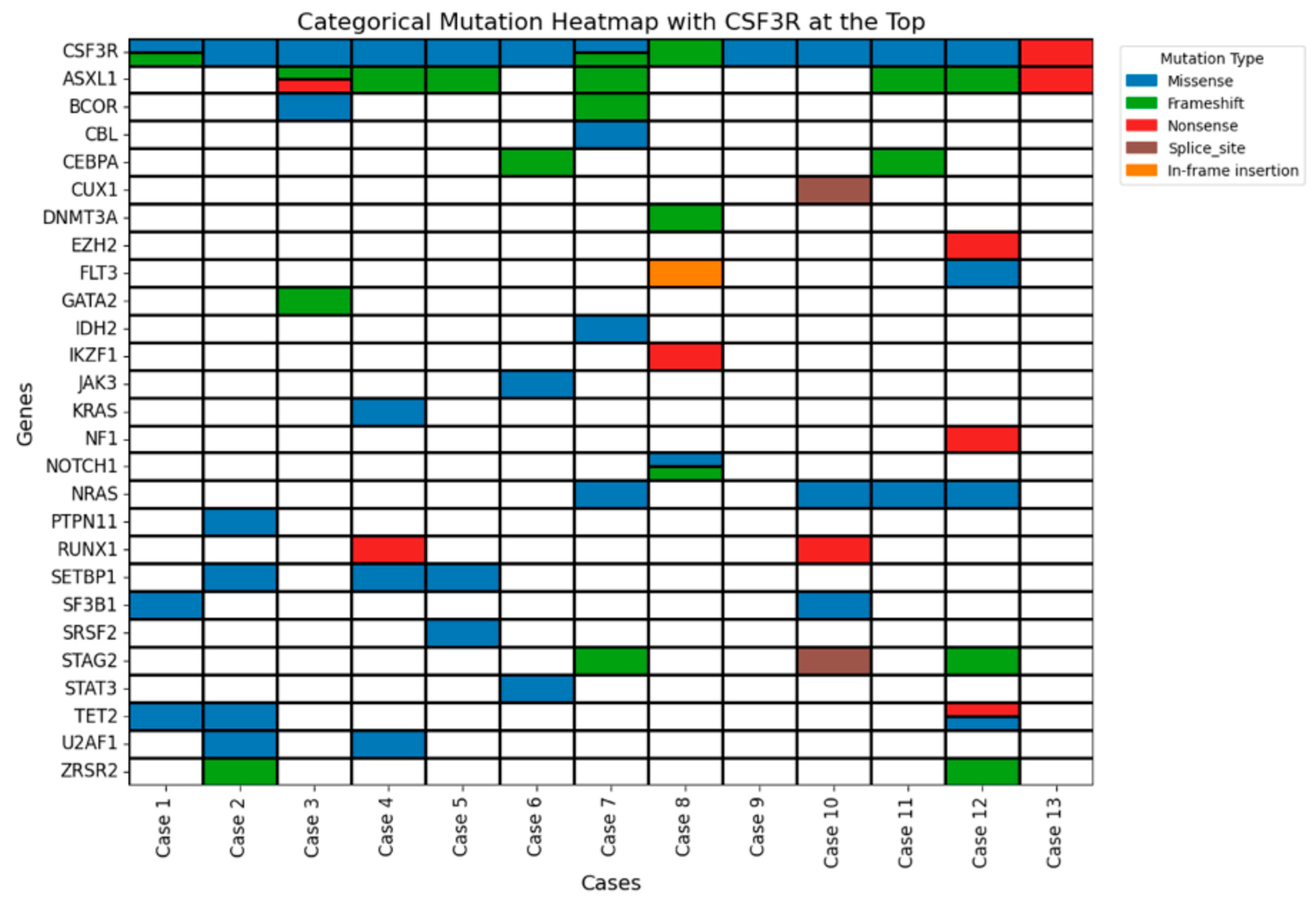
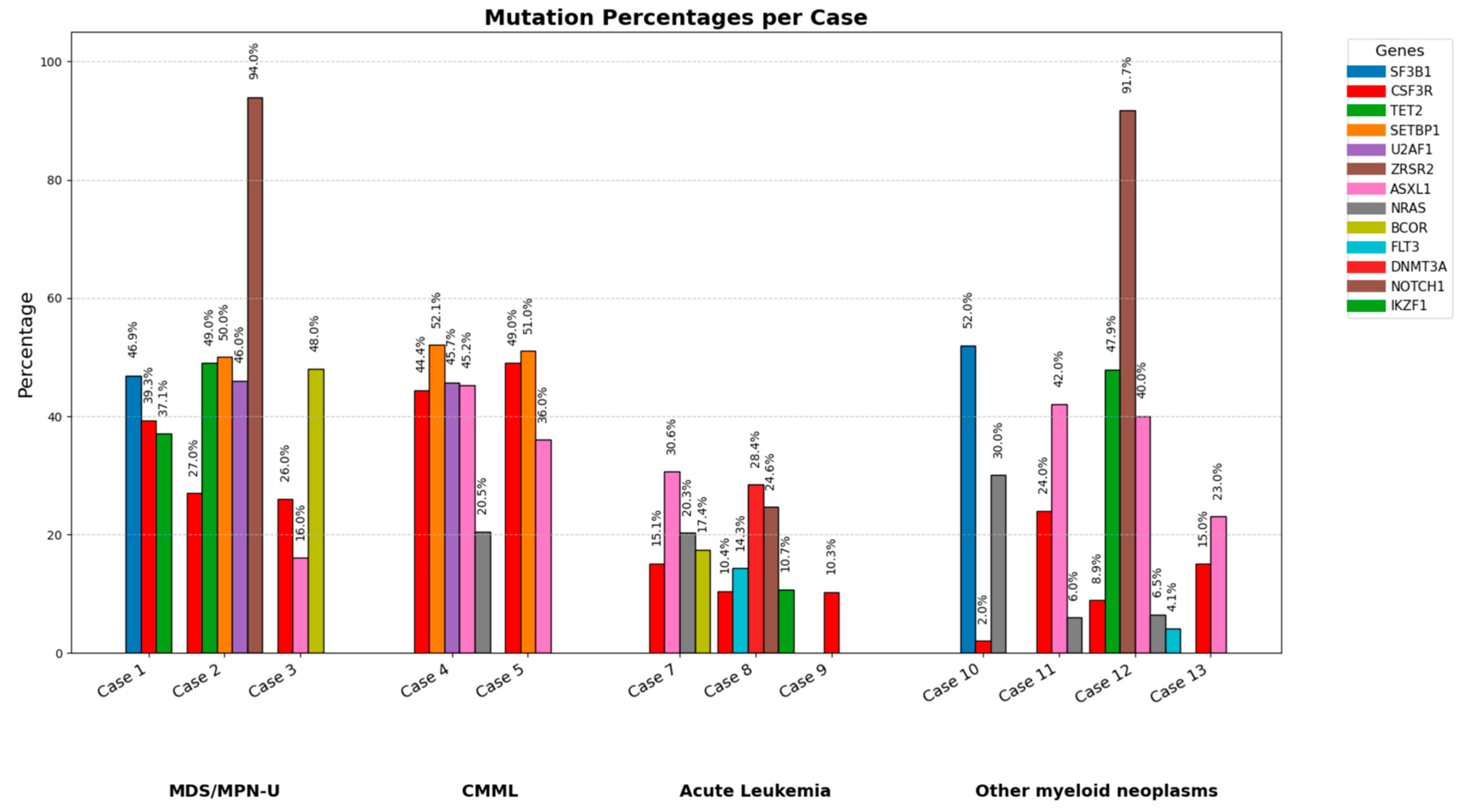
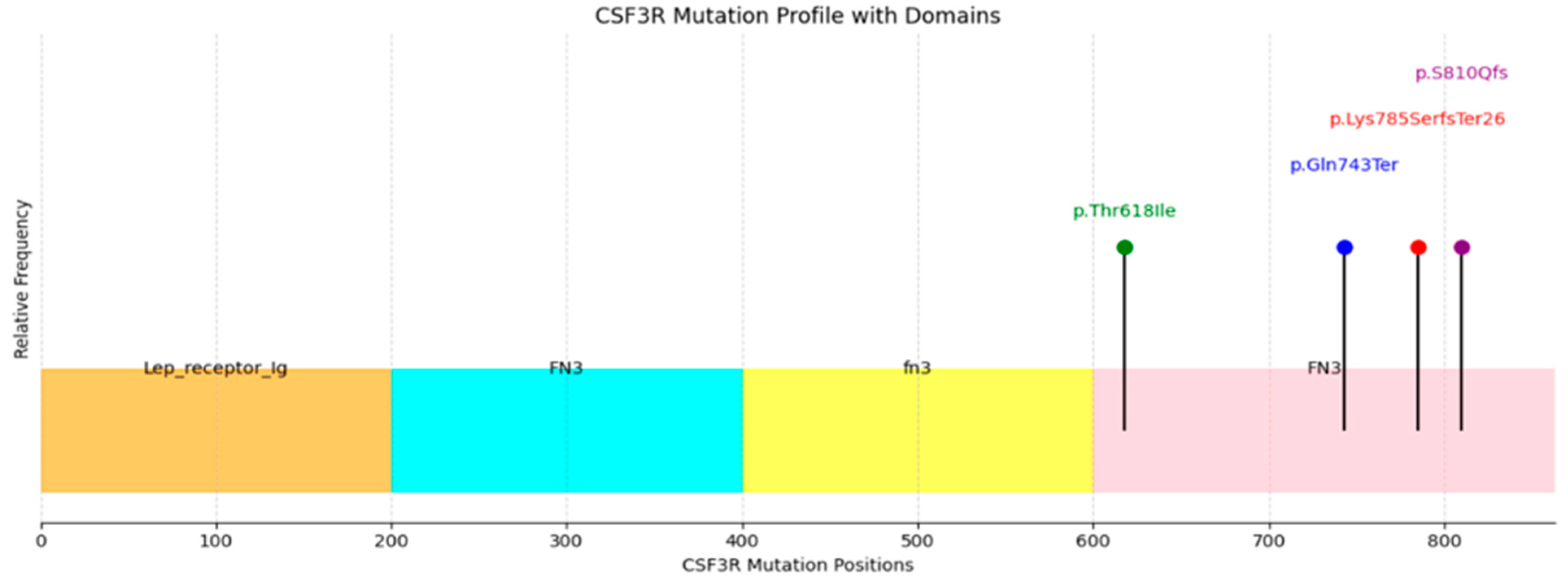
3.5. Molecular Findings
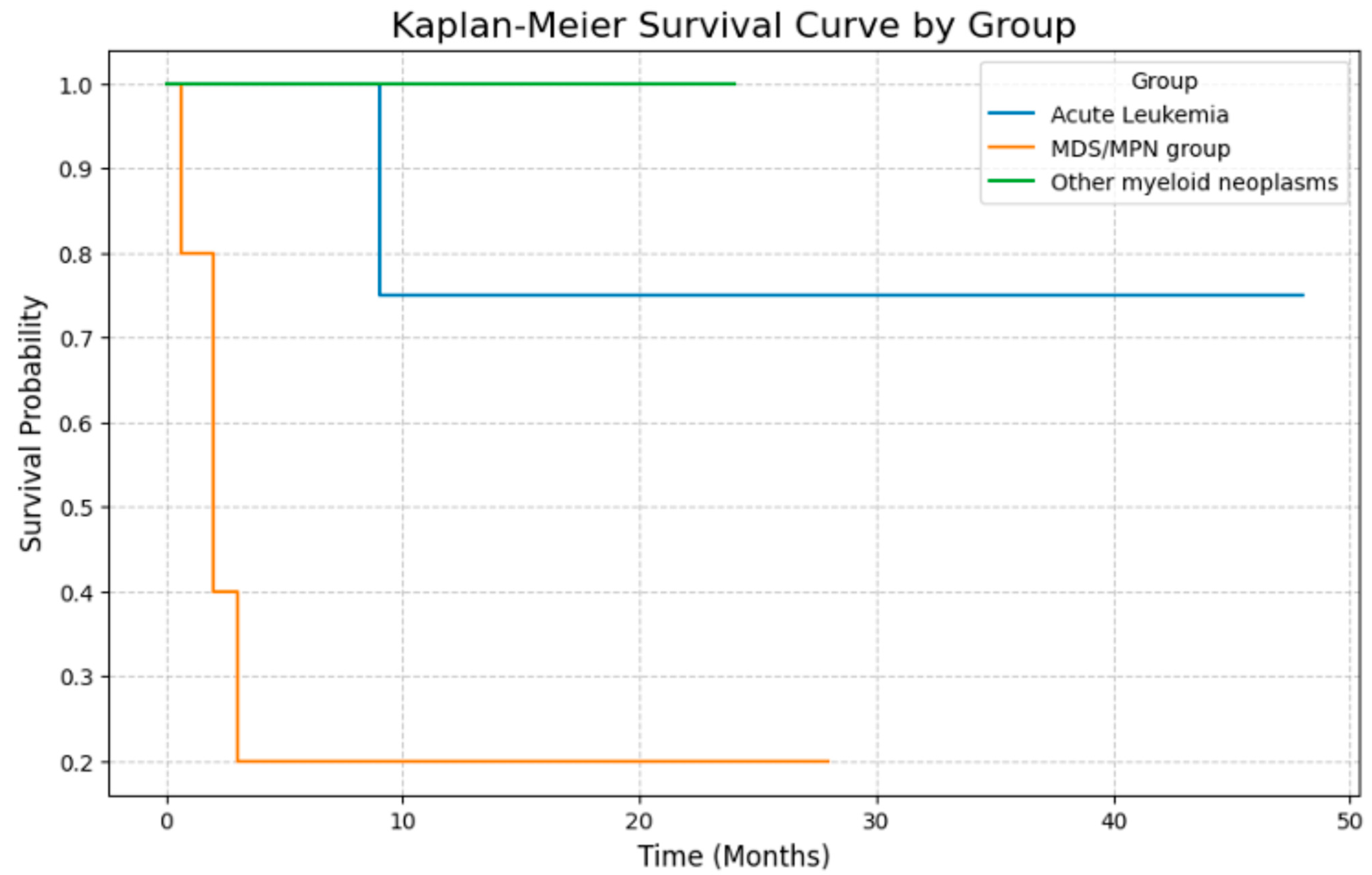
3.6. Clinical Outcomes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beekman, R.; Touw, I.P. G-CSF and its receptor in myeloid malignancy. Blood 2010, 115, 5131–5136. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, H.Y.; Wesselschmidt, R.; Kornaga, T.; Link, D.C. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity 1996, 5, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.S.; Xia, L.; Mills, G.B.; Lowell, C.A.; Touw, I.P.; Corey, S.J. G-CSF induced reactive oxygen species involves Lyn-PI3-kinase-Akt and contributes to myeloid cell growth. Blood 2006, 107, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Price, A.; Druhan, L.J.; Lance, A.; Clark, G.; Vestal, C.G.; Zhang, Q.; Foureau, D.; Parsons, J.; Hamilton, A.; Steuerwald, N.M.; et al. T618I CSF3R mutations in chronic neutrophilic leukemia induce oncogenic signals through aberrant trafficking and constitutive phosphorylation of the O-glycosylated receptor form. Biochem. Biophys. Res. Commun. 2020, 523, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Coblentz, C.; Watanabe-Smith, K.; Means, S.; Means, J.; Maxson, J.E.; Tyner, J.W. Gain-of-function mutations in granulocyte colony-stimulating factor receptor (CSF3R) reveal distinct mechanisms of CSF3R activation. J. Biol. Chem. 2018, 293, 7387–7396. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.A. Chronic neutrophilic leukemia: A contemporary review. Curr. Hematol. Rep. 2004, 3, 210–217. [Google Scholar] [PubMed]
- Reilly, J.T. Chronic neutrophilic leukaemia: A distinct clinical entity? Br. J. Haematol. 2002, 116, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wilmot, B.; Bottomly, D.; Dao, K.-H.T.; Stevens, E.; Eide, C.A.; Khanna, V.; Rofelty, A.; Savage, S.; Schultz, A.R.; et al. Genomic landscape of neutrophilic leukemias of ambiguous diagnosis. Blood 2019, 134, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Langabeer, S.E.; Haslam, K.; Kelly, J.; Quinn, J.; Morrell, R.; Conneally, E. Targeted next-generation sequencing identifies clinically relevant mutations in patients with chronic neutrophilic leukemia at diagnosis and blast crisis. Clin. Transl. Oncol. 2018, 20, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Faisal, M.; Stark, H.; Büsche, G.; Schlue, J.; Teiken, K.; Kreipe, H.H.; Lehmann, U.; Bartels, S. Comprehensive mutation profiling and mRNA expression analysis in atypical chronic myeloid leukemia in comparison with chronic myelomonocytic leukemia. Cancer Med. 2019, 8, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Maxson, J.E.; Gotlib, J.; Pollyea, D.A.; Fleischman, A.G.; Agarwal, A.; Eide, C.A.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; Tognon, C.E.; et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N. Engl. J. Med. 2013, 368, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Meggendorfer, M.; Haferlach, T.; Alpermann, T.; Jeromin, S.; Haferlach, C.; Kern, W.; Schnittger, S. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica 2014, 99, e244–e246. [Google Scholar] [CrossRef] [PubMed]
- Szuber, N.; Finke, C.M.; Lasho, T.L.; Elliott, M.A.; Hanson, C.A.; Pardanani, A.; Tefferi, A. CSF3R-mutated chronic neutrophilic leukemia: Long-term outcome in 19 consecutive patients and risk model for survival. Blood Cancer J. 2018, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Dao, K.H.; Solti, M.B.; Maxson, J.E.; Winton, E.F.; Press, R.D.; Druker, B.J.; Tyner, J.W. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leuk. Res. Rep. 2014, 3, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Dao, K.T.; Gotlib, J.; Deininger, M.M.; Oh, S.T.; Cortes, J.E.; Collins, R.H.; Winton, E.F.; Parker, D.R.; Lee, H.; Reister, A.; et al. Efficacy of Ruxolitinib in Patients With Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia. J. Clin. Oncol. 2020, 38, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Gao, J.; Chen, Y.-H.; Abaza, Y.; Altman, J.; Jennings, L.; Vormittag-Nocito, E.; Sukhanova, M.; Lu, X.; Chen, Q. CSF3R mutated myeloid neoplasms: Beyond chronic neutrophilic leukemia. Hum. Pathol. 2024, 149, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Maxson, J.E.; Luty, S.B.; MacManiman, J.D.; Abel, M.L.; Druker, B.J.; Tyner, J.W. Ligand independence of the T618I mutation in the colony-stimulating factor 3 receptor (CSF3R) protein results from loss of O-linked glycosylation and increased receptor dimerization. J. Biol. Chem. 2014, 289, 5820–5827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Schultz, A.R.; Luty, S.; Rofelty, A.; Su, Y.; Means, S.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; Tyner, J.W. Characterization of the leukemogenic potential of distal cytoplasmic CSF3R truncation and missense mutations. Leukemia 2017, 31, 2752–2760. [Google Scholar] [CrossRef] [PubMed]
- Mehta, H.M.; Futami, M.; Glaubach, T.; Lee, D.W.; Andolina, J.R.; Yang, Q.; Whichard, Z.; Quinn, M.; Lu, H.F.; Kao, W.M.; et al. Alternatively spliced, truncated GCSF receptor promotes leukemogenic properties and sensitivity to JAK inhibition. Leukemia 2014, 28, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Rohrabaugh, S.; Kesarwani, M.; Kincaid, Z.; Huber, E.; Leddonne, J.; Siddiqui, Z.; Khalifa, Y.; Komurov, K.; Grimes, H.L.; Azam, M. Enhanced MAPK signaling is essential for CSF3R-induced leukemia. Leukemia 2017, 31, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Adam, F.C.; Szybinski, J.; Halter, J.P.; Cantoni, N.; Wenzel, F.; Leonards, K.; Brkic, S.; Passweg, J.R.; Touw, I.; Maxson, J.E.; et al. Co-Occurring CSF3R W791* Germline and Somatic T618I Driver Mutations Induce Early CNL and Clonal Progression to Mixed Phenotype Acute Leukemia. Curr. Oncol. 2022, 29, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.A.; Pardanani, A.; Hanson, C.A.; Lasho, T.L.; Finke, C.M.; Belachew, A.A.; Tefferi, A. ASXL1 mutations are frequent and prognostically detrimental in CSF3R-mutated chronic neutrophilic leukemia. Am. J. Hematol. 2015, 90, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Alawieh, D.; Cysique-Foinlan, L.; Willekens, C.; Renneville, A. RAS mutations in myeloid malignancies: Revisiting old questions with novel insights and therapeutic perspectives. Blood Cancer J. 2024, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Qiao, C.; Chen, Y.; Zhang, S.-J. Clinical significance of CSF3R, SRSF2 and SETBP1 mutations in chronic neutrophilic leukemia and chronic myelomonocytic leukemia. Oncotarget 2017, 8, 20834–20841. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Yoshida, K.; Nguyen, N.; Przychodzen, B.; Sanada, M.; Okuno, Y.; Ng, K.P.; Gudmundsson, K.O.; Vishwakarma, B.A.; Jerez, A.; et al. Somatic SETBP1 mutations in myeloid malignancies. Nat. Genet. 2013, 45, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, A.S.; Arfelli, V.C.; Danese, A.; Windisch, R.; Kerbs, P.; Monte, E.R.; Bagnoli, J.W.; Chen-Wichmann, L.; Caroleo, A.; Cusan, M.; et al. CSF3R T618I Collaborates With RUNX1-RUNX1T1 to Expand Hematopoietic Progenitors and Sensitizes to GLI Inhibition. Hemasphere 2023, 7, e958. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wen, L.; Wang, Z.; Chen, S.; Qiu, H. Differential Implications of CSF3R Mutations in t(8;21) and CEBPA Double Mutated Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2022, 22, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Chen, Y.; Li, X. CSF3R T618I, SETBP1 G870S, SRSF2 P95H, and ASXL1 Q780* tetramutation co-contribute to myeloblast transformation in a chronic neutrophilic leukemia. Ann. Hematol. 2021, 100, 1459–1461. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, A.S.; McLornan, D.P.; Wilkins, B.; Waghorn, K.; Hoade, Y.; Cross, N.C.P.; Harrison, C.N. Ruxolitinib, a potent JAK1/JAK2 inhibitor, induces temporary reductions in the allelic burden of concurrent CSF3R mutations in chronic neutrophilic leukemia. Haematologica 2017, 102, e238–e240. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Case 10 | Case 11 | Case 12 | Case 13 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | 89 | 81 | 87 | 84 | 78 | 22 | 69 | 55 | 56 | 57 | 77 | 63 | 79 |
| Gender | F | M | F | M | M | M | F | M | M | F | M | M | M |
| WBC | 58.5 | 14.6 | 44 | 16.2 | 51.9 | 25.5 | 4.2 | 6.1 | 1.4 | 10.4 | 2.6 | 25.3 | 2.5 |
| Hb | 10.6 | 7.3 | 7.4 | 9.2 | 11.7 | 11.7 | 12 | 11 | 7.6 | 9.4 | 10.6 | 7.7 | 11.2 |
| MCV | 99.7 | 100.4 | 103.9 | 85.5 | 105.5 | 94.6 | 81 | 86.2 | 92.6 | 90.9 | 84.3 | 98.7 | 96.3 |
| Plt | 216 | 27 | 141 | 32 | 158 | 47 | 184 | 95 | 44 | 117 | 280 | 77 | 65 |
| Mono(%) | 3 | 6 | NA | 10 | 12.1 | 0 | 9 | 1 | 4 | 5.2 | NA | 2.9 | 9 |
| Mono# | 1.2 | 0.9 | 2.3 | 1.8 | 6.3 | 0 | 0.4 | 0 | 0.1 | 0.54 | NA | 0.7 | 0.22 |
| PB blasts (%) | 0 | 0 | 0 | 0 | 0 | 85 | 0 | 63 | 14 | 0 | NA | 0 | 0 |
| LDH | 385 | 220 | 531 | 418 | 888 | 406 | 337 | 314 | 666 | 416 | 1269 | 1176 | ND |
| Cellularity | 90 | 85–90 | 95 | 70–85 | 100 | 70–85 | Limited | 95–100 | 70 | 95 | 90 | 95–100 | 30 |
| M/E | 10:1 | MP | MP | 1:1 | 2:1 | 0.8:1 | 3:1 | MP | 5.2:1 | 6.5:1 | 1.6:1 | 10.8:1 | MP |
| Erythroid | Dys+ | Dys+ | Dec | Dys+ | Dec | Dys+ | Dec | Dec | Dec | Dys+ | Dys+ | Inc | Mild dec |
| Myeloid | NA | NA | Inc | Dys+ | Inc | Minimal | Dec | Dec | Dec | Dys+ | Dys+ | Rare | Dys+ |
| Megakaryocytes | NA | Dys+ | Dec | Dys+ | Inc, Dys+ | No | Rare | Dec | Present | Dys+ | Dys+ | Dys+ | Dec |
| BM blasts (%) | <3 | <3 | <3 | 7 | 1 | 66 | 4 | 83 | 56 | 8 | 8 | 8 | 5 |
| Fibrosis | 0 | Gr 2-3 | Gr 2-3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | Gr 1-2 | 0 |
| Karyotype | 46, XX | 45,XY,-7 | 46, XX | 46,XY del(7)(q22) | 46, XX | 46, XY | 46, XX | 46,XY | Complex | 46,XX | 46,XY | 46,XY,der(13;14)9q10;q10) | 45, -X,-Y |
| FISH | Normal | Monosomy 7 | NA | Del(7q) | Normal | RUNX1 (3 copies) | Normal | Normal | RUNX1T1/RUNX1 | Normal | Normal | NA | Normal |
| Diagnosis | MDS/MPN-U | MDS/MPN-U | MDS/MPN-U | CMML | CMML | AML | MS | MPAL | AML | MDS-EB1 | MDS-EB1 | ET/MPN transformed to AML | MDS-EB1 |
| Treatment | Hy 500 mg OD | Azax1 cycle | NA | Supportive care only | Hy 500 mg OD | 7+3+GO, Consolidation HiDAC + GO × 4 | 7+3; HiDAC + Aza × 2; Ven + Dec × 2 | 7+3 | 7+3 | No Rx | Ven + Dec × 4; GO+ LD Ara-C | Ven + Dec; ASCT | No Rx |
| Prognosis | Died, 33 months | Died, 2 months | 2 months | Died, 19 days | Alive, 28 months | CR, 4 years | Relapse, 14 months | Died, 9 months | Relapse, 19 months | Alive, 1 year | Transform into AML, alive, 2 years | CR, 1 year | Alive, 5 months |
| Clinical | None | Hepatitis, cirrhosis | Breast Ca, colon Ca | Prostate Ca | B-ALL, remission | None | RA | DVT | None | OA knee | MG | None | Prostate Ca |
| Splenomegaly | No | Yes | Yes | No | No | No | No | No | No | No | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seth, N.; Brody, J.; Hsu, P.; Kolitz, J.; Deb, P.Q.; Zhang, X. Expanding the Spectrum of CSF3R-Mutated Myeloid Neoplasm Beyond Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia: A Comprehensive Analysis of 13 Cases. J. Clin. Med. 2025, 14, 5174. https://doi.org/10.3390/jcm14155174
Seth N, Brody J, Hsu P, Kolitz J, Deb PQ, Zhang X. Expanding the Spectrum of CSF3R-Mutated Myeloid Neoplasm Beyond Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia: A Comprehensive Analysis of 13 Cases. Journal of Clinical Medicine. 2025; 14(15):5174. https://doi.org/10.3390/jcm14155174
Chicago/Turabian StyleSeth, Neha, Judith Brody, Peihong Hsu, Jonathan Kolitz, Pratik Q. Deb, and Xinmin Zhang. 2025. "Expanding the Spectrum of CSF3R-Mutated Myeloid Neoplasm Beyond Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia: A Comprehensive Analysis of 13 Cases" Journal of Clinical Medicine 14, no. 15: 5174. https://doi.org/10.3390/jcm14155174
APA StyleSeth, N., Brody, J., Hsu, P., Kolitz, J., Deb, P. Q., & Zhang, X. (2025). Expanding the Spectrum of CSF3R-Mutated Myeloid Neoplasm Beyond Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia: A Comprehensive Analysis of 13 Cases. Journal of Clinical Medicine, 14(15), 5174. https://doi.org/10.3390/jcm14155174