

Genotype–Phenotype Relationship in Hereditary Hemorrhagic Telangiectasia: Quality of Life and Cardiovascular Risk Evaluation
 ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. The Mutational Analysis
2.3. Variables and Definitions
- Epistaxis Severity Score (ESS): This was used to assess the frequency and intensity of epistaxis episodes (0–10). ESSs were then stratified according to severity. This created categories consisting of no epistaxis (an ESS < 1), mild epistaxis (an ESS between 1 and 4), and moderate epistaxis (an ESS ≥ 4) [5]
- Arteriovenous malformations (AVMs): These were evaluated based on location (pulmonary, hepatic, cerebral, spinal, or digestive) and age at diagnosis.
- Anemia: Hemoglobin and iron levels were measured.
- Renal and hepatic function: Serum creatinine; glomerular filtration rates (GFRs) calculated using the MDRD and CKD-EPI; liver enzymes (AST, ALT, GGT); and lipid profiles were analyzed.
- Cardiovascular risk assessment: The SCORE2 model was used to estimate 10-year cardiovascular risk, stratifying patients into low-, intermediate-, and high-risk categories.
- The quality of life assessment was carried out using the EQ-5D-5L survey. The investigators personally asked the patients the questions during the consultation and completed the surveys themselves. This scale is based on 5 main components: ‘mobility’, ‘self-care’, ‘activities of daily living’, ‘pain/discomfort’, and ‘anxiety/depression’.
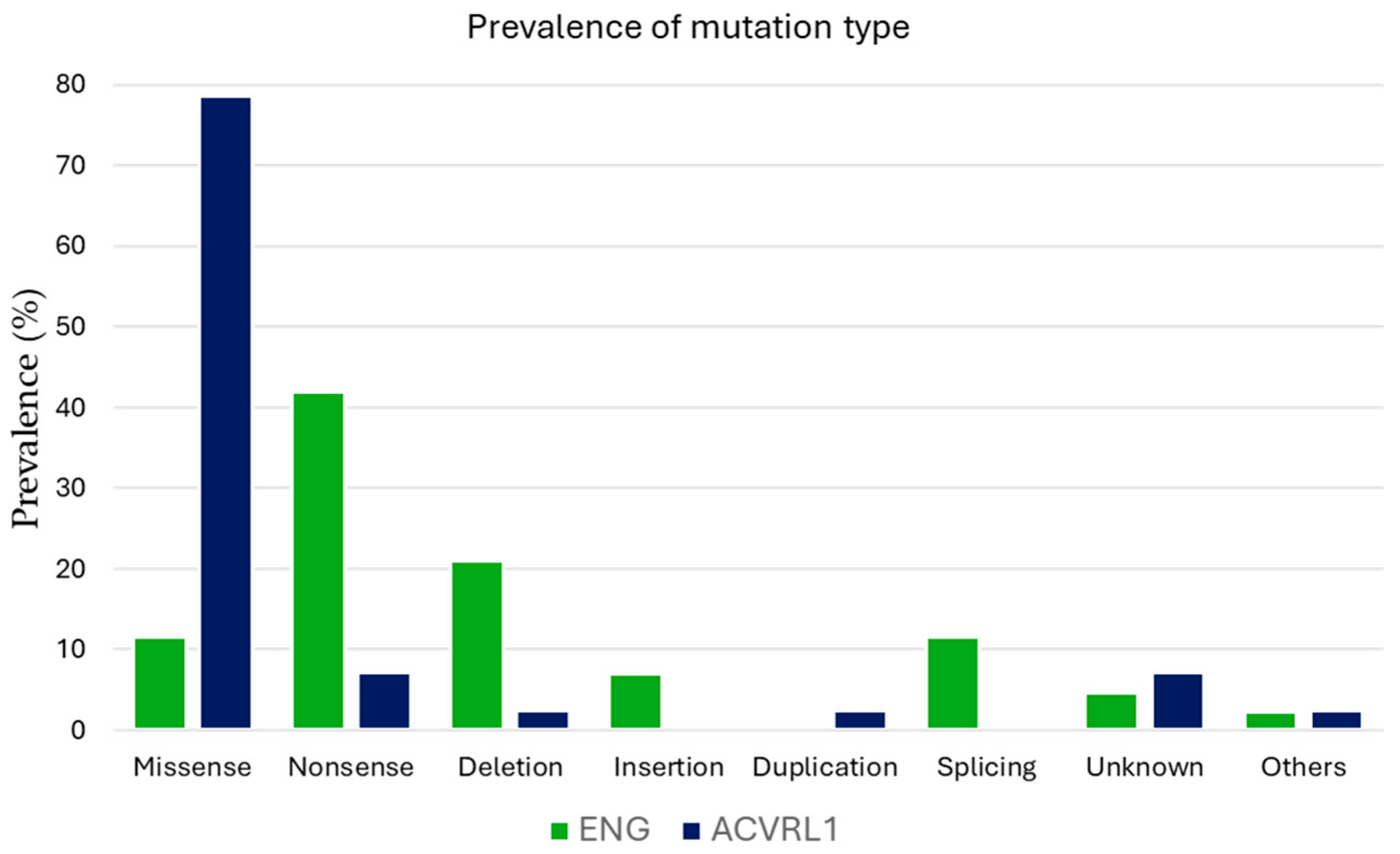
- Genetic analysis: The classification of pathogenic variants included missense, nonsense, deletion, insertion, duplication, splicing, and other rare mutations. To assess the impact of mutation type on phenotype expression, genetic variants were categorized into truncating mutations (including nonsense and deletion/insertion mutations) and non-truncating mutations (including missense and splicing mutations).
2.4. The Statistical Analysis
- Student’s t-test or the Mann–Whitney U test for quantitative variables, depending on the distribution.
- The paired Student’s t-test or the Wilcoxon test for repeated measures.
- The Chi-square test for associations between categorical variables.
- Pearson’s correlation coefficient for linear associations.
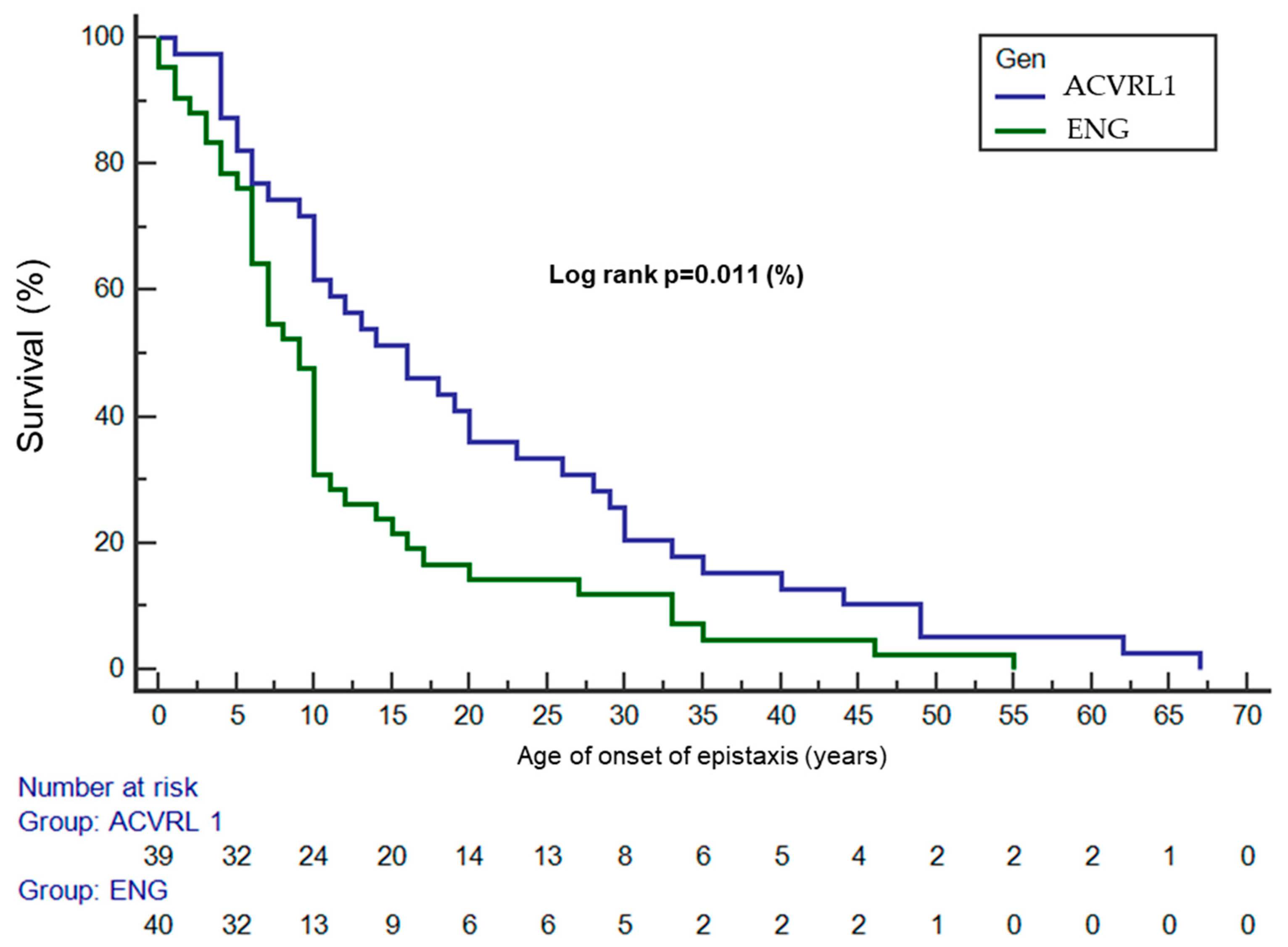
- Survival analyses were performed using the Kaplan–Meier method for estimation of the survival curves, and the log-rank test was used to compare the age of onset of epistaxis between genotypes, assessing the differences in the epistaxis-free survival curves.
- A logistic regression analysis was used to assess the association of truncated variants with anemia, adjusting for age, sex, and genotype. A p-value < 0.05 was considered statistically significant. The statistical analyses were performed using SPSS version 22 stadistical software.
3. Results
3.1. The Baseline Characteristics and Genotype Implications
3.2. Epistaxis
3.3. Telangiectasias
3.4. Vascular Malformations
3.5. Family History and Mortality
3.6. Biochemical Differences
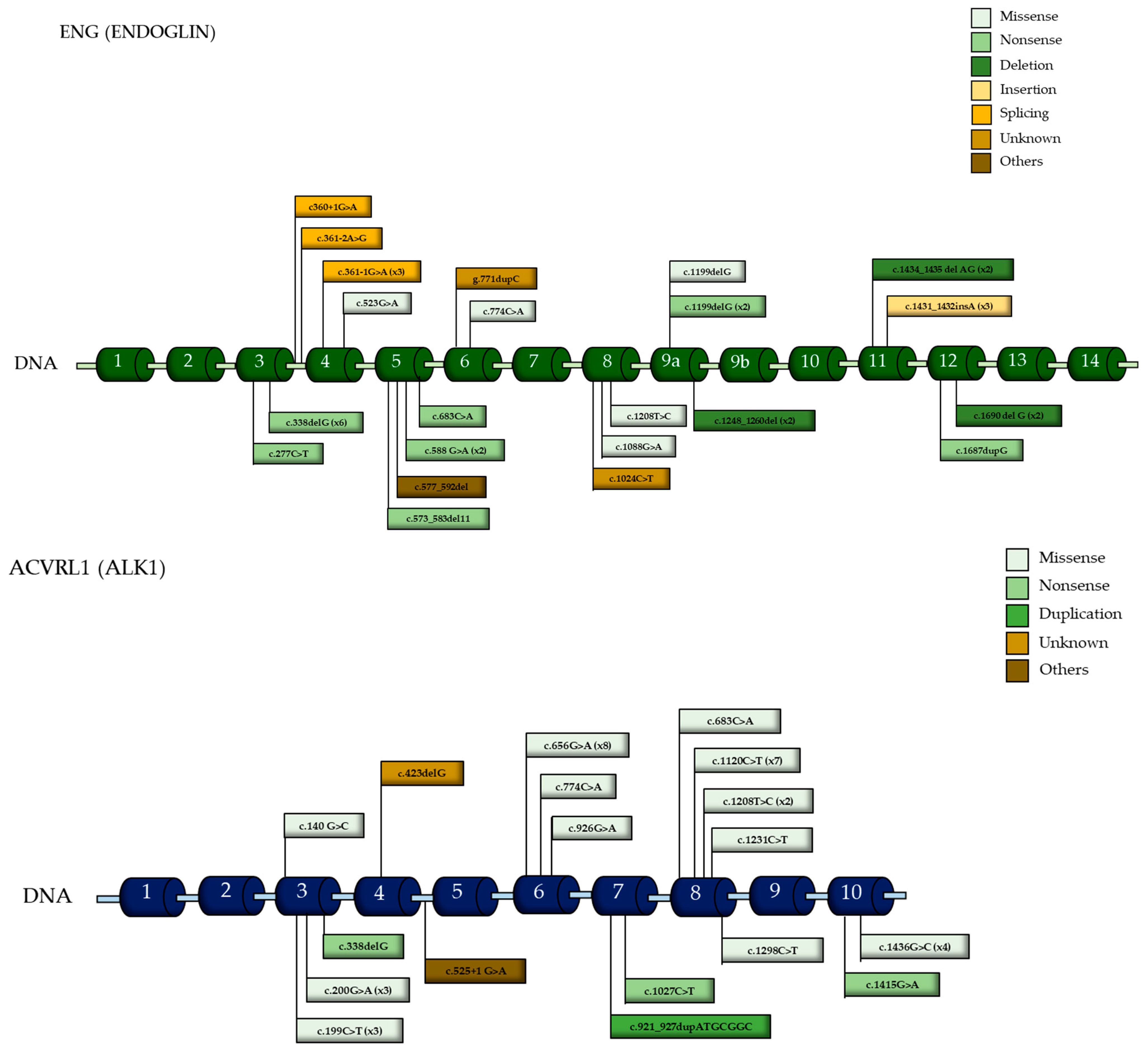
3.7. Mutational Variants and Their Implications
3.8. Cardiovascular Risk
3.9. Quality of Life and Phenotype Correlations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HHT | Hemorrhagic Hereditary Telangiectasia |
| ESS | Epistaxis Severity Score |
| GFR | Glomerular Filtration Rate |
| VM | Vascular Malformation |
| AVM | Arteriovenous Malformation |
| CNS | Central Nervous System |
References
- Federación Española de Enfermedades Raras s. f. Available online: https://enfermedades-raras.org/index.php/enfermedades-raras (accessed on 6 July 2021).
- Shovlin, C.L. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Rev. 2010, 24, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Zarrabeitia, R.; Fariñas-Álvarez, C.; Santibáñez, M.; Señaris, B.; Fontalba, A.; Botella, L.M.; Parra, J.A. Quality of life in patients with hereditary haemorrhagic telangiectasia (HHT). Health Qual. Life Outcomes 2017, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Faughnan, M.E.; Mager, J.J.; Hetts, S.W.; Palda, V.A.; Lang-Robertson, K.; Buscarini, E.; Deslandres, E.; Kasthuri, R.S.; Lausman, A.; Poetker, D.; et al. Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. Ann. Intern. Med. 2020, 173, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Hoag, J.B.; Terry, P.; Mitchell, S.; Reh, D.; Merlo, C.A. An Epistaxis Severity Score for Hereditary Hemorrhagic Telangiectasia. Laryngoscope 2010, 120, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Guttmacher, A.E.; Marchuk, D.A.; White, R.I., Jr. Hereditary Hemorrhagic Telangiectasia. N. Engl. J. Med. 1995, 333, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Khalid, S.K.; Garcia-tsao, G. Hepatic Vascular Malformations in Hereditary Hemorrhagic Telangiectasia. Semin. Liver Dis. 2008, 28, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Buscarini, E.; Gandolfi, S.; Alicante, S.; Londoni, C. Liver involvement in hereditary hemorrhagic telangiectasia. Abdom. Radiol. 2018, 43, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Canzonieri, C.; Centenara, L.; Ornati, F.; Pagella, F.; Matti, E.; Alvisi, C.; Danesino, C.; Perego, M.; Olivieri, C. Endoscopic evaluation of gastrointestinal tract in patients with hereditary hemorrhagic telangiectasia and correlation with their genotypes. Genet. Med. 2014, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, R.; Iriarte, A.; Mora-Luján, J.M.; Patier, J.L.; López-Wolf, D.; Ojeda, A.; Torralba, M.A.; Juyol, M.C.; Gil, R.; Añón, S.; et al. Current HHT genetic overview in Spain and its phenotypic correlation: Data from RiHHTa registry. Orphanet J. Rare Dis. 2020, 15, 138. [Google Scholar] [CrossRef]
- Lesca, G.; Olivieri, C.; Burnichon, N.; Pagella, F.; Carette, M.F.; Gilbert-Dussardier, B.; Goizet, C.; Roume, J.; Rabilloud, M.; Saurin, J.C.; et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: Data from the French-Italian HHT network. Genet. Med. 2007, 9, 14–22. [Google Scholar] [CrossRef]
- Kasthuri, R.S.; Montifar, M.; Nelson, J.; Helen, K.; Lawton, M.T.; Faughnan, M.E. Prevalence and Predictors of Anemia in Hereditary Hemorrhagic Telangiectasia. Am. J. Hematol. 2017, 92, E591–E593. [Google Scholar] [CrossRef]
- Rozenberg, D.; Leek, E.; Faughnan, M.E. Prevalence and nature of dyspnea in patients with hereditary hemorrhagic telangiectasia (HHT). Respir. Med. 2015, 109, 768–777. [Google Scholar] [CrossRef]
- Bodilsen, J. Hereditary haemorrhagic telangiectasia and pulmonary arteriovenous malformations in brain abscess patients: A nationwide, population-based matched cohort study. Clin. Microbiol. Infect. 2020, 26, 1093.e1–1093.e3. [Google Scholar] [CrossRef]
- Tellapuri, S.; Park, H.S.; Kalva, S.P. Pulmonary arteriovenous malformations. Int. J. Cardiovasc. Imaging 2019, 35, 1421–1428. [Google Scholar] [CrossRef]
- Bernabeu, C.; Bayrak-toydemir, P.; Mcdonald, J.; Letarte, M. Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. J Clin. Med. 2020, 9, 3571. [Google Scholar] [CrossRef]
- Eker, O.F.; Boccardi, E.; Sure, U.; Patel, M.C.; Alicante, S.; Alsafi, A.; Coote, N.; Droege, F.; Dupuis, O.; Fialla, A.D.; et al. European Reference Network for Rare Vascular Diseases (VASCERN) position statement on cerebral screening in adults and children with hereditary haemorrhagic telangiectasia (HHT). Orphanet J. Rare Dis. 2020, 15, 165. [Google Scholar] [CrossRef] [PubMed]
- Curnes, N.R.; Hung, M.L.; DePietro, D.M.; Ferrari, V.A.; Drivas, T.G.; Chittams, J.; Quinn, R.; Trerotola, S.O. Comparison of Transthoracic Contrast Echocardiography with High-Resolution Chest CT after Embolization of Pulmonary Arteriovenous Malformation. J. Vasc. Interv. Radiol. 2023, 34, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Schleer, H.; Park, H.; Jang, E.; Boyer, M.; Tao, B.; Gamez-Mendez, A.; Singh, A.; Folta-Stogniew, E.; Zhang, X.; et al. Genetic or therapeutic neutralization of ALK1 reduces LDL transcytosis and atherosclerosis in mice. Nat. Cardiovasc. Res. 2023, 2, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Zinellu, A.; Paliogiannis, P.; Mangoni, A.A. A systematic review and meta-analysis of the diagnostic accuracy of the neutrophil-to-lymphocyte ratio and the platelet-to-lymphocyte ratio in systemic lupus erythematosus. Clin. Exp. Med. 2024, 24, 170. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.M.; Kallogjeri, D.; Spitznagel, E.; Chakinala, M.M.; Schneider, J.S.; Piccirillo, J.F. Development and Validation of the Nasal Outcome Score for Epistaxis in Hereditary Hemorrhagic Telangiectasia (NOSE HHT). JAMA Otolaryngol. 2020, 146, 999–1005. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 85) | ENG (HHT1) (n = 43) | ACVRL1 (HHT2) (n = 42) | ORs * [IC95%] | p | |
|---|---|---|---|---|---|
| Age, years | - | 0.352 | |||
| Mean ± SD | 48.6 ± 14.3 | 47.2 ± 14.4 | 50.1 ± 14.3 | ||
| Range (min–max) | 20–76 | 20–74 | 22–76 | ||
| Women, n (%) | 50 (58.8%) | 25 (58.1%) | 25 (59.5%) | 0.94 [0.39 to 2.24] | 0.897 |
| Family history, n (%) | |||||
| Relatives affected by HHT | 78 (91.8%) | 38 (88.4%) | 40 (95.2%) | 0.38 [0.06 to 2.07] | 0.250 |
| Brother/sister | 48 (56.5%) | 20 (46.5%) | 28 (66.7%) | 0.43 [0.18 to 1.04] | 0.061 |
| Son/daughter | 28 (32.9%) | 16 (37.2%) | 12 (28.6%) | 1.48 [0.59 to 3.68] | 0.397 |
| Father/mother | 66 (77.6%) | 31 (72.1%) | 35 (83.3%) | 0.51 [0.18 to 1.47] | 0.214 |
| Grandfather/mother | 27 (31.2%) | 12 (27.9%) | 15 (35.7%) | 0.69 [0.27 to 1.74] | 0.440 |
| Grandson/daughter | 2 (2.4%) | 1 (2.3%) | 1 (2.4%) | 0.97 [0.05 to 16.1] | 0.987 |
| Deceased relatives related to the disease | 24 (28.2%) | 16 (18.8%) | 8 (9.4%) | 2.51 [0.93 to 6.76] | 0.063 |
| Causes of death of relatives | |||||
| Digestive bleeding | 10 (11.8%) | 6 (14.0%) | 4 (9.5%) | 1.54 [0.40 to 5.9] | 0.526 |
| Hemorrhagic stroke | 7 (8.2%) | 6 (14.0%) | 1 (2.4%) | 6.64 [0.76 to 57.8] | 0.052 * |
| Brain abscess | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | - | - |
| Other 1 | 11 (12.9%) | 7 (16.7%) | 4 (9.5%) | 1.84 [0.49 to 6.84] | 0.354 |
| Total (n = 85) | ENG (HHT1) (n = 43) | ACVRL1 (HHT2) (n = 42) | ORs * [IC95%] | p | |
|---|---|---|---|---|---|
| Epistaxis | 81 (95.3%) | 42 (97.7%) | 39 (92.9%) | 3.23 [0.32 to 32.3] | 0.294 |
| Clinical symptoms related to epistaxis, n (%) | |||||
| ESS | 2.49 ± 1.79 | 2.40 ± 1.66 | 2.68 ± 2.08 | - | 0.645 |
| Severity of epistaxis | 0.142 | ||||
| Severe (ESS ≥ 4) | 13 (15.3%) | 4 (9.3%) | 9 (21.4%) | 0.376 | |
| Mild/none (ESS < 4) | 72 (84.7%) | 39 (90.7%) | 33 (78.6%) | [0.106 to 1.333] | |
| Frequency of epistaxis | - | 0.531 | |||
| At least once a day | 21 (24.7%) | 10 (23.3%) | 11 (26.2%) | ||
| At least once a week | 39 (45.9%) | 18 (41.9%) | 21 (50.0%) | ||
| At least once a month | 25 (29.4%) | 15 (34.9%) | 10 (23.8%) | ||
| Duration of epistaxis | - | 0.006 | |||
| More than 5 min | 16 (18.8%) | 7 (16.3%) | 9 (21.4%) | ||
| 1–5 min | 41 (48.2%) | 15 (34.9%) | 26 (61.9%) | ||
| Less than 1 min | 28 (32.9%) | 21 (48.8%) | 7 (16.7%) | ||
| Intensity of epistaxis | 0.676 [0.258 to 1.772] | 0.425 | |||
| Sheets of blood | 23 (27.1%) | 10 (23.3%) | 13 (31.0%) | ||
| Drips of blood | 62 (72.9%) | 33 (76.7%) | 29 (69.0%) | ||
| Needs to visit the emergency room due to epistaxis | 3 (3.5%) | 2 (4.7%) | 1 (2.4%) | 2000 [0.174 to 22.927] | 1.000 |
| Currently anemic | 10 (11.8%) | 5 (11.6%) | 5 (11.9%) | 0.974 [0.260 to 3.644] | 0.968 |
| Needs transfusions | 1 (1.2%) | 0 (0.0%) | 1 (2.4%) | - | 0.494 |
| Total (n = 85) | ENG (HHT1) (n = 43) | ACVRL1 (HHT2) (n = 42) | ORs * [IC95%] | p | |
|---|---|---|---|---|---|
| Internal organ involvement, n (%) | |||||
| Pulmonary AVMs | 29 (34.1%) | 25 (58.1%) | 4 (9.5%) | 13.19 [3.9 to 43.5] | <0.001 |
| Age at diagnosis of pulmonary AVMs, years, median [IQR] | 39.0 [24.0] | 39.0 [25.0] | 41.5 [20.3] | - | 0.635 |
| CNS VMs | 12 (14.1%) | 9 (20.9%) | 3 (7.1%) | 3.44 [0.86 to 3.75] | 0.068 |
| Brain VMs | 11 (12.9%) | 8 (18.6%) | 3 (7.1%) | 2.97 [0.7 to 12.08] | 0.115 |
| Spinal VMs | 1 (1.2%) | 1 (2.3%) | 0 (0.0%) | - | 0.320 |
| Age at diagnosis of CNS VMs, years, median [IQR] | 28.0 [10.0] | 26.0 [8.0] | 32.0 [6.0] | - | 0.630 |
| Hepatic VMs | 15 (17.6%) | 5 (11.6%) | 10 (23.8%) | 0.42 [0.13 to 1.35] | 0.141 |
| Age at diagnosis of hepatic VMs, years, median [IQR] | 48.5 [19.5] | 41.0 [24.0] | 49.0 [17.8] | - | 0.304 |
| Digestive VMs | 10 (11.8%) | 4 (9.3%) | 6 (14.3%) | 0.61 [0.16 to 2.35] | 0.476 |
| Age at diagnosis of digestive VMs, years, median [IQR] | 57.5 [19.3] | 56.5 [13.0] | 58.0 [29.0] | - | 0.610 |
| All types of VMs | 44 (51.8%) | 27 (62.8%) | 17 (40.5%) | 2.48 [1.03 to 5.94] | 0.040 |
| Age at diagnosis of VMs, years, median [IQR] | 39.0 [25.0] | 37.0 [26.0] | 41.0 [22.0] | - | 0.198 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viteri-Noël, A.; Patier, J.L.; Bara-Ledesma, N.; González-García, A.; Fabregate, M.; Fernández-San Jose, P.; López-Rodriguez, M.; Manzano, L.; Gómez del Olmo, V. Genotype–Phenotype Relationship in Hereditary Hemorrhagic Telangiectasia: Quality of Life and Cardiovascular Risk Evaluation. J. Clin. Med. 2025, 14, 4409. https://doi.org/10.3390/jcm14134409
Viteri-Noël A, Patier JL, Bara-Ledesma N, González-García A, Fabregate M, Fernández-San Jose P, López-Rodriguez M, Manzano L, Gómez del Olmo V. Genotype–Phenotype Relationship in Hereditary Hemorrhagic Telangiectasia: Quality of Life and Cardiovascular Risk Evaluation. Journal of Clinical Medicine. 2025; 14(13):4409. https://doi.org/10.3390/jcm14134409
Chicago/Turabian StyleViteri-Noël, Adrián, José Luis Patier, Nuria Bara-Ledesma, Andrés González-García, Martin Fabregate, Patricia Fernández-San Jose, Mónica López-Rodriguez, Luis Manzano, and Vicente Gómez del Olmo. 2025. "Genotype–Phenotype Relationship in Hereditary Hemorrhagic Telangiectasia: Quality of Life and Cardiovascular Risk Evaluation" Journal of Clinical Medicine 14, no. 13: 4409. https://doi.org/10.3390/jcm14134409
APA StyleViteri-Noël, A., Patier, J. L., Bara-Ledesma, N., González-García, A., Fabregate, M., Fernández-San Jose, P., López-Rodriguez, M., Manzano, L., & Gómez del Olmo, V. (2025). Genotype–Phenotype Relationship in Hereditary Hemorrhagic Telangiectasia: Quality of Life and Cardiovascular Risk Evaluation. Journal of Clinical Medicine, 14(13), 4409. https://doi.org/10.3390/jcm14134409