

Clinical Phenotypes of a Pediatric Cohort with GDF2-Related Hereditary Hemorrhagic Telangiectasia
 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
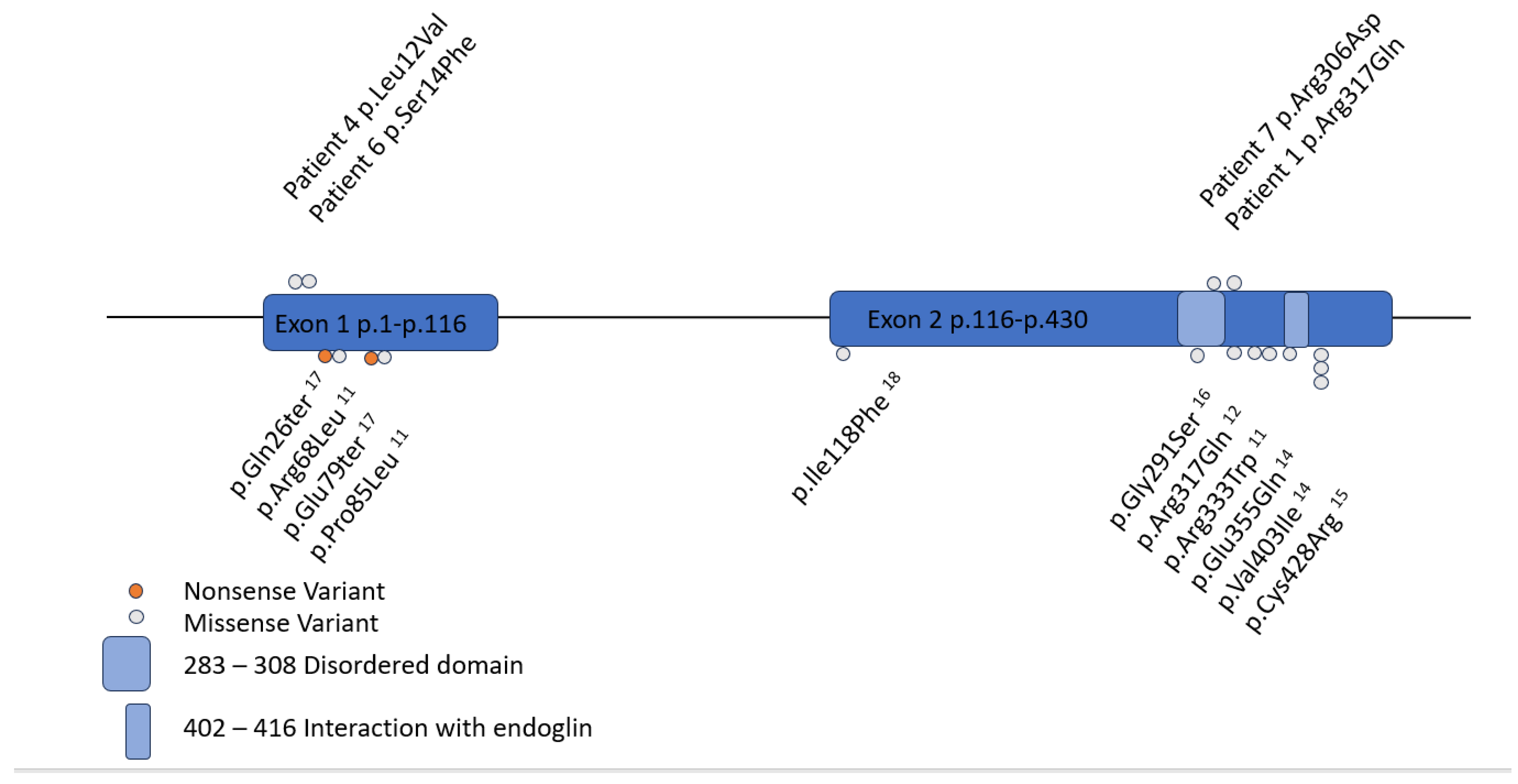
3. Results
3.1. Case Series
3.1.1. Patient 1
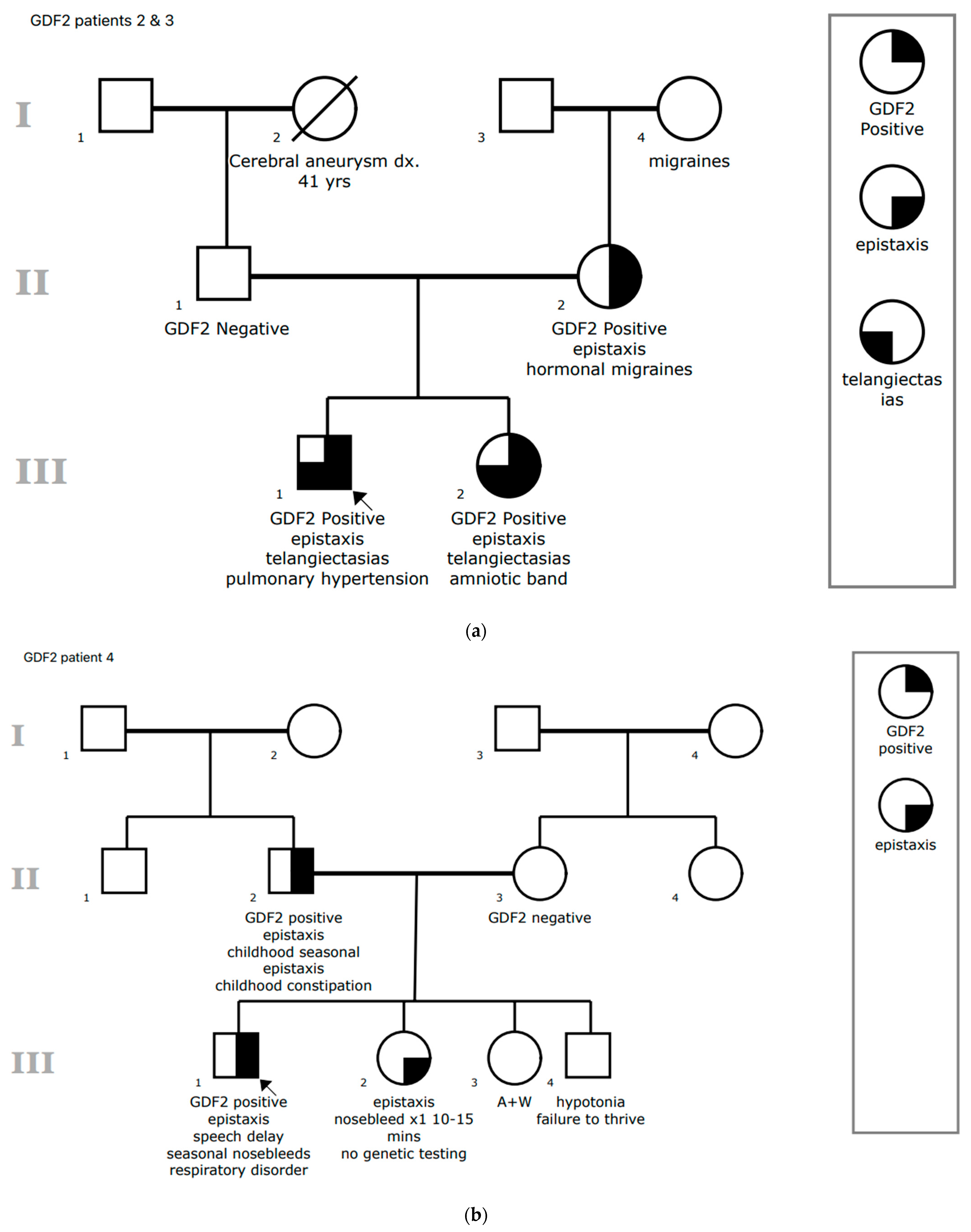
3.1.2. Patient 2
3.1.3. Patient 3
3.1.4. Patient 4
3.1.5. Patient 5
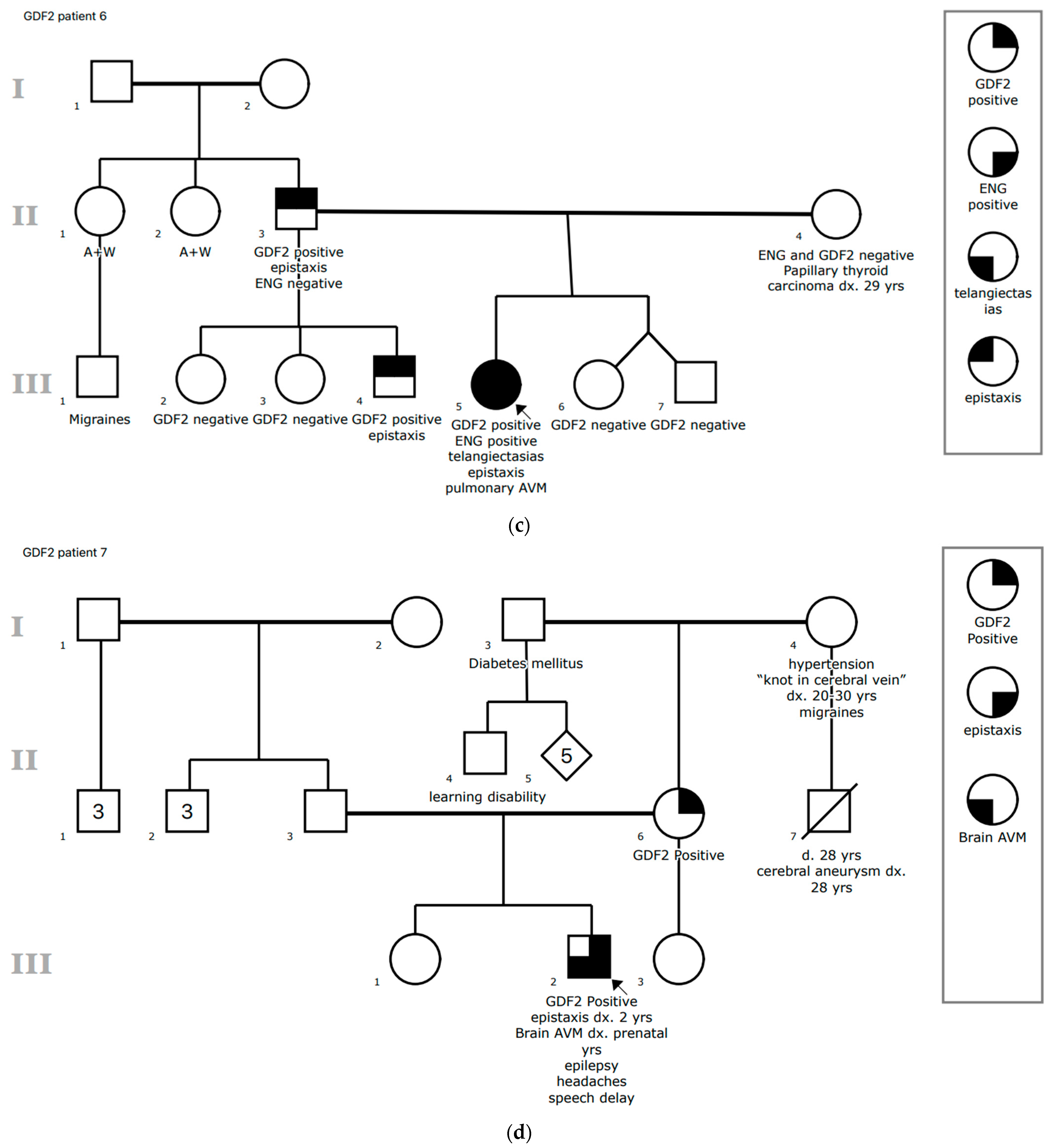
3.1.6. Patient 6
3.1.7. Patient 7
3.2. Literature Review
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HHT | Hereditary hemorrhagic telangiectasia |
| AVM | Arteriovenous malformation |
| VUS | Variant of uncertain significance |
| BMP9 | Bone morphogenetic protein |
| CHOP | Children’s Hospital of Philadelphia |
| EHR | Electronic health record |
| ASD | Atrial septal defect |
| NR | Not reported |
| SVT | Supraventricular tachycardia |
| VSD | Ventricular septal defect |
| VM | Vascular malformation |
References
- Faughnan, M.E.; Mager, J.J.; Hetts, S.W.; Palda, V.A.; Lang-Robertson, K.; Buscarini, E.; Deslandres, E.; Kasthuri, R.S.; Lausman, A.; Poetker, D.; et al. Second international guidelines for the diagnosis and management of hereditary hemorrhagic telangiectasia. Ann. Intern. Med. 2020, 173, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Hetts, S.W.; Shieh, J.T.; Ohliger, M.A.; Conrad, M.B. Hereditary hemorrhagic telangiectasia: The convergence of genotype, phenotype, and imaging in modern diagnosis and management of a multisystem disease. Radiology 2021, 300, 17–30. [Google Scholar] [CrossRef]
- McDonald, J.; Wooderchak-Donahue, W.; VanSant Webb, C.; Whitehead, K.; Stevenson, D.A.; Bayrak-Toydemir, P. Hereditary hemorrhagic telangiectasia: Genetics and molecular diagnostics in a new era. Front. Genet. 2015, 6, 1. [Google Scholar] [CrossRef]
- McDonald, J.; Pyeritz, R.E. Hereditary hemorrhagic telangiectasia. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1351/ (accessed on 5 April 2020).
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am. J. Med. Genet. 2000, 91, 66–67. [Google Scholar] [CrossRef]
- Shovlin, C.L. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Rev. 2010, 24, 203–219. [Google Scholar] [CrossRef]
- Govani, F.S.; Shovlin, C.L. Hereditary haemorrhagic telangiectasia: A clinical and scientific review. Eur. J. Hum. Genet. 2009, 17, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Audiger, C.; Popovic, N.; Akla, N.; Lanthier, K.; Legault-Navarrete, I.; Melichar, H.; Costantino, S.; Lesage, S.; Larrivée, B. BMP9 signaling promotes the normalization of tumor blood vessels. Oncogene 2020, 39, 2996–3014. [Google Scholar] [CrossRef] [PubMed]
- Medina-Jover, F.; Riera-Mestre, A.; Viñals, F. Rethinking growth factors: The case of BMP9 during vessel maturation. Vasc. Biol. 2022, 4, R1–R14. [Google Scholar] [CrossRef]
- Robert, F.; Desroches-Castan, A.; Bailly, S.; Dupuis-Girod, S.; Feige, J.-J. Future treatments for hereditary hemorrhagic telangiectasia. Orphanet J. Rare Dis. 2020, 15, 4. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.L.; McDonald, J.; O’Fallon, B.; Upton, P.D.; Li, W.; Roman, B.L.; Young, S.; Plant, P.; Fülöp, G.T.; Langa, C.; et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 2013, 93, 530–537. [Google Scholar] [CrossRef]
- Hernandez, F.; Huether, R.; Carter, L.; Johnston, T.; Thompson, J.; Gossage, J.R.; Chao, E.; Elliott, A.M. Mutations in RASA1 and GDF2 identified in patients with clinical features of hereditary hemorrhagic telangiectasia. Hum. Genome Var. 2015, 2, 15040. [Google Scholar] [CrossRef] [PubMed]
- Richards-Yutz, J.; Grant, K.; Chao, E.C.; Walther, S.E.; Ganguly, A. Update on molecular diagnosis of hereditary hemorrhagic telangiectasia. Hum. Genet. 2010, 128, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Farhan, A.; Yuan, F.; Partan, E.; Weiss, C.R. Clinical manifestations of patients with GDF2 mutations associated with hereditary hemorrhagic telangiectasia type 5. Am. J. Med. Genet. A 2022, 188, 199–209. [Google Scholar] [CrossRef]
- Balachandar, S.; Graves, T.J.; Shimonty, A.; Kerr, K.; Kilner, J.; Xiao, S.; Slade, R.; Sroya, M.; Alikian, M.; Curetean, E.; et al. Identification and validation of a novel pathogenic variant in GDF2 (BMP9) responsible for hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations. Am. J. Med. Genet. A 2022, 188, 959–964. [Google Scholar] [CrossRef]
- Topiwala, K.K.; Patel, S.D.; Nouh, A.M.; Alberts, M.J. Novel GDF2 gene mutation associated with pulmonary arteriovenous malformation. J. Stroke Cerebrovasc. Dis. 2020, 29, 105301. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.; Ruiz-Llorente, L.; McDonald, J.; Quarrell, O.; Ugonna, K.; Bentham, J.; Mason, R.; Martin, J.; Moore, D.; Bergstrom, K.; et al. Homozygous GDF2 nonsense mutations result in a loss of circulating BMP9 and BMP10 and are associated with either PAH or an “HHT-like” syndrome in children. Mol. Genet. Genom. Med. 2021, 9, e1685. [Google Scholar] [CrossRef]
- Ma, L.; Peng, X.; Gong, Q. A GDF2 missense mutation potentially involved in the pathogenesis of hereditary hemorrhagic telangiectasia: A case report. J. Int. Med. Res. 2023, 51, 1–8. [Google Scholar] [CrossRef]
- Yang, M.; Liang, Z.; Yang, M.; Jia, Y.; Yang, G.; He, Y.; Li, X.; Gu, H.F.; Zheng, H.; Zhu, Z.; et al. Role of bone morphogenetic protein-9 in the regulation of glucose and lipid metabolism. FASEB J. 2019, 33, 10077–10088. [Google Scholar] [CrossRef]
- Hodgson, J.; Swietlik, E.M.; Salmon, R.M.; Hadinnapola, C.; Nikolic, I.; Wharton, J.; Guo, J.; Liley, J.; Haimel, M.; Bleda, M.; et al. Characterization of GDF2 mutations and levels of BMP9 and BMP10 in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, 575–585. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Pahl, K.S.; Choudhury, A.; Wusik, K.; Hammill, A.; White, A.; Henderson, K.; Pollak, J.; Kasthuri, R.S. Applicability of the curaçao criteria for the diagnosis of hereditary hemorrhagic telangiectasia in the pediatric population. J. Pediatr. 2018, 197, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Kilian, A.; Latino, G.A.; White, A.J.; Clark, D.; Chakinala, M.M.; Ratjen, F.; McDonald, J.; Whitehead, K.J.; Gossage, J.R.; Lin, D.; et al. Genotype–phenotype correlations in children with HHT. J. Clin. Med. 2020, 9, 2714. [Google Scholar] [CrossRef] [PubMed]
- Sabbà, C.; Pasculli, G.; Lenato, G.M.; Suppressa, P.; Lastella, P.; Memeo, M.; Dicuonzo, F.; Guanti, G. Hereditary hemorrhagic telangiectasia: Clinical features in ENG and ALK1 mutation carriers. J. Thromb. Haemost. 2007, 5, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Hunter, B.N.; Timmins, B.H.; McDonald, J.; Whitehead, K.J.; Ward, P.D.; Wilson, K.F. An evaluation of the severity and progression of epistaxis in hereditary hemorrhagic telangiectasia 1 versus hereditary hemorrhagic telangiectasia 2. Laryngoscope 2016, 126, 786–790. [Google Scholar] [CrossRef]
- Gonzalez, C.D.; Cipriano, S.D.; Topham, C.A.; Stevenson, D.A.; Whitehead, K.J.; Vanderhooft, S.; Presson, A.P.; McDonald, J. Localization and age distribution of telangiectases in children and adolescents with hereditary hemorrhagic telangiectasia: A retrospective cohort study. J. Am. Acad. Dermatol. 2019, 81, 950–955. [Google Scholar] [CrossRef]
- McDonald, J.; Kornish, J.; Stevenson, D.A.; Hanson-Kahn, A.; Balch, H.; James, J.; Naik, H.; Whitehead, K.J. Frequency of epistaxis and telangiectasia in patients with hereditary hemorrhagic telangiectasia (HHT) in comparison with the general population: Curaçao diagnostic criteria revisited. Genet. Med. 2023, 25, 100865. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.L.; McDonald, J.; Farrell, A.; Akay, G.; Velinder, M.; Johnson, P.; VanSant-Webb, C.; Margraf, R.; Briggs, E.; Whitehead, K.J.; et al. Genome sequencing reveals a deep intronic splicing ACVRL1 mutation hotspot in Hereditary Haemorrhagic Telangiectasia. J. Med. Genet. 2018, 55, 824–830. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Patient | Age at Clinical Presentation | GDF2 Genetic Variant | Additional Genetic Variation | Curaçao Criteria (#) | Pulmonary Hypertension | Other Clinical Features |
|---|---|---|---|---|---|---|
| 1 | 3 | c.950C>A p.Arg317Gln VUS heterozygous | None | Epistaxis, mucocutaneous telangiectasias (2) | No | None |
| 2 | 6 | Pathogenic deletion (entire coding sequence) | BMPR1B: c.1355C>T | Epistaxis, mucocutaneous telangiectasia (single) (1) | Yes | Asthma, heart murmur |
| 3 | 6 | Pathogenic deletion (entire coding sequence) | None | Epistaxis, mucocutaneous telangiectasias (2) | No | Left distal foot truncated and abnormal digits from amniotic band syndrome in utero, dysplastic pulmonary valve |
| 4 | 9 | c.34C>G p.Leu12Val VUS heterozygous | None | Epistaxis (1) | No | Asthma, chronic pulmonary symptoms, pneumothorax |
| 5 | 4 | Pathogenic deletion (entire GDF2 gene deleted) 1 | SLC2A1 c.1387A>T p.Ile463Phe MT-ND4, m.11580C>A p.S274&, 2% heteroplasmy VUS | Epistaxis (1) | No | Possible seizures, behavioral problems, intellectual disability |
| 6 | 6 | c.41C>T p.Ser14Phe VUS heterozygous | ENG deletion (Exons 9–14) | Epistaxis, mucocutaneous telangiectasias, lung AVMs (3) | No | ADHD, celiac disease, insomnia, closed lip schizencephaly |
| 7 | Prenatal | c.917G>A p.Gly306Asp VUS heterozygous | None | Epistaxis, vein of Galen malformation (2) | Yes (Secondary to shunting) | Epilepsy, headaches, speech delay |
| Patient (Reference) | Age at First Symptoms (Years) | GDF2 Genetic Variant Classification | Curaçao Criteria (#) * | First-Degree Family Member HHT Symptoms | Pulmonary Hypertension | Other Clinical Features |
|---|---|---|---|---|---|---|
| 1 [14] | 37 | c.1063G>C p.Glu355Gln missense VUS | Epistaxis, mucocutaneous telangiectasias, liver AVM (4) * | Mother with same VUS—epistaxis, telangiectasias, colonic AVM | No | Diabetes, asthma, paroxysmal SVT, mitral valve regurgitation |
| 2 [14] | 9 | c.1207G>A p.Val403Ile missense VUS | Epistaxis, brain AVMs (2) | Mother—epistaxis | No | None |
| Son—spider veins/possible telangiectasias, epistaxis | ||||||
| 3 [14] | 5 | 5.10 Mb deletion from 10q11.22 to 10q11.23 deletion likely pathogenic | Epistaxis (1) | Unknown—adopted | No | Headaches/migraines, ADHD, SVT |
| 4 [14] | 2 | 5.50 Mb deletion from 10q11.22 to 10q11.23 likely pathogenic | Epistaxis, mucocutaneous telangiectasias, brain capillary malformations, developmental venous anomaly (3) | Not reported | No | Right ventricular hypoplasia, large posterior VSD, secundum ASD |
| 5 [11] | Early childhood | c.254C>T p.Pro85Leu missense pathogenic | Epistaxis, mucocutaneous telangiectasias (2) | Father—autopsy findings suggested HHT but no report available | No | None |
| Sibling—epistaxis, stroke at age 43 of unknown cause | ||||||
| 6 [11] | 30 | c.203G>T p.Arg68Leu missense pathogenic | Epistaxis, mucocutaneous telangiectasias (2) | Father with same VUS—epistaxis | No | Hepatopulmonary syndrome—hepatic findings consistent with likely hepatic VM but not confirmed |
| Sister with same VUS—epistaxis | ||||||
| 7 [11] | 3 | c.997C>T p.Arg333Trp missense VUS | Epistaxis, mucocutaneous telangiectasias (2) | Father—epistaxis | No | None |
| 8 [15] ± | Early childhood | c.1282T>C p.Cys428Arg missense VUS | Epistaxis, mucocutaneous telangiectasias, lung AVM (3) | Mother with same VUS—epistaxis, telangiectasias | No | Cirrhosis |
| Sister with same VUS—epistaxis, telangiectasias | ||||||
| 9 [15] ± | 30s | c.1282T>C p.Cys428Arg missense VUS | Epistaxis, mucocutaneous telangiectasias (2) * | Mother with same VUS—epistaxis, telangiectasias | No | None |
| Brother with same VUS—epistaxis, telangiectasias, lung AVM | ||||||
| 10 [15] ± | Childhood | c.1282T>C p.Cys428Arg missense VUS | Epistaxis, mucocutaneous telangiectasias (2) * | Son with same VUS—epistaxis, telangiectasias, lung AVM | No | None |
| Daughter with same VUS—epistaxis, telangiectasias | ||||||
| 11 [12] | Not reported | c.950G>A p.Arg317Gln missense VUS | Epistaxis, mucocutaneous telangiectasias, lung AVM (3) | Sons (2)—epistaxis | No | None |
| 12 [18] | Likely childhood | c.352A>T p.Ile118Phe missense VUS | Epistaxis, mucocutaneous telangiectasias, GI AVMs with anemia (3) | Brother and sister—“HHT-related symptoms” that were not specified, both died of anemia | Yes | None |
| 13 [16] | 43 | c.871G>A p.Gly291Ser missense VUS | Lung AVM with stroke (1) | No | No | None |
| 14 [17] | 3 | c.76C>T; p.Gln26Ter nonsense homozygous pathogenic | Skin telangiectasias (1) | Parents heterozygous with no symptoms or clinical signs suggestive of HHT | Yes | Severely dilated right ventricle |
| 15 [17] | 9 | c.835G>T p.Glu279Ter nonsense homozygous VUS | Epistaxis, skin telangiectasias, lung AVM (3) | Parents heterozygous with no symptoms or clinical signs suggestive of HHT, both parents had negative testing for lung AVMs | No | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliver, O.; Britt, A.D.; Borst, A.J.; Goldmuntz, E.; Bakeer, N.; Lang, S.-s.; Fuller, S.; Vossough, A.; Beslow, L.A. Clinical Phenotypes of a Pediatric Cohort with GDF2-Related Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2025, 14, 3359. https://doi.org/10.3390/jcm14103359
Oliver O, Britt AD, Borst AJ, Goldmuntz E, Bakeer N, Lang S-s, Fuller S, Vossough A, Beslow LA. Clinical Phenotypes of a Pediatric Cohort with GDF2-Related Hereditary Hemorrhagic Telangiectasia. Journal of Clinical Medicine. 2025; 14(10):3359. https://doi.org/10.3390/jcm14103359
Chicago/Turabian StyleOliver, Owen, Allison D. Britt, Alexandra J. Borst, Elizabeth Goldmuntz, Nihal Bakeer, Shih-shan Lang, Stephanie Fuller, Arastoo Vossough, and Lauren A. Beslow. 2025. "Clinical Phenotypes of a Pediatric Cohort with GDF2-Related Hereditary Hemorrhagic Telangiectasia" Journal of Clinical Medicine 14, no. 10: 3359. https://doi.org/10.3390/jcm14103359
APA StyleOliver, O., Britt, A. D., Borst, A. J., Goldmuntz, E., Bakeer, N., Lang, S.-s., Fuller, S., Vossough, A., & Beslow, L. A. (2025). Clinical Phenotypes of a Pediatric Cohort with GDF2-Related Hereditary Hemorrhagic Telangiectasia. Journal of Clinical Medicine, 14(10), 3359. https://doi.org/10.3390/jcm14103359