Abstract
Fabry disease (FD) is a lysosomal storage disorder caused by pathogenic variants in the gene encoding alpha-galactosidase A (GLA). Deficiency of GLA results in the progressive accumulation of glycosphingolipids in virtually all organs, resulting in a progressive multisystem disease. Due to multi-organ involvement in FD, a comprehensive, multidisciplinary approach to diagnosis and treatment with regular follow-ups is essential. The Pusan National University Yangsan Hospital (PNUYH) multidisciplinary care model of FD aims to provide detailed practice guidelines and evidence-based recommendations for the diagnosis, screening, and treatment of FD according to specialty. This guideline focuses on the “quarterback” type of multidisciplinary team (MDT) operation and is limited in its applicability to the Korean insurance system. However, it reflects our team’s extensive experience and insights into optimizing MDT operations within these constraints and is expected to be highly beneficial for centers initiating MDTs for the effective treatment of FD.
1. Introduction
Fabry disease (FD) is a lysosomal storage disorder caused by pathogenic variants in the gene encoding alpha-galactosidase A (GLA) [1]. Deficiency of GLA results in the inability of cells to catabolize glycosphingolipids with terminal α-D-galactosyl residues [2]. Glycosphingolipids, particularly globotriaosylceramide (Gb3), progressively accumulate in virtually all organs, resulting in a progressive multisystem disease. The cardinal features of the disease are acroparesthesia (burning pain in hands and feet), progressive proteinuric renal insufficiency, and cardiac disease consisting of rhythm and conduction disturbances and progressive cardiac hypertrophy, as well as cerebrovascular stroke.
FD is characterized by gradually deteriorating functions over time as the disease invades various organs of the body. The involvement of vital organs such as the heart, kidneys, and brain leads to critical and potentially fatal complications, including organ failure and stroke. Furthermore, peripheral neuropathies and non-specific symptoms, such as gastrointestinal or otorhinolaryngological symptoms, may significantly impair the quality of life of patients with FD.
A multidisciplinary team (MDT) approach to treatment involving experts in various fields is crucial for managing FD, considering its complexity, rarity, and multi-systemic nature. The MDT approach is important for an accurate and comprehensive diagnosis, personalized and holistic care, improved coordination and efficacy, enhanced collaboration and innovation, and to address systemic challenges [3,4]. It also ensures patient access to cutting-edge treatments while reducing the burden on families. This collaborative treatment model is essential for improving outcomes in a field where individual experts alone often fall short.
For the first time, a Fabry center of excellence was started in 2021 by the Pusan National University Yangsan Hospital (PNUYH) in Korea to discuss challenging patient cases with diverse clinical courses of FD. Since then, specialists from cardiology, nephrology, neurology, ophthalmology, dermatology, otolaryngology, pathology, pain medicine, digestive medicine, and psychiatry have joined the team for in-depth discussion of research and case studies of FD and to collate their experience and insights into a management guideline for Korean patients with FD.
This guideline aims to provide practical guidance for an MDT approach to the treatment of FD and to improve communication between experts involved in diagnosing and treating FD. Recommendations in the guideline are based on comprehensive literature reviews and clinical treatment experiences. For specialties where sufficient data or evidence regarding the diagnosis and treatment of FD is not available, the level of recommendations is not specified.
Summary of Key Recommendations according to Clinical Specialty
| Genetic Medicine Diagnosis
|
Cardiology
|
Nephrology
|
| Neurology Patients with FD are at high risk for atherosclerotic cardiovascular disease and should follow a comprehensive stroke prevention strategy, especially in ischemic stroke or transient ischemic attack (TIA). Recommendations for secondary prevention of stroke follow American Heart Association/American Stroke Association (AHA/ASA) guidelines. Management of risk factors for stroke
|
Gastroenterology
|
Otorhinolaryngology
|
Ophthalmology
|
Pain Management
|
2. Diagnosis and Follow-Up Assessments
2.1. Genetics
Effective diagnosis and follow-up of FD in a medical genetic department requires a comprehensive multidisciplinary approach. Early diagnosis through enzyme activity testing and genetic analysis, combined with regular monitoring of organ function and biomarkers, is essential for optimal patient management. By implementing a structured follow-up protocol, clinicians can reliably track disease progression, adjust treatments, and improve the quality of life of patients with FD.
FD manifests with a wide spectrum of symptoms, including acroparesthesia, angiokeratoma (small, dark red spots on the skin), hypohidrosis (decreased ability to sweat), corneal opacity, gastrointestinal issues, tinnitus and hearing loss, progressive kidney failure, heart problems, and stroke. Symptoms occur widely across the body, necessitating a multidisciplinary approach to recognizing and managing them.
The diagnostic approach for FD is gender-specific due to its X-linked inheritance pattern. The diagnostic algorithm can be summarized as follows:
For males:
- Measure GLA enzyme activity in blood, with GLA activity < 1% being highly suggestive of classic FD.
- Genetic testing involves sequencing the GLA gene to confirm the diagnosis.
For females:
- Genetic testing by sequencing the GLA gene is mandatory.
- Measurement of GLA enzyme activity is optional, as the activity may be within the normal range due to random X-chromosome inactivation.
Assessment of biomarkers such as globotriaosylsphingosine (lyso-Gb3) is important for identifying atypical FD variants. High-sensitive troponin T (hsTNT) may be useful for identifying cardiac involvement.
The Korean reimbursement system provides the following diagnostic criteria for initiating enzyme replacement therapy (ERT) (Table 1). In our center, patient eligibility for treatment is verified based on these criteria and the following tests are conducted to accurately assess the involvement of various organs.

Table 1.
Reimbursement criteria for enzyme replacement therapy (ERT) in South Korea [5].
Initial evaluation of FD should include the following items:
- Comprehensive medical history and physical examination.
- Family history assessment.
- Baseline organ function tests:
- ✓
- Renal function: proteinuria, microalbuminuria, estimated glomerular filtration rate (eGFR)
- ✓
- Cardiac function: echocardiography, electrocardiography (ECG), cardiac magnetic resonance imaging (CMRI)
- ✓
- Neurological assessment: Brain magnetic resonance imaging (MRI), nerve conduction studies
- ✓
- Ophthalmological examination
- ✓
- Audiology assessment
For follow-up monitoring of FD, the following should be included:
- Regular follow-up visits (frequency depends on disease severity and treatment status)
- Periodic organ function assessments:
- ✓
- Renal function: Every 6–12 months
- ✓
- Cardiac function: Annually or more frequently if indicated
- ✓
- Neurological assessment: Annually
- ✓
- Ophthalmological and audiology examinations: Annually
- Biomarker monitoring:
- ✓
- Lyso-Gb3 levels: To assess treatment response and disease progression
- ✓
- hsTNT of TNI: For cardiac involvement
- Genetic counseling:
- ✓
- Discuss inheritance pattern and implications for family members
- ✓
- Genetic testing should be offered to at-risk relatives
Detailed guidance for initial evaluation and monitoring is discussed below according to specialty.
2.2. Cardiology
The prevalence of FD in patients with unexplained LVH ranges from 12 to 18% in highly selected cohorts, but most studies suggest a prevalence of approximately 0.5% to 1% in adult patients [6,7,8]. To evaluate cardiac function in patients with FD, various test modalities can be utilized:
- ECG and Holter monitoring: A short PR interval due to increased atrioventricular (AV) conduction without evidence of an accessory pathway is the earliest ECG feature in the subclinical stage of FD cardiomyopathy [9]. Voltage signs of LVH, strain pattern, and T-wave inversion in the precordial leads are the usual findings when overt FD cardiomyopathy has developed. In progressed overt FD cardiomyopathy, sinus bradycardia and advanced conduction abnormalities in the AV node/His bundle and distal conduction system are common and suggest an adverse prognosis [10]. Therefore, serial ECG follow-up and side-by-side comparison of ECGs are necessary for assessing disease progression. According to a study by Shah et al. published in 2005, non-sustained ventricular tachycardia (NSVT), defined as three or more consecutive ventricular premature beats at a rate of ≥100 beats per minute lasting less than 30 s, was a common finding on Holter monitoring in patients with Fabry disease—observed in 38% of males and 39% of females aged 60 years or older [11]. The prevalence of malignant ventricular arrhythmias increases with age and is associated with the extent of late gadolinium enhancement (LGE) on cardiac magnetic resonance imaging (CMRI) [12]. Although, as in hypertrophic cardiomyopathy, the relationship between NSVT and the risk of sudden cardiac death (SCD) remains unclear, prophylactic strategies for SCD should be considered in cases of NSVT detected in Holter monitoring, accompanied by LGE [7].
- Echocardiography is the most useful tool for diagnosing and monitoring cardiac involvement of FD. The typical findings are left ventricular (LV), right ventricular (RV), or papillary muscle hypertrophy, sometimes mimicking diffuse-type hypertrophic cardiomyopathy. However, asymmetric thickening of the interventricular septum or apical hypertrophy cannot exclude FD cardiomyopathy [7]. Myocardial strain and strain rate are useful for detecting early changes in non-overt FD cardiomyopathies. In treatment naïve patients with FD, early reduction of LV longitudinal strain (particularly basal segments) can develop without clear-cut LVH [13]. Valves can also be involved, as well as the myocardium. The mitral and aortic valves are often thickened, with mild-to-moderate regurgitation, whereas stenotic lesions are rare. As myocardial fibrosis develops during disease progression, paradoxical thinning and hypokinetic or akinetic movement of the involved segments can be observed. These findings suggest that severe and irreversible changes are associated with poor prognosis. According to a study conducted in Taiwan, 38.1% of males and 16.7% of females with late-onset FD had cardiac fibrosis, even without LVH [14]. The echocardiographic parameters that must be assessed are LV mass/mass index, velocity of the septal mitral annulus in early diastole (septal e’), ratio between early mitral inflow velocity and mitral annular early diastolic velocity (E/e’), maximal tricuspid regurgitation velocity (TR Vmax), right ventricular systolic excursion velocity (RV s’), tricuspid annular plane systolic excursion (TAPSE), pulmonary artery systolic pressure (PASP), global longitudinal strain (GLS), and isovolumic relaxation time (IVRT), in addition to routine parameters.
- CMRI provides an accurate assessment of LV size, mass, and geometry. The gadolinium contrast images visualize the localization and extent of myocardial fibrosis. The presence of extensive fibrosis is associated with a reduced response to ERT and an increased risk of arrhythmia [7]. CMRI can be used for differential diagnosis as well as for the prediction of prognosis. CMRI can also be used safely in patients with advanced chronic kidney disease and in patients undergoing hemodialysis. Native T1 mapping values (i.e., measured without contrast media such as gadolinium) are lower in patients with FD than in patients with normal myocardium, possibly due to sphingolipid accumulation [15].
- Although endomyocardial biopsy (EMB, Figure 1) is not recommended to determine treatment efficacy or to follow up cardiac involvement, it can be considered as a confirmatory diagnostic tool in patients with a variant of uncertain significance (VUS) or suspected cardiac variant of FD [7]. In addition, for patients who are not able to discontinue anticoagulation, EMB could be a good alternative for kidney biopsy. Lamellar bodies and intracellular inclusions found in electron microscopy provide strong evidence to diagnose FD.
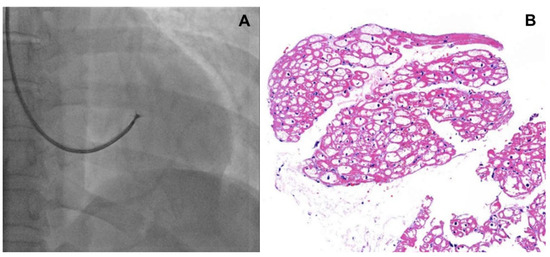 Figure 1. (A) Fluoroscopic image of endomyocardial biopsy, (B) H&E stain of endomyocardium, intracellular inclusions are found.
Figure 1. (A) Fluoroscopic image of endomyocardial biopsy, (B) H&E stain of endomyocardium, intracellular inclusions are found. - Coronary angiography: Damage to the coronary vascular bed by accumulation of glycolipids and secondary inflammations may lead to angina pectoris, variant angina, and myocardial infarction that do not necessarily follow the typical patterns observed in atherosclerosis [7]. Endothelial dysfunction with vasospasm and thrombotic events predominantly involves small, penetrating vessels. Therefore, angina symptoms in patients with FD need to be clarified by a coronary angiogram.
Follow-up of cardiac involvement in FD should be carried out regularly and the recommended schedule is based on outpatient visits every 3–6 months, as in Table 2.
Table 2.
Recommended tests and schedules of cardiac evaluations in classical and non-classical types of Fabry disease.
2.3. Nephrology
Screening for Fabry nephropathy should focus on high-risk populations and involve both clinical evaluation and diagnostic testing. High-risk populations include individuals with unexplained proteinuria or chronic kidney disease of unknown origin, relatives of patients diagnosed with FD (especially male family members), and patients presenting with early-onset stroke or cardiomyopathy without identifiable causes [16].
Laboratory findings in urine are crucial for the diagnosis of FD. Urinalysis typically reveals proteinuria, often in the nephrotic range, along with possible microscopic hematuria and lipiduria [17]. Blood tests may indicate elevated serum creatinine levels and reduced eGFR. Low GLA enzyme activity in males strongly suggests FD. Imaging studies, such as renal ultrasound or magnetic MRI, may show enlarged kidneys in the early disease stages, progressing to atrophic kidneys as the disease advances [18]. Histopathological examination through renal biopsy can provide definitive evidence with characteristic findings like podocyte inclusions (zebra bodies) and glomerulosclerosis [19].
Effective monitoring of Fabry nephropathy necessitates the incorporation of specific biomarkers and assessment tools to assess disease progression and therapeutic response. In addition to the key biomarkers, including GLA activity and lyso-Gb3 levels, proteinuria and albuminuria serve as indicators of renal involvement, while serum creatinine and eGFR are crucial for evaluating renal function [20]. Assessment tools such as renal biopsy offer detailed histological insights, albeit invasively. Non-invasive imaging studies such as renal ultrasound and MRI aid in assessing kidney structure and identifying complications. Additionally, 24 h urine collection and regular urinalysis are essential for quantifying protein excretion and identifying renal pathology.
A structured visit schedule is vital for effective management. Initial evaluations at diagnosis should include comprehensive biomarker analysis and imaging studies. Routine follow-ups every 3 to 6 months, along with a thorough annual review, ensure continuous monitoring and timely adjustments to therapy [21]. This systematic approach enables healthcare providers to mitigate renal damage and enhance patient outcomes by closely monitoring disease progression and treatment response [21].
2.4. Neurology
The buildup of Gb3 disrupts normal endothelial function, contributing to a pro-inflammatory and pro-thrombotic state. This dysfunction can lead to vascular occlusions, increased vascular resistance, and a higher risk of ischemic events. Clinical manifestations that may raise suspicion and ultimately lead to the diagnosis of FD are as follows:
- Ischemic stroke: Ischemic strokes are more common in patients with FD than in the general population, often occurring at a younger age [22]. Strokes in Fabry patients are often due to small vessel disease, but larger vessel involvement and cardioembolism can also occur due to atrial fibrillation (AF) or other cardiac abnormalities related to FD [23].
- Transient Ischemic Attacks (TIAs): TIAs are characterized by brief episodes of neurological dysfunction caused by temporary ischemia, lasting less than 24 h. TIAs are often a warning sign of a future stroke and require immediate medical attention and preventive treatment.
- Cerebral small vessel disease: The accumulation of Gb3 in endothelial cells and smooth muscle cells of small blood vessels leads to their dysfunction and damage. This can result in thickening of the vessel walls, narrowing of the lumen, and reduced blood flow. These lesions are often detected incidentally on MRI scans such as white matter lesions and lacunar infarcts, even in asymptomatic patients [24]. They indicate chronic microvascular damage and are more prevalent in patients with FD. White matter lesions are associated with cognitive decline, memory impairment, and other neuropsychological deficits, contributing to the overall neurological burden in patients with FD.
- Cerebral large vessel disease: While small vessel disease is more common in FD, some patients may also present with large cerebral vessel involvement due to Gb3 accumulation. In some cases, these patients may develop stenosis, aneurysms, or dolichoectasia, or present with large vessel dissection, contributing to cerebrovascular events. Large vessel disease can lead to ischemic strokes from either atherosclerotic changes or emboli.
- Chronic neurologic symptoms: Patients may experience chronic headaches, dizziness, vertigo, and cognitive difficulties over time. Some patients may also present with silent strokes, which are small ischemic events that go unnoticed but can accumulate and lead to significant neurological impairment. Cognitive decline is common due to repeated ischemic events and the presence of white matter lesions. Patients may experience memory loss, difficulty concentrating, and other executive function deficits [25,26].
2.5. Gastroenterology
Gastrointestinal presentations in patients with FD are heterogeneous. Abdominal pain, diarrhea, constipation, bloating, vomiting, and nausea are common. Abdominal pain and diarrhea are the most common gastrointestinal symptoms [27]. The prevalence of gastrointestinal symptoms in patients with FD is unclear. Registry data in the Fabry Outcome Survey reported that 60% of untreated children and 50% of female adults had gastrointestinal symptoms [28,29].
The clinical features of FD are similar to those of functional gastrointestinal disorders. In addition, FD is rare, and non-specific gastrointestinal symptoms are often the initial presentation of the disease. Misdiagnosis due to non-specific gastrointestinal symptoms is common in patients with FD. Therefore, if FD is suspected, it is important to evaluate the patient’s history and determine the cause of the unexplained gastrointestinal symptoms. The presence of other FD-associated symptoms, including burning pain in the extremities, abnormal sweating, and heat intolerance, should raise suspicion of FD. Clinicians should evaluate the presence of other organ involvement, including the skin, eye, kidney, and heart, and assess the family history of FD-associated symptoms.
After diagnosis of FD, the 24 h and 7-day FABry Disease Patient-Reported Outcome-Gastrointestinal (FABPRO-GI) is a useful instrument for assessing and monitoring FD-related gastrointestinal symptoms in clinical trials and real-world settings [30]. Prokinetics, acid-suppressants, and loperamide are used to control gastrointestinal symptoms. Although no specific diet intervention was established as non-pharmacological therapy, a low Fermentable Oligosaccharides, Disaccharides, Monosaccharides, and Polyols (FODMAPs) diet is suggested as an effective alternative approach to control gastrointestinal symptoms in FD patients [31].
2.6. Otorhinolaryngology
A nationwide population-based study utilizing data from Taiwan’s National Health Insurance database showed that among patients diagnosed with FD, 16.7% experienced tinnitus, 7.5% had hearing disorders, and 1.7% suffered from sudden sensorineural hearing loss (SSNHL). These prevalence rates were significantly higher than those in the general control group [32]. Another study involving 57 patients with FD (mean age 46.2 years) found that 73.7% of patients exhibited SSNHL of ≥25 dB, 54.4% complained of dizziness, and 71.9% showed abnormal nystagmus responses [33]. Research focused on pediatric patients with FD reported tinnitus in 31.8–40% of patients, hearing loss in 12.8–21.8% of cases with onset around 4 years of age, and vertigo in 25.5–30.4% of cases with onset typically after 10 years of age; all symptoms occurred earlier in boys [34].
A thorough review of symptoms such as hearing loss, tinnitus, and dizziness is essential for newly diagnosed patients with FD and their family members who are suspected of having this condition. For a more systematic approach, validated questionnaires can be utilized, such as the Hearing Handicap Inventory for the Elderly [35], the Tinnitus Handicap Inventory [36], and the Dizziness Handicap Inventory [37,38].
After evaluating medical history, hearing tests should be conducted as part of the initial assessment [39]. A multidisciplinary management protocol for FD published in the Netherlands also recommends hearing tests in the initial examination [40]. For adult and pediatric patients with FD, the hearing tests that can be considered are detailed below:
- For adults: The initial otolaryngological hearing tests should include pure-tone audiometry (PTA), tympanometry, and Otoacoustic Emission (OAE) [39]. Depending on the patient’s reported symptoms, speech audiometry (SA) and tinnitogram (tinnitus pitch and loudness matching) can be added. PTA is the most widelyused subjective hearing test for assessinghearing thresholds across different frequencies. The OAE, particularly Distortion Product-OAE (DPOAE), is an objective hearing test that can detect early dysfunction of the outer hair cells in the cochlea. It can be used as a screening tool for patients with normal hearing who present with otological symptoms [41].
- For pediatric patients: PTA can be used reliably from 5 years of age onwards, under the guidance of a familiar audiologist. For younger children, visual reinforcement audiometry or play audiometry can be employed. However, these tests are subjective and may not always be accurate [42]. To overcome these limitations, objective hearing threshold measurements using DPOAE, Auditory Brain Response, and Auditory Steady State Response should be conducted.
Vestibular function tests are primarily conducted for patients reporting vestibular abnormalities and are not routinely recommended for initial assessment or periodic follow-up. For patients complaining of dizziness or vertigo, available tests include cranial nerve examination, nystagmus testing using electronystagmography or video nystagmography, positional and positioning tests, and video head impulse tests. For a more detailed examination of the peripheral vestibular organs, caloric tests and rotation chair tests can be performed.
There is no unified recommendation for regular monitoring of hearing in patients with FD. Some guidelines suggest conducting hearing tests when pediatric [34] or adult [40] patients complain of hearing loss. Conversely, other pieces of literature recommend annual follow-ups with PTA, tympanometry, and OAE testing [39]. For the regular monitoring of patients with FD, we recommend the following:
- Adult patients: For adults with normal hearing, there are no established principles for periodic hearing tests. However, hearing thresholds tend to increase with age [43], and hearing tests are recommended for adults with risk factors such as a family history of FD, genetic disorders associated with hearing loss, or noise exposure [44]. We recommend that patients with FD undergo periodic hearing tests for the early detection of hearing loss, even in the absence of reported symptoms.
- Pediatric patients: According to the newborn screening guidelines in South Korea, it is recommended that infants at high-risk for hearing loss should undergo regular otolaryngological examinations every 6 months to 1 year until school age, even if they pass the newborn hearing screening. These examinations should include an assessment of language development, otoscopy, and detailed hearing tests [45]. In line with these recommendations, we recommend that pediatric patients with FD undergo age-appropriate hearing tests at least once a year.
- Symptomatic patients: For patients complaining of dizziness or vertigo, we recommend conducting vestibular function tests as needed. In cases of sudden onset of symptoms such as SSNHL or acute vertigo, prompt otolaryngological consultations and appropriate treatment should be provided.
Table 3 summarizes the initial and follow-up assessments for otorhinolaryngological involvement in FD and recommends a schedule for outpatient visits.
Table 3.
Recommended tests and schedules of otorhinolaryngological evaluations in Fabry disease patients.
2.7. Ophthalmology
Ocular manifestations of FD result from sphingomyelin accumulation within the lysosomes of eye tissues [46].
Vortex keratopathy (also called cornea verticillate), characterized by grayish or golden-brown deposits arranged in a vortex pattern in the corneal epithelial and subepithelial layers, is a common and diagnostically significant finding in patients with FD [46,47]. This manifestation occurs in most affected males and asymptomatic female carriers. Although it does not impact vision, the presence of vortex keratopathy is a highly sensitive and specific marker and crucial for early diagnosis of FD. It can be detected early and is present in about 70% of patients with FD. This condition can also occur due to amiodarone, chloroquine, indomethacin, and tamoxifen drug deposits, necessitating a systemic evaluation for differential diagnosis [47,48].
Another ocular manifestation of FD is the appearance of round intracellular inclusions in the conjunctival tissue, which may be accompanied by vascular changes such as tortuosity, microaneurysms, and dilation. These vascular changes can be seen in any conjunctival area but occur most commonly in the inferior bulbar area. Vessel tortuosity is more common in males than in females and has a significant correlation with the disease severity score [46,49].
Cataracts are the most common finding in the lens, particularly in the form of anterior capsular or posterior subcapsular opacities. Cataracts may have a detrimental effect on visual function, often requiring surgical intervention [46].
In the retina, the most common ocular finding is vascular tortuosity that is rarely accompanied by optic disc edema [46,49]. These changes can be detected by funduscopic examination.
Since the corneal wound healing process differs in FD, as shown in Figure 2., caution is required to prevent the progression of corneal opacity before and after surgery, such as Epi-LASIK. In FD, it is suspected that the wound healing process leads to intracellular Gb3 deposits in the surrounding tissue, exacerbating inflammation and opacity. Nevertheless, the specific processes causing the progression of corneal lesions are not known, necessitating further research.
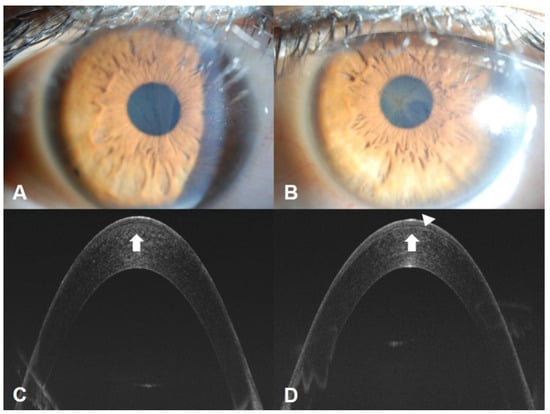
Figure 2.
A 32-year-old woman with FD presented with decreased vision in her left eye, which had previously undergone Epi-LASIK. Photograph of the cornea showing the extent and density of vortex keratopathy or cornea verticillata, which was less significant in the right eye (A) compared to the left eye (B). Optical coherence tomography showed deposits (white arrows) in the anterior corneas of both eyes (C,D) and an irregular border between the corneal subepithelium and the anterior stroma layer (arrowhead, D). This suggests that prior refractive surgery may exacerbate corneal lesions in FD through altered wound healing.
2.8. Dermatology
Angiokeratomas are a representative skin symptom of FD [50]. They are reported in 70% of male patients and 39% of female patients [50], typically manifesting between the ages of 5 and 15 years in males, and between the ages of 8 and 25 years in females [29]. Angiokeratomas appear as dark red to bluish-black macules and papules ranging from pinpoint size to 4 mm in diameter, and their color does not blanch under pressure. Hyperkeratosis overlying the lesions varies and is rarely seen outside the genital and umbilical areas. They can be widespread or clustered. In males, angiokeratomas are mainly observed in the “bathing trunk” area, including the lower abdomen, umbilicus, groin, genitals, buttocks, and inner thighs. They are also commonly found on the proximal limbs, especially the medial aspects, and can appear in protruding areas such as the elbows and knees, as well as on the palms and soles, and the distal phalanges of digits. They can also be observed on the lips, particularly the vermilion border, and rarely on mucosal surfaces, but almost never on other parts of the face. In females, angiokeratomas are generally less common and can sometimes be found distributed in a dermatomal pattern. The most common areas are the trunk and proximal limbs, with genital lesions being relatively rare.
Histopathologically, angiokeratomas consist of dilated capillaries in papillary dermis surrounded by elongated rete ridges. Older lesions may exhibit varying degrees of hyperkeratosis. In skin lesions, lipid storage can be observed in endothelial cells, pericytes, arteriolar smooth muscle, and arrector pili muscle. These findings can be confirmed with periodic acid-Schiff or Sudan black B staining. Lipid accumulation may also be observed in perineural cells and eccrine gland epithelium.
Up to one-third of males and two-thirds of females do not present with angiokeratomas [51]. Some have no notable skin vascular lesions, while others have bright erythematous macular angiomatous lesions of 1–2 mm in diameter, which are thought to be early lesions of angiokeratomas [52]. There are also patients with widely distributed papular angiomas that clinically and histologically resemble cherry angiomas prevalent in up to 50% of the general population, making their clinical significance in FD unclear. Telangiectasias are also common and can be distinguished from angiokeratomas and angiomas by the loss of color under diascopy. They are reported more frequently in males (23%) than in females (9%). Telangiectasias appear later than angiokeratomas, with an average onset age of 26 years (range: 3–70 years) in males and 42 years (range: 5–73 years) in females. They are commonly observed in Caucasians and typically occur in sun-exposed areas such as the face and “V” area of the neck, but can also be seen on the flanks, groin, elbows, and knees. Dermoscopy reveals well-defined, dilated, and tortuous vessels in the upper dermis, particularly in patients with widely distributed angiokeratomas and a typical phenotype of the disease.
Hypohidrosis is a typical skin symptom of FD, which is thought to result from autonomic neuropathy, although structural accumulation in sweat glands may also play a role. In total, 53% of males and 28% of females reported reduced sweating, with earlier onset in males (average age: males 23 years, females 26 years). Anhidrosis was reported by 25% of males and 4% of females [53]. Heat intolerance, often accompanied by other symptoms, can lead to decreased exercise tolerance, nausea, dyspnea, dizziness, headaches, or loss of consciousness. Previous studies have confirmed improvement in sweating in patients with FD undergoing ERT [54]. Hyperhidrosis may occur more frequently in females than in males, with one study reporting the occurrence of hyperhidrosis in 44 of 369 females (11.9%) and 22 of 345 males (6.4%) [55].
2.9. Pathology
In FD, podocyte cytoplasm appears expanded, foamy, pale, and lacy by light microscopy due to lipid deposits dissolved during processing of renal biopsy tissue [56,57] (Figure 3). With progressive disease, mesangial expansion and glomerulosclerosis develop, with proportional interstitial fibrosis and tubular atrophy. Segmental glomerular basement membrane double contours may be observed [56]. Inclusions are also present early in Henle’s loop and in the distal tubule, and occasionally in the proximal tubule [57]. Vascular sclerosis can be prominent even in early diseases. Standard immunofluorescence may show IgM and C3 in mesangial areas [56,57].
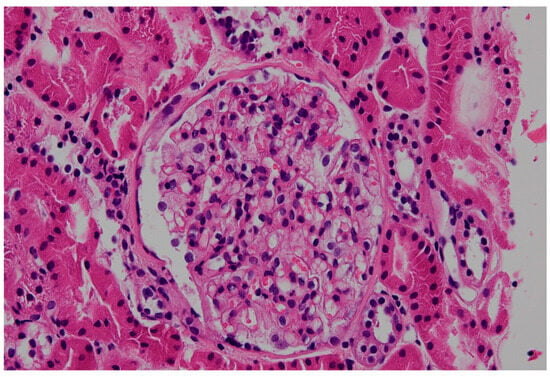
Figure 3.
Podocytes and parietal epithelial cells show a vacuolated, honeycomb appearance resulting from accumulation of abnormal glycosphingolipid in Fabry disease and extraction of lipids during processing for standard light microscopy (hematoxylin and eosin, ×200).
By electron microscopy (EM), inclusions in lysosomes are widespread and may be present in all renal cells. The processing of tissue for EM does not extract the lipids; therefore, the inclusions can be viewed directly on the toluidine blue-stained scout sections assessed for these studies. The inclusions vary in size and structure and have been called myelin bodies, whorled lamellated inclusions, or zebra bodies (Figure 4) [56,57]. Lamellated inclusions show alternating dark and light layers [56]. These inclusions are most prominent in podocytes but are also variably present in parietal epithelial cells, endothelium, peritubular capillaries, and interstitial cells [56]. Vascular smooth muscle cells also exhibit inclusions, with distal tubules frequently affected. Mosaic appearance of podocytes by EM, with lipid inclusions in some and none in other podocytes, may shed light on structural changes in affected versus nonaffected podocytes in female heterozygotes.
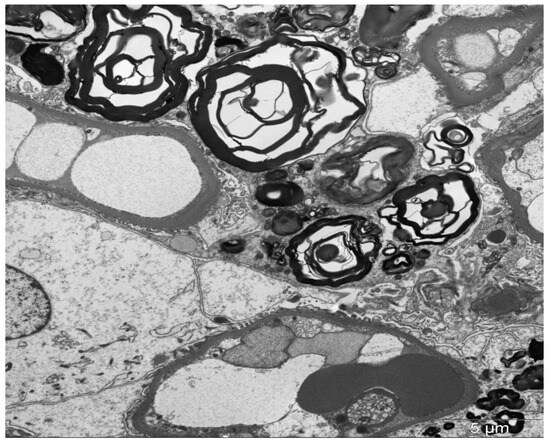
Figure 4.
Lysosomal inclusion with a so-called “myelin body appearance” in the podocyte (transmission electron microscopy, ×12,000).
3. Diagnostic Challenges in Cases with a Variant of Uncertain Significance of the GLA Gene
Interpreting novel mutations and determining their pathogenicity can be problematic because of genetic complexity. While over 1000 GLA variants have been identified, many are classified as VUS, presenting significant diagnostic challenges. Screening may identify individuals with VUS or later-onset phenotypes, leading to diagnostic dilemmas [58]. High-throughput next-generation sequencing-based screening programs in high-risk populations and newborns have identified several novel GLA variants [59]. Understanding variant pathogenicity is the key to accurate prevalence estimation, diagnosis, and management of FD. Distinguishing disease-causing variants from benign bystanders is probably one of the biggest challenges in contemporary clinical genetics [59].
Screening studies have revealed a high prevalence of individuals with GLA genetic VUS. In newborn populations, the prevalence of GLA variants is approximately 0.04%, while in high-risk populations, it reaches 0.62% [60]. However, only a fraction of these cases (0.12% in high-risk populations) receive a definite diagnosis of classical FD [61].
Several factors contribute to the diagnostic challenges associated with VUS in FD:
- Heterogeneous clinical presentation: FD manifestations are highly variable, even among individuals with the same variant. This variability complicates the correlation between genotype and phenotype.
- Residual enzyme activity: Some VUS may result in partial enzyme activity, leading to milder or late-onset forms of the disease that are harder to diagnose.
- Lack of specific biomarkers: In some individuals with VUS, FD-specific biomarkers and imaging findings may not uncover evidence of organ involvement.
- Coexisting conditions: FD often coexists with other nephrotic disorders, such as IgA nephropathy, further complicating diagnosis [62].
To address these challenges, a comprehensive diagnostic approach including genetic tests, enzymatic activity assays, biomarker analysis, tissue biopsy, comprehensive clinical investigation, and family history and pedigree analysis is necessary. While essential, genetic testing alone is insufficient for the diagnosis of FD when a VUS is identified. Enzyme activity assays are crucial but may have varying cut-off values (10–55% of the mean reference value for men, and up to 80% for women). Biomarker analyses, including measurement of Gb3 or lyso-Gb3, can provide additional evidence, although levels may be normal in some VUS cases [63]. The kidney, skin, heart, or sural nerve biopsy can reveal characteristic features such as myeloid bodies, aiding in diagnosis. Furthermore, a thorough assessment of organ involvement, including renal, cardiac, gastrointestinal, and neurological examinations, is essential. Lastly, family history and pedigree assay can provide valuable context for interpreting VUS.
4. FD Treatment and Management of Associated Symptoms and Risk Factors
4.1. Genetics: ERT or Chaperone Therapy
There is currently no cure for FD, but treatments can alleviate symptoms, prevent progression, and improve patient quality of life [64,65]. Management involves a multidisciplinary approach with regular follow-ups with specialists, including nephrologists, cardiologists, neurologists, gastroenterologists, pain medicine specialists, and genetic counselors [34]. In South Korea, the management of FD encompasses both ERT and pharmacological chaperone therapy, tailored to the patient’s specific genetic mutation and clinical presentation.
ERT options comprise agalsidase alfa and agalsidase beta, administered every two weeks via intravenous infusion [34]. Clinical evidence has shown that these therapies are effective in stabilizing or improving renal function, reducing left ventricular mass index (LVMi), alleviating neuropathic pain, and enhancing overall quality of life [65,66,67,68,69,70,71]. Notably, early commencement of ERT is associated with improved outcomes, particularly in preserving organ function and delaying disease progression [72]. Adverse events are typically mild, with some patients experiencing infusion-related reactions that can be managed with premedication [73].
Recently, analyses of up to 20 years of data from the Fabry Outcome Survey (FOS) revealed that patients treated with Replagal (agalsidase alfa) experienced improvement or stabilization of renal, cardiac, morbidity, and mortality outcomes compared with untreated external Fabry disease cohorts [74]. Fabrazyme (agalsidase beta) has also been reported to reduce the risk of major clinical events (cardiac and cerebrovascular events) significantly compared to placebo over the last 20 years [75]. The Fabry Disease International Registry (Fabry Registry) obtained medical records from 1944 to 2002 for 447 patients from five countries, and also confirmed a preservation of kidney function and delays progression to severe clinical events among patients with FD following Fabrazyme treatment [76].
For patients aged 16 years and older with amenable mutations of the GLA gene, migalastat presents an oral treatment option. Clinical trials, such as the phase 3 ATTRACT study, have demonstrated that migalastat is comparable to ERT in maintaining renal function and reducing LVMi over a 30-month period [77,78]. Furthermore, migalastat improves gastrointestinal symptoms and stabilizes overall quality of life [71,77,79]. The safety profile of migalastat is favorable, with most adverse events being mild or moderate in severity [78]. However, its use is restricted to patients with specific amenable mutations [80] and in Korea, health insurance currently only covers reimbursement of migalastat after 12 months of ERT use.
For patients receiving either ERT or chaperone therapy, treatment adherence and tolerability should be assessed and monitoring for adverse effects is needed [80,81]. Treatment efficacy can be evaluated through improvement of clinical symptoms, change in biomarker levels (e.g., lyso-Gb3), and organ function tests [80].
4.2. Cardiology and Neurology
Given the overlapping and interrelated risk factors, an integrated and multidisciplinary approach to cardiovascular and neurologic risk factor management is critical in the care of patients with Fabry disease.
4.2.1. Hypertension
Male patients with classical FD rarely have hypertension. If high blood pressure is measured at the outpatient clinic, consider 24 h blood pressure monitoring before starting treatment. Aim for a normal blood pressure of <130/80 mmHg in all patients according to current hypertension management guidelines [7,82]. Antihypertensive agents such as angiotensin-converting enzyme inhibitors (ACE inhibitors), angiotensin II receptor blockers (ARBs), and calcium channel blockers are commonly used, and more aggressive blood pressure control is often required after a stroke [83].
4.2.2. Dyslipidemia
Low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol are commonly elevated in FD. FD can be considered a high-risk disease in which endothelial dysfunction and vascular glycolipid accumulation develop frequently. While there are no official guideline recommendations, in our Fabry expert center, the risk for cardiovascular disease in patients with FD is considered similar to that of diabetes. Therefore, LDL cholesterol targets are stratified based on individual risk profiles: <100 mg/dL for young patients (<50 years) without additional risk factors, <70 mg/dL for those with comorbidities such as hypertension or smoking, and <55 mg/dL for patients with established atherosclerotic cardiovascular or cerebrovascular disease. Statins are the primary lipid-lowering agents, with high-intensity therapy recommended for both primary and secondary prevention [84].
4.2.3. Coexisting Diabetes
In patients with coexisting diabetes, which significantly increases the risk of ischemic stroke, strict glycemic control is critical. A target HbA1c level of <7% is advised, achieved through lifestyle modification and pharmacologic therapy such as metformin or insulin [84].
4.2.4. Atrial Fibrillation (AF)
While not common in all FD patients, AF may occur particularly in those with cardiomyopathy or advanced age [7]. If AF is detected, stroke prevention with anticoagulation is indicated. In patients with non-valvular AF, direct oral anticoagulants (DOACs)—including dabigatran, rivaroxaban, apixaban, and edoxaban—are preferred due to superior safety profiles and ease of use [83,85]. Warfarin remains the treatment of choice in patients with valvular AF, such as those with moderate-to-severe mitral stenosis or mechanical heart valves, with an INR target of 2.0–3.0 [83].
4.2.5. Antiplatelet Therapy
In patients with a history of ischemic stroke, antiplatelet therapy such as aspirin or clopidogrel is essential for secondary prevention, with short-term dual therapy considered in certain subtypes [83,85]. For cardioembolic strokes, anticoagulation therapy is mandatory. Furthermore, in patients with significant carotid artery stenosis, carotid endarterectomy or stenting may be considered to prevent recurrent stroke [83].
4.3. Nephrology
Numerous studies have shown that ERT with agalsidase α or agalsidase β can stabilize renal function in patients with Fabry nephropathy, especially when initiated early in disease progression [86]. While ERT and chaperone therapy are effective, additional treatments such as renin–angiotensin–aldosterone system inhibitors are beneficial once kidney damage has occurred. Since ERT alone has not been shown to reduce proteinuria consistently, complementary treatments like angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are vital for managing proteinuria in Fabry nephropathy patients [87]. Newer drugs, such as sodium-glucose cotransporter-2 inhibitors, hold promise as additional therapies for Fabry nephropathy.
4.4. Gastroenterology
Certain medications can provide symptom improvement for specific gastrointestinal symptoms. The use of metoclopramide showed symptom relief in some patients with gastroparesis [88]. Proton pump inhibitors are used to improve upper gastrointestinal symptoms. Anti-diarrhea medications such as loperamide may be useful to control lower gastrointestinal symptoms. Probiotics and antibiotics can be a potential treatment to prevent or control bacterial overgrowth [89]. The use of gabapentin has demonstrated a beneficial effect in patients with neuropathic pain [90].
4.5. Otorhinolaryngology
In patients with FD with multi-organ involvement, otolaryngological symptoms can be easily overlooked by both patients and healthcare providers. However, these symptoms significantly impact patients’ quality of life and, if left untreated at the appropriate time, can lead to a greater burden on the patient. It affects not only communication and language development but also cognitive function, educational attainment, employment prospects, and mental health. Hearing loss can lead to social isolation, loneliness, and a reduced quality of life. In children, it can impair speech and language development, while in adults, it is associated with an increased risk of dementia [91]. For patients with FD who are at risk of progressive hearing loss, timely and appropriate management of hearing impairment is crucial to mitigate these potential adverse effects and maintain overall quality of life.
Tinnitus can significantly impact various aspects of a person’s life. It affects listening and communication, often making it difficult to understand speech, especially in noisy environments. In children, tinnitus can impair language and speech development. Educationally and professionally, it can lead to reduced performance and lower achievement. Tinnitus frequently contributes to social isolation and loneliness, particularly in women and older adults. Mental health can be adversely affected, with higher rates of depression and lower quality of life reported among tinnitus sufferers [92].
There is currently no clear evidence that ERT can improve auditory symptoms [93]. Appropriate follow-up and testing through collaboration among various departments are necessary, and immediate specialist consultation is recommended when related symptoms occur [34,39]. Symptomatic treatment is provided for hearing loss, tinnitus, and vertigo, depending on the situation. Hearing rehabilitation for patients with hearing loss most commonly involves the use of hearing aids, while cochlear implants may be considered for severe hearing loss [39].
Furthermore, in patients with FD with multi-organ involvement and a predisposition to cerebrovascular and cardiovascular diseases, the possibility of acute emergencies such as SSNHL or central vertigo should always be considered, necessitating immediate intervention when required. SSNHL is, in particular, an otological emergency requiring prompt diagnosis and appropriate treatment [94]. Similarly, vertigo cases may require appropriate testing and differential diagnosis between peripheral and central vertigo to guide treatment.
4.6. Pain Management
Neuropathic pain is one of the most debilitating manifestations of FD, significantly impacting the quality of life of affected individuals [95]. Current strategies for managing neuropathic pain in patients with FD are discussed here.
A recent survey found that frequently used acute pain medications among patients with FD include non-steroidal anti-inflammatory drugs (NSAIDs) and non-opioid analgesics, with few patients using neuropathic pain-control agents [96]. This could be because there are limited comparative clinical data for the use of neuropathic pain-control agents specifically in patients with FD. Nevertheless, case reports of small numbers of patients with FD have shown that pain can be reduced with the administration of carbamazepine, gabapentin, phenytoin, and intravenous lidocaine.
For chronic pain management, current pain-management strategies are founded on clinical experience or based on national and international guidelines for the management of neuropathic pain. Based on a systematic literature review and meta-analysis of pharmacotherapy for the management of neuropathic pain in adults, tricyclic antidepressants, serotonin and norepinephrine reuptake inhibitors (e.g., duloxetine, venlafaxine), carbamazepine, and gabapentinoids (e.g., gabapentin, pregabalin) should be considered as first-line options. Intravenous lidocaine, topical capsaicin (8%) patches, and tramadol should be considered as second-line options. Strong opioids should be considered as third-line options, with controlled-release opioids being preferred over short-acting opioids. Fourth-line treatments may include methadone (with both N-methyl-D-aspartate and opioid receptor effects), tapentadol, and anticonvulsants with lesser evidence of efficacy (e.g., lamotrigine, lacosamide) [96,97,98,99,100,101].
Tricyclic antidepressants and serotonin–norepinephrine reuptake inhibitors enhance the levels of neurotransmitters that inhibit pain signals, providing relief from neuropathic pain [102]. Carbamazepine and phenytoin work by stabilizing the inactivated state of sodium channels, thereby reducing neuronal excitability and decreasing the transmission of pain signals [103]. This makes them effective options for treating neuropathic pain, which is characterized by abnormal nerve function and heightened pain sensitivity. The use of carbamazepine in FD-related neuropathic pain is supported by its efficacy in treating other forms of neuropathic pain, such as trigeminal neuralgia and diabetic neuropathy [104]. While specific studies evaluating carbamazepine for FD are limited, the drug’s established role in managing neuropathic pain suggests it can be beneficial for patients with FD experiencing similar symptoms. There is some evidence to suggest that phenytoin may be useful during pain attacks [105].
Gabapentinoids such as gabapentin and pregabalin are commonly used to manage neuropathic pain in FD. These medications work by modulating calcium channels and reducing neuronal excitability, thereby alleviating pain. Clinical trials have demonstrated their efficacy in reducing pain severity in patients with FD [99].
Lidocaine and capsaicin plasters may be useful to control localized pain, as well as during pain crises. However, the mostly episodic nature of pain in FD reduces the appropriateness of these drugs. Furthermore, in FD, the area affected by pain crises is too large for plasters. Based on clinical experience, lidocaine or capsaicin cream may be used as acute-pain prophylaxis before physical activity. There is some evidence to suggest that phenytoin may be useful during pain attacks [96,105]. More recently, intravenous lidocaine has been used to successfully treat FD pain crisis [99].
Opioids may be considered for severe neuropathic pain that is unresponsive to other treatments. However, their use is generally limited due to the risk of dependence and side effects. Controlled-release opioids are preferred over short-acting opioids. Opioids may also be effective during pain crises, but to avoid worsening gut motility problems and to reduce the risk of dependency, they should be used only when anticonvulsant agents are ineffective. A multimodal approach, combining opioids with other pain management strategies and close patient monitoring, is essential to optimize pain relief while minimizing risks [106].
Key characteristics of neuropathic pain control agents specifically in patients with FD are summarized in Table 4.
Table 4.
Recommended analgesic drugs for supportive treatment of chronic neuropathic pain in Fabry disease.
Ketamine has been used to effectively treat pain with a neuropathic component; however, because of its adverse events and potential addiction risks, its use should be restricted to therapy-resistant severe neuropathic pain until definite proof is obtained that its benefits outweigh the risks and costs of treatment [107]. Although the potential of alpha-2 adrenoceptor agonists for the management of chronic neuropathic pain has also been explored, further clinical trials in this area are probably required [108].
Because of the distinct phenotype and comorbidities in patients with FD and since evidence for the use of analgesic treatment in FD is scarce, individualized recommendations will be necessary in most cases.
A multidisciplinary approach is important when forming an analgesic care plan, involving not only metabolic and pain specialists but also specialist pharmacists, pain psychologists, and physiotherapists. The management of pain in FD is challenging due to the unique interplay between several factors. Neuropathic pain, which is normally challenging to treat, occurs with multi-organ dysfunction, often limiting pharmacological management. Additionally, the most severe symptoms are often found in males starting in their youth, a time of significant psychosocial development.
5. Discussion
- Evidence supports the role of a multidisciplinary team approach in managing Fabry disease
There are currently no randomized controlled trials (RCTs) specifically designed to evaluate the effectiveness of the MDT model in Fabry disease. Most evidence supporting MDT care comes from expert consensus, registry data, and observational studies rather than from RCTs [109]. The MDT approach has been demonstrated to provide better outcomes in terms of disease stability after ERT and clinical regression, including patients with mild organ impairment [110]. In addition, a recent study in a Chinese hospital highlighted that the MDT assessment allowed for the identification of FD in 35 high-risk children, whereas none were identified before the establishment of the MDT [111].
- Our experiences of the MDT approach for FD diagnosis and management
MDTs are characterized by two distinct models. The first, a concurrent consultation model, involves all specialists convening simultaneously for a unified patient assessment. This approach offers heightened efficiency, expedited inter-professional communication, enhanced educational opportunities, and a reduction in overall patient consultation time. The second, a “quarterback” physician model, features a designated primary physician who coordinates consultations with organ-specific specialists, who must be designated team members. While this model may extend the patient’s overall consultation time due to sequential departmental visits, it minimizes individual specialist consultation time, thereby facilitating MDT implementation by lowering the barrier to initial team formation. Both models necessitate a comprehensive understanding of FD among all team members and require regular inter-specialist meetings and communication.
Our team, recognizing the pragmatic advantages of the latter model within the South Korean healthcare landscape, initiated a collaborative effort in 2021. This involved five specialties engaging in regular case discussions pertaining to diagnosis and treatment, thereby fostering team development. By 2024, the team had evolved into an FD Center of Excellence (COE) comprising individual specialists from ten disciplines. This expanded team convenes for biannual to triannual meetings, facilitating discussions, peer education, and the exploration of optimized treatment strategies. Figure 5 schematically illustrates our quarterback system. The genetic specialist assumes the role of the primary physician for FD patients, spearheading initial diagnosis and ERT. MDT activation occurs at diagnosis and during follow-up, with variations in involved specialties. Following confirmatory diagnosis and ERT initiation, follow-up intervals for participating specialties range from 3 to 6 months, tailored to individual patient disease status.
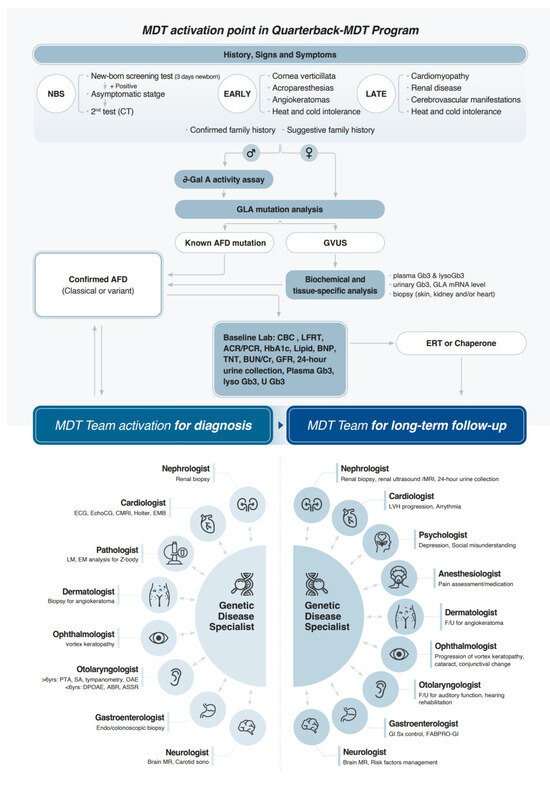
Figure 5.
MDT activation points in Quarterback-MDT program for Fabry disease. MDT: multidisciplinary team; NBS: newborn screening; GLA: alpha-galactosidase A; AFD: Anderson–Fabry disease; GVUS: genetic variants of unknown significance; Gb3: globotriaosylceramide; lysoGb3: globotriaosylsphingosine; mRNA: messenger ribonucleic acid; CBC: complete blood count; LFRT: liver function test; ACR/PCR: albumin/protein-to-creatinine ratio; HbA1c: hemoglobin A1c; BNP: brain natriuretic peptide; TNT: troponin T; BUN: blood urea nitrogen; Cr: creatinine; GFR: glomerular filtration rate; U: urine; ERT: enzyme replacement therapy; ECG: electrocardiography; EchoCG: echocardiography; CMR: cardiac magnetic resonance imaging; EMB: endomyocardial biopsy; LM: light microscopy; EM: electron microscopy; PTA: Pure Tone Audiometry; SA: Speech Audiometry; OAE: Otoacoustic Emission; DPOAE: distortion product OAE; ABR: auditory brainstem response; ASSR: auditory steady-state response; MR: magnetic resonance imaging; sono: sonography; LVH: left ventricular hypertrophy.
While the MDT model for FD faces substantial barriers in resource-limited areas, these can be mitigated through targeted training, telemedicine and remote MDT, international partnerships, advocacy for national health policy change, and the gradual introduction of emerging therapy. Early diagnosis and coordinated care remain critical to improving outcomes for FD patients everywhere.
5.1. Future Direction for Improved Diagnosis and Management of VUS Cases
In cases where patients with variants of uncertain significance (VUS) do not meet current treatment indications, identifying multi-organ involvement and treatment needs in Fabry disease underscores the essential role of the multidisciplinary team (MDT) in diagnosis and management. Since Fabry disease symptoms may not manifest simultaneously across all organ systems—and in some cases may progress more severely in a specific organ—long-term monitoring by an MDT is crucial to determining the pathogenicity of VUS. Over time, such monitoring can help elucidate the clinical significance of these variants. Functional research, including both in vitro and in vivo studies assessing the impact of VUS on enzyme activity and cellular processes, will further support early diagnosis and intervention. Additionally, data sharing and collaboration—not only within institutions and at the national level but also internationally—are vital for building robust evidence on rare variants.
5.2. Areas of Improvement in Reimbursement Criteria of FD Treatment in South Korea
According to the current Korean reimbursement guideline (Table 1), the cost for ERT cannot be reimbursed for patients with cardiac variants who do not meet the LVH criteria (LV wall thickness > 12 mm) despite a clear family history and genotype. However, subtle histologic and echocardiographic changes (shown by tissue Doppler image or longitudinal strain) are known to start before the development of overt LVH in treatment-naive patients with FD [13,82,112]. Frustaci et al. proved that in the prehypertrophic stage (left ventricular maximal wall thickness ≤ 10.5 mm), ERT stabilizes storage deposits and myocyte dimensions regardless of sex [113]. In the study by Langeveld et al. investigating the optimal timing of ERT initiation, starting ERT before the age of 16 years in male patients with classic FD reduced the occurrence of renal and cardiac manifestations [114]. Therefore, it is necessary to refine reimbursement conditions so that ERT can be started early if the genotype is typical, thereby improving long-term prognosis.
In South Korea, the reimbursement criteria for ERT (Table 1) for renal management of FD necessitates the presence of proteinuria and include the following conditions: a reduced eGFR (15 ≤ eGFR < 90 mL/min/1.73 m2, adjusted for age > 40) measured on at least two occasions; microalbuminuria detected more than twice over a minimum 24 h interval and albumin > 30 mg/g (in males); albuminuria detected more than twice over a minimum 24 h interval and albumin > 20 µg/min (in males); proteinuria > 150 mg/24 h (in males); and proteinuria > 300 mg/24 h with clinical evidence of progression (in females).
However, our previous research showed that segmental foot process effacement and Gb3 deposits can persist in Fabry nephropathy despite ERT [19]. Additionally, these deposits are observed in various kidney cell types in normoalbuminuric patients with FD, demonstrating the limitations of using albuminuria alone to detect early renal damage. These findings highlight the need for baseline and follow-up kidney biopsies for the appropriate initiation and monitoring of ERT in Fabry nephropathy. Therefore, we suggest that the reimbursement criteria for ERT should also account for the presence of Gb3 deposits in kidney biopsy specimens, even in normoalbuminuric patients with FD.
To contextualize Korea’s reimbursement framework within a broader global perspective, Table 5 summarizes ERT eligibility criteria across representative countries. While Korea offers a clearly structured and standardized approach, its strict organ-damage thresholds—such as requiring overt proteinuria or LVH—may limit timely access to treatment, particularly for female patients or those with late-onset phenotypes. These individuals may not meet reimbursement criteria despite histopathological or imaging evidence of progressive disease. In contrast, many international guidelines have evolved toward preventive treatment strategies, recommending ERT initiation based on confirmed genotype, early signs, or tissue-level involvement. Such approaches aim to optimize long-term outcomes through earlier intervention. These international trends highlight the potential benefit of adopting more flexible, genotype- and biomarker-driven reimbursement models in Korea.
Table 5.
International comparison of reimbursement criteria for enzyme replacement therapy (ERT) in Fabry disease.
MDT members must recognize the importance of not only vital organ problems but also symptoms from other functional areas, including the intestine, eyes, ears, and skin, that significantly affect the quality of life and overall health of patients with FD. A group of specialists gathered as an MDT should conduct regular assessments and monitoring in various clinical specialties, and a prompt and effective interdisciplinary consultation system should be implemented to address possible emergencies such as sudden hearing loss or central vertigo. Regular MDT meetings for collaborative clinical decisions and education will greatly contribute to improving health management and quality of life for patients with FD and thus deserve financial and operational support at the national level.
To complement the discussion in Section 5.2, Table 5 presents a cross-national comparison of ERT reimbursement systems for Fabry disease, focusing on South Korea, Japan, Australia, and select European countries. While South Korea’s reimbursement criteria are highly structured and organ-damage-oriented, they are considered relatively restrictive. Patients often require overt clinical manifestations—such as reduced eGFR, proteinuria, or established left ventricular hypertrophy (LVH)—to initiate ERT, making early intervention difficult, especially in female or late-onset patients [1]. In contrast, Australia provides subsidized ERT through the Life Saving Drugs Program (LSDP), which permits treatment based on histological or imaging findings of organ involvement and includes chronic pain or ischemic vascular events as eligible criteria. However, the process requires reapplication and clinical data submission every year to maintain eligibility [2]. European countries, including Italy and the UK, largely follow the EMA indication for Replagal®, reimbursing ERT for patients with confirmed Fabry diagnosis regardless of sex or phenotype. These systems emphasize the importance of early intervention and broader access, with fewer bureaucratic restrictions [3,4,5]. Japan’s practical guidelines recommend ERT initiation in classical male patients upon the onset of characteristic symptoms such as acroparesthesia. For late-onset male and female patients, ERT is advised when there is clear evidence of organ involvement, confirmed by regular monitoring of renal, cardiac, and neurologic parameters [6]. This international landscape underscores the need for Korean reimbursement policies to evolve toward a more genotype-driven and biomarker-guided model, aligning with global trends favoring early and preventive treatment strategies.
6. Conclusions
The diagnosis and long-term follow-up of FD, a multi-organ systemic disorder, necessitate an MDT approach to ensure high-quality management. Given the rarity of FD, it is unrealistic to expect all physicians to possess comprehensive knowledge of this condition. Therefore, PNUYH has established an MDT, which has been operational for over 4 years and comprises designated specialists from each relevant department who possess a thorough understanding of FD. This team is coordinated by a “quarterback” physician who maintains a comprehensive overview of each patient’s condition and medical history. The team has progressively expanded over time, and the sophistication of case discussions has significantly improved, enabling the successful management of numerous challenging cases, including those involving VUS. It is anticipated that this review report on FD diagnosis and management from an MDT perspective will serve as a valuable resource for centers initiating MDTs for the optimized management of FD.
Author Contributions
S.Y.L. and I.Y.K.: investigation, data curation, formal analysis and visualization, and manuscript writing—original draft preparation and editing. J.E.L., H.-M.L. and S.J.K.: investigation, visualization and formal analysis. G.-J.B., H.J.L. and H.-C.K.: project administration, formal analysis and validation. S.C.: project administration, writing—review and editing. C.K.C. and S.-H.A.: conceptualization, supervision, funding acquisition, manuscript writing—original draft preparation and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We thank Da-eun Jeong for her comprehensive review and operational support in manuscript development.
Conflicts of Interest
Songhwa Choi is a full-time employee of Takeda Pharmaceuticals Korea Co., Ltd. All other authors declare no competing interests.
Abbreviations
The following abbreviations are used in this manuscript:
| AHA | American Heart Association |
| ASA | American Stroke Association |
| CEA | Carotid endarterectomy |
| CMRI | Cardiac magnetic resonance imaging |
| DPOAE | Distortion Product Otoacoustic Emission |
| ECG | Electrocardiogram |
| EMB | Endomyocardial biopsy |
| FD | Fabry disease |
| Gb3 | Globotriaosylceramide |
| eGFR | estimated glomerular filtration rate |
| GLA | Alpha-galactosidase A |
| HbA1c | Glycated hemoglobin |
| HDL | High-density cholesterol |
| hsTNT | High-sensitive troponin T |
| LDL | Low density lipoprotein |
| LV | Left ventricular |
| LVH | Left ventricular hypertrophy |
| MDT | Multidisciplinary team |
| MRI | Magnetic resonance imaging |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| OAE | Otoacoustic Emission |
| PNUYH | Pusan National University Yangsan Hospital |
| PTA | Pure Tone Audiometry |
| RV | Right ventricular |
| SA | Speech Audiometry |
| TIA | Transient ischemic attack |
| VUS | Variants of uncertain significance |
References
- Schiffmann, R.; Fuller, M.; Clarke, L.A.; Aerts, J.M.F.G. Is it Fabry disease? Genet. Med. 2016, 18, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Sweeley, C.C.; Klionsky, B. Fabry’s disease: Classification as a sphingolipidosis and partial characterization of a novel glycolipid. J. Biol. Chem. 1963, 238, 3148–3150. [Google Scholar] [CrossRef] [PubMed]
- Torralba-Cabeza, M.A.; Olivera-Gonzalez, S.; Sierra-Monzon, J.L. The Importance of a Multidisciplinary Approach in the Management of a Patient with Type I Gaucher Disease. Diseases 2018, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Macken, W.L.; Falabella, M.; McKittrick, C.; Pizzamiglio, C.; Ellmers, R.; Eggleton, K.; Woodward, C.E.; Patel, Y.; Labrum, R.; Genomics England Research Consortium; et al. Specialist multidisciplinary input maximises rare disease diagnoses from whole genome sequencing. Nat. Commun. 2022, 13, 6324. [Google Scholar] [CrossRef]
- Ministry of Health and Welfare of South Korea. Details on the Application Criteria and Methods of Nursing Care Benefits; Notice No. 2019-313, effective date: 2020.01.01; Ministry of Health and Welfare of South Korea: Sejong, Republic of Korea, 2020.
- Doheny, D.; Srinivasan, R.; Pagant, S.; Chen, B.; Yasuda, M.; Desnick, R.J. Fabry Disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J. Med. Genet. 2018, 55, 261–268. [Google Scholar] [CrossRef]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagege, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef]
- Seo, J.; Kim, M.; Hong, G.R.; Kim, D.S.; Son, J.W.; Cho, I.J.; Shim, C.Y.; Chang, H.J.; Ha, J.W.; Chung, N. Fabry disease in patients with hypertrophic cardiomyopathy: A practical approach to diagnosis. J. Hum. Genet. 2016, 61, 775–780. [Google Scholar] [CrossRef]
- Namdar, M.; Steffel, J.; Vidovic, M.; Brunckhorst, C.B.; Holzmeister, J.; Luscher, T.F.; Jenni, R.; Duru, F. Electrocardiographic changes in early recognition of Fabry disease. Heart 2011, 97, 485–490. [Google Scholar] [CrossRef]
- O’Mahony, C.; Coats, C.; Cardona, M.; Garcia, A.; Calcagnino, M.; Murphy, E.; Lachmann, R.; Mehta, A.; Hughes, D.; Elliott, P.M. Incidence and predictors of anti-bradycardia pacing in patients with Anderson-Fabry disease. Europace 2011, 13, 1781–1788. [Google Scholar] [CrossRef]
- Shah, J.S.; Hughes, D.A.; Sachdev, B.; Tome, M.; Ward, D.; Lee, P.; Mehta, A.B.; Elliott, P.M. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am. J. Cardiol. 2005, 96, 842–846. [Google Scholar] [CrossRef]
- Kramer, J.; Niemann, M.; Stork, S.; Frantz, S.; Beer, M.; Ertl, G.; Wanner, C.; Weidemann, F. Relation of burden of myocardial fibrosis to malignant ventricular arrhythmias and outcomes in Fabry disease. Am. J. Cardiol. 2014, 114, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Esposito, R.; Galderisi, M.; Santoro, C.; Imbriaco, M.; Riccio, E.; Maria Pellegrino, A.; Sorrentino, R.; Lembo, M.; Citro, R.; Angela Losi, M.; et al. Prominent longitudinal strain reduction of left ventricular basal segments in treatment-naïve Anderson-Fabry disease patients. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.R.; Hung, S.C.; Chang, F.P.; Yu, W.C.; Sung, S.H.; Hsu, C.L.; Dzhagalov, I.; Yang, C.F.; Chu, T.H.; Lee, H.J.; et al. Later Onset Fabry Disease, Cardiac Damage Progress in Silence: Experience with a Highly Prevalent Mutation. J. Am. Coll. Cardiol. 2016, 68, 2554–2563. [Google Scholar] [CrossRef] [PubMed]
- Sado, D.M.; White, S.K.; Piechnik, S.K.; Banypersad, S.M.; Treibel, T.; Captur, G.; Fontana, M.; Maestrini, V.; Flett, A.S.; Robson, M.D.; et al. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ. Cardiovasc. Imaging 2013, 6, 392–398. [Google Scholar] [CrossRef]
- de Haan, A.; Morel, C.F.; Eijgelsheim, M.; de Jong, M.F.C.; Broekroelofs, J.; Vogt, L.; Knoers, N.; de Borst, M.H. Fabry disease with atypical phenotype identified by massively parallel sequencing in early-onset kidney failure. Clin. Kidney J. 2023, 16, 722–726. [Google Scholar] [CrossRef]
- Fervenza, F.C.; Torra, R.; Lager, D.J. Fabry disease: An underrecognized cause of proteinuria. Kidney Int. 2008, 73, 1193–1199. [Google Scholar] [CrossRef]
- Weidemann, F.; Sanchez-Nino, M.D.; Politei, J.; Oliveira, J.P.; Wanner, C.; Warnock, D.G.; Ortiz, A. Fibrosis: A key feature of Fabry disease with potential therapeutic implications. Orphanet J. Rare Dis. 2013, 8, 116. [Google Scholar] [CrossRef]
- Kim, I.Y.; Lee, H.J.; Cheon, C.K. Fabry nephropathy before and after enzyme replacement therapy: Important role of renal biopsy in patients with Fabry disease. Kidney Res. Clin. Pract. 2021, 40, 611–619. [Google Scholar] [CrossRef]
- Levstek, T.; Vujkovac, B.; Trebusak Podkrajsek, K. Biomarkers of Fabry Nephropathy: Review and Future Perspective. Genes 2020, 11, 1091. [Google Scholar] [CrossRef]
- Choi, S.J.; Kim, S.H.; Lee, M.S.; Park, S.; Cho, E.; Han, S.S.; Koh, E.S.; Chung, B.H.; Jeong, K.H.; Bae, E.; et al. A questionnaire survey on the diagnosis and treatment of Fabry nephropathy in clinical practice. Kidney Res. Clin. Pract. 2023, 42, 628–638. [Google Scholar] [CrossRef]
- Rolfs, A.; Fazekas, F.; Grittner, U.; Dichgans, M.; Martus, P.; Holzhausen, M.; Böttcher, T.; Heuschmann, P.U.; Tatlisumak, T.; Tanislav, C.; et al. Acute cerebrovascular disease in the young: The Stroke in Young Fabry Patients study. Stroke 2013, 44, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Chen, J.; Pongmoragot, J.; Lanthier, S.; Saposnik, G. Prevalence of Fabry disease in stroke patients--a systematic review and meta-analysis. J. Stroke Cerebrovasc. Dis. 2014, 23, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Fazekas, F.; Enzinger, C.; Schmidt, R.; Dichgans, M.; Gaertner, B.; Jungehulsing, G.J.; Hennerici, M.G.; Heuschmann, P.; Holzhausen, M.; Kaps, M.; et al. MRI in acute cerebral ischemia of the young: The Stroke in Young Fabry Patients (sifap1) Study. Neurology 2013, 81, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Körver, S.; Geurtsen, G.J.; Hollak, C.E.M.; van Schaik, I.N.; Longo, M.G.F.; Lima, M.R.; Dijkgraaf, M.G.W.; Langeveld, M. Cognitive functioning and depressive symptoms in Fabry disease: A follow-up study. J. Inherit. Metab. Dis. 2020, 43, 1070–1081. [Google Scholar] [CrossRef]
- Sigmundsdottir, L.; Tchan, M.C.; Knopman, A.A.; Menzies, G.C.; Batchelor, J.; Sillence, D.O. Cognitive and psychological functioning in Fabry disease. Arch. Clin. Neuropsychol. 2014, 29, 642–650. [Google Scholar] [CrossRef]
- Hilz, M.J.; Arbustini, E.; Dagna, L.; Gasbarrini, A.; Goizet, C.; Lacombe, D.; Liguori, R.; Manna, R.; Politei, J.; Spada, M.; et al. Non-specific gastrointestinal features: Could it be Fabry disease? Dig. Liver Dis. 2018, 50, 429–437. [Google Scholar] [CrossRef]
- Deegan, P.B.; Baehner, A.F.; Barba Romero, M.A.; Hughes, D.A.; Kampmann, C.; Beck, M. Natural history of Fabry disease in females in the Fabry Outcome Survey. J. Med. Genet. 2006, 43, 347–352. [Google Scholar] [CrossRef]
- Ramaswami, U.; Whybra, C.; Parini, R.; Pintos-Morell, G.; Mehta, A.; Sunder-Plassmann, G.; Widmer, U.; Beck, M. Clinical manifestations of Fabry disease in children: Data from the Fabry Outcome Survey. Acta Paediatr. 2006, 95, 86–92. [Google Scholar] [CrossRef]
- Shields, A.L.; Lamoureux, R.E.; Taylor, F.; Barth, J.A.; Mulberg, A.E.; Kessler, V.; Skuban, N. FABry Disease Patient-Reported Outcome-GastroIntestinal (FABPRO-GI): A new Fabry disease-specific gastrointestinal outcomes instrument. Qual. Life Res. 2021, 30, 2983–2994. [Google Scholar] [CrossRef]
- Gugelmo, G.; Vitturi, N.; Francini-Pesenti, F.; Fasan, I.; Lenzini, L.; Valentini, R.; Carraro, G.; Avogaro, A.; Spinella, P. Gastrointestinal Manifestations and Low-FODMAP Protocol in a Cohort of Fabry Disease Adult Patients. Nutrients 2023, 15, 658. [Google Scholar] [CrossRef]
- Cheng, Y.F.; Xirasagar, S.; Chen, C.S.; Niu, D.M.; Lin, H.C. Association of Fabry Disease with Hearing Loss, Tinnitus, and Sudden Hearing Loss: A Nationwide Population-Based Study. J. Clin. Med. 2022, 11, 7396. [Google Scholar] [CrossRef] [PubMed]
- Köping, M.; Shehata-Dieler, W.; Schneider, D.; Cebulla, M.; Oder, D.; Müntze, J.; Nordbeck, P.; Wanner, C.; Hagen, R.; Schraven, S.P. Characterization of vertigo and hearing loss in patients with Fabry disease. Orphanet J. Rare Dis. 2018, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Fouilhoux, A.; Decramer, S.; Tardieu, M.; Pillet, P.; Fila, M.; Rivera, S.; Deschênes, G.; Lacombe, D. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin. Genet. 2019, 96, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Park, S.N.; Han, G.C.; Cho, Y.S.; Byun, J.Y.; Shin, J.E.; Chu, H.S.; Cheon, B.C.; Chung, J.W.; Chae, S.W.; Choi, J.Y. Standardization for a Korean Version of Hearing Handicap Inventory for the Elderly. Korean J. Otorhinolaryngol.-Head Neck Surg. 2011, 54, 828–834. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.Y.; Kim, C.H.; Lim, S.L.; Shin, J.N.; Chung, W.H.; Yu, B.H.; Hong, S.H. Reliability and Validity of a Korean Adaptation of the Tinnitus Handicap Inventory. Korean J. Otorhinolaryngol.-Head Neck Surg. 2002, 45, 328–334. [Google Scholar]
- Han, G.C.; Lee, E.Y.; Lee, J.H.; Park, S.; Lee, H.; Jeon, E.; Lee, H.; Cheon, B.C.; Kim, K.S.; Gon, E.; et al. The Study of Standardization for a Korean Adaptation of Self-report Measures of Dizziness. Res. Vestib. Sci. 2004, 3, 307–325. [Google Scholar]
- Kim, T.-H.; Cha, H.E.; Lee, J.-G.; Im, G.J.; Song, J.-J.; Kim, S.H.; Moon, I.J.; Kim, Y.H.; Byun, J.Y.; Chae, S.-W. The Study of Standardization for a Korean Dizziness Handicap Inventory for Patient Caregivers. Korean J. Otorhinolaryngol.-Head Neck Surg. 2019, 62, 442–447. [Google Scholar] [CrossRef]
- Eng, C.M.; Germain, D.P.; Banikazemi, M.; Warnock, D.G.; Wanner, C.; Hopkin, R.J.; Bultas, J.; Lee, P.; Sims, K.; Brodie, S.E.; et al. Fabry disease: Guidelines for the evaluation and management of multi-organ system involvement. Genet. Med. 2006, 8, 539–548. [Google Scholar] [CrossRef]
- Langeveld, M.; Hollak, C.E.; Klein van Loon, S.; van der Veen, S.; el Sayed, M.; Eskes, E. Protocol: Diagnosis, Evaluation and Treatment of Fabry Disease in the Netherlands; Amsterdam UMC: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Wang, F.; Yamamoto, H.; Yoshida, T.; Sugimoto, S.; Teranishi, M.; Tsuboi, K.; Sone, M. Otological aspects of Fabry disease in patients with normal hearing. Nagoya J. Med. Sci. 2019, 81, 469–475. [Google Scholar] [CrossRef]
- Sommerfeldt, J.; Kolb, C.M. Hearing Loss Assessment in Children. In StatPearls [Internet]; StatPearls Publishing: Treasure Island FL, USA, 2023. [Google Scholar]
- Park, Y.H.; Shin, S.H.; Byun, S.W.; Kim, J.Y. Age- and Gender-Related Mean Hearing Threshold in a Highly-Screened Population: The Korean National Health and Nutrition Examination Survey 2010–2012. PLoS ONE 2016, 11, e0150783. [Google Scholar] [CrossRef]
- American Speech-Language-Hearing Association (ASHA). Adult Hearing Screening (Practice Portal). Available online: www.asha.org/Practice-Portal/Professional-Issues/Adult-Hearing-Screening/ (accessed on 19 February 2025).
- Newborn Hearing Screening Special Committee of the Korean Audiological Society. Newborn Hearing Screening Guideline Update in Korea. 2018. Available online: https://hearingscreening.or.kr/ (accessed on 10 May 2025).
- Gambini, G.; Scartozzi, L.; Giannuzzi, F.; Carlà, M.M.; Boselli, F.; Caporossi, T.; De Vico, U.; Baldascino, A.; Rizzo, S. Ophthalmic Manifestations in Fabry Disease: Updated Review. J. Pers. Med. 2023, 13, 904. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Choi, B.S.; Park, Y.M.; Lee, J.S. In Vivo Confocal Microscopic Findings of Corneal Tissue in Amiodarone-Induced Vortex Keratopathy. J. Korean Ophthalmol. Soc. 2015, 56, 127–133. [Google Scholar] [CrossRef]
- Panthagani, J.; MacDonald, T.; Bruynseels, A.; Madathilethu, S.C.; Jenyon, T. Deposition keratopathy. Br. J. Hosp. Med. 2022, 83, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kalkum, G.; Pitz, S.; Karabul, N.; Beck, M.; Pintos-Morell, G.; Parini, R.; Rohrbach, M.; Bizjajeva, S.; Ramaswami, U. Paediatric Fabry disease: Prognostic significance of ocular changes for disease severity. BMC Ophthalmol. 2016, 16, 202. [Google Scholar] [CrossRef]
- Orteu, C.H.; Jansen, T.; Lidove, O.; Jaussaud, R.; Hughes, D.A.; Pintos-Morell, G.; Ramaswami, U.; Parini, R.; Sunder-Plassman, G.; Beck, M.; et al. Fabry disease and the skin: Data from FOS, the Fabry outcome survey. Br. J. Dermatol. 2007, 157, 331–337. [Google Scholar] [CrossRef]
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J. Med. Genet. 2001, 38, 769–775. [Google Scholar] [CrossRef]
- Anderson, W. A case of “angio-keratoma”. Br. J. Dermatol. 1898, 10, 113–117. [Google Scholar] [CrossRef]
- Wilcox, W.R.; Oliveira, J.P.; Hopkin, R.J.; Ortiz, A.; Banikazemi, M.; Feldt-Rasmussen, U.; Sims, K.; Waldek, S.; Pastores, G.M.; Lee, P.; et al. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Mol. Genet. Metab. 2008, 93, 112–128. [Google Scholar] [CrossRef]
- Hilz, M.J.; Brys, M.; Marthol, H.; Stemper, B.; Dütsch, M. Enzyme replacement therapy improves function of C-, Adelta-, and Abeta-nerve fibers in Fabry neuropathy. Neurology 2004, 62, 1066–1072. [Google Scholar] [CrossRef]
- Lidove, O.; Ramaswami, U.; Jaussaud, R.; Barbey, F.; Maisonobe, T.; Caillaud, C.; Beck, M.; Sunder-Plassmann, G.; Linhart, A.; Mehta, A. Hyperhidrosis: A new and often early symptom in Fabry disease. International experience and data from the Fabry Outcome Survey. Int. J. Clin. Pract. 2006, 60, 1053–1059. [Google Scholar] [CrossRef]
- Fischer, E.G.; Moore, M.J.; Lager, D.J. Fabry disease: A morphologic study of 11 cases. Mod. Pathol. 2006, 19, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.A.B.; Moura-Neto, J.A.; Dos Reis, M.A.; Vieira Neto, O.M.; Barreto, F.C. Renal Manifestations of Fabry Disease: A Narrative Review. Can. J. Kidney Health Dis. 2021, 8, 2054358120985627. [Google Scholar] [CrossRef] [PubMed]
- Gragnaniello, V.; Burlina, A.P.; Commone, A.; Gueraldi, D.; Puma, A.; Porcù, E.; Stornaiuolo, M.; Cazzorla, C.; Burlina, A.B. Newborn Screening for Fabry Disease: Current Status of Knowledge. Int. J. Neonatal Screen. 2023, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Levade, T.; Hachulla, E.; Knebelmann, B.; Lacombe, D.; Seguin, V.L.; Nguyen, K.; Noël, E.; Rabès, J.P. Challenging the traditional approach for interpreting genetic variants: Lessons from Fabry disease. Clin. Genet. 2022, 101, 390–402. [Google Scholar] [CrossRef]
- van der Tol, L.; Smid, B.E.; Poorthuis, B.J.; Biegstraaten, M.; Deprez, R.H.; Linthorst, G.E.; Hollak, C.E. A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J. Med. Genet. 2014, 51, 1–9. [Google Scholar] [CrossRef]
- Vieitez, I.; Souto-Rodriguez, O.; Fernandez-Mosquera, L.; San Millan, B.; Teijeira, S.; Fernandez-Martin, J.; Martinez-Sanchez, F.; Aldamiz-Echevarria, L.J.; Lopez-Rodriguez, M.; Navarro, C.; et al. Fabry disease in the Spanish population: Observational study with detection of 77 patients. Orphanet J. Rare Dis. 2018, 13, 52. [Google Scholar] [CrossRef]
- Esposito, P.; Caputo, C.; Repetto, M.; Somaschini, A.; Pietro, B.; Colomba, P.; Zizzo, C.; Parodi, A.; Zanetti, V.; Canepa, M.; et al. Diagnosing Fabry nephropathy: The challenge of multiple kidney disease. BMC Nephrol. 2023, 24, 344. [Google Scholar] [CrossRef]
- Burlina, A.; Brand, E.; Hughes, D.; Kantola, I.; Krämer, J.; Nowak, A.; Tøndel, C.; Wanner, C.; Spada, M. An expert consensus on the recommendations for the use of biomarkers in Fabry disease. Mol. Genet. Metab. 2023, 139, 107585. [Google Scholar] [CrossRef]
- van der Veen, S.J.; Hollak, C.E.M.; van Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 43, 908–921. [Google Scholar] [CrossRef]
- Umer, M.; Kalra, D.K. Treatment of Fabry Disease: Established and Emerging Therapies. Pharmaceuticals 2023, 16, 320. [Google Scholar] [CrossRef]
- Arends, M.; Biegstraaten, M.; Wanner, C.; Sirrs, S.; Mehta, A.; Elliott, P.M.; Oder, D.; Watkinson, O.T.; Bichet, D.G.; Khan, A.; et al. Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: An international cohort study. J. Med. Genet. 2018, 55, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Banikazemi, M.; Bultas, J.; Waldek, S.; Wilcox, W.R.; Whitley, C.B.; McDonald, M.; Finkel, R.; Packman, S.; Bichet, D.-G.; Warnock, D.G.; et al. Agalsidase-Beta Therapy for Advanced Fabry Disease. Ann. Intern. Med. 2007, 146, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, C.; Perrin, A.; Beck, M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: Cardiac outcomes after 10 years’ treatment. Orphanet J. Rare Dis. 2015, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Li, P.; Zhang, S.; Zhiqing, Z. Effectiveness and safety of enzyme replacement therapy in the treatment of Fabry disease: A Chinese monocentric real-world study. Orphanet J. Rare Dis. 2024, 19, 422. [Google Scholar] [CrossRef]
- Schiffmann, R.; Pastores, G.M.; Lien, Y.H.; Castaneda, V.; Chang, P.; Martin, R.; Wijatyk, A. Agalsidase alfa in pediatric patients with Fabry disease: A 6.5-year open-label follow-up study. Orphanet J. Rare Dis. 2014, 9, 169. [Google Scholar] [CrossRef]
- Mignani, R.; Biagini, E.; Cianci, V.; Pieruzzi, F.; Pisani, A.; Tuttolomondo, A.; Pieroni, M. Effects of Current Therapies on Disease Progression in Fabry Disease: A Narrative Review for Better Patient Management in Clinical Practice. Adv. Ther. 2025, 42, 597–635. [Google Scholar] [CrossRef]
- Germain, D.P.; Charrow, J.; Desnick, R.J.; Guffon, N.; Kempf, J.; Lachmann, R.H.; Lemay, R.; Linthorst, G.E.; Packman, S.; Scott, C.R.; et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J. Med. Genet. 2015, 52, 353–358. [Google Scholar] [CrossRef]
- Barbey, F.; Livio, F. Safety of enzyme replacement therapy. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Ramaswami, U.; Pintos-Morell, G.; Kampmann, C.; Nicholls, K.; Niu, D.M.; Reisin, R.; West, M.L.; Anagnostopoulou, C.; Botha, J.; Jazukeviciene, D.; et al. Two decades of experience of the Fabry Outcome Survey provides further confirmation of the long-term effectiveness of agalsidase alfa enzyme replacement therapy. Mol. Genet. Metab. Rep. 2025, 43, 101215. [Google Scholar] [CrossRef]
- Oudit, G.Y.; DasMahapatra, P.; Lyn, N.; Wilson, F.R.; Adeyemi, A.; Lee, C.S.; Crespo, A.; Namdar, M. A systematic literature review to evaluate the cardiac and cerebrovascular outcomes of patients with Fabry disease treated with agalsidase Beta. Front. Cardiovasc. Med. 2024, 11, 1415547. [Google Scholar] [CrossRef]
- Batista, J.L.; Hariri, A.; Maski, M.; Richards, S.; Gudivada, B.; Raynor, L.A.; Ponce, E.; Wanner, C.; Desnick, R.J. Reduction in kidney function decline and risk of severe clinical events in agalsidase beta-treated Fabry disease patients: A matched analysis from the Fabry Registry. Clin. Kidney J. 2024, 17, sfae194. [Google Scholar] [CrossRef]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in Fabry disease: 30-month results from the open-label extension of the randomized, phase 3 ATTRACT study. Mol. Genet. Metab. 2020, 131, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Bichet, D.G.; Hopkin, R.J.; Aguiar, P.; Allam, S.R.; Chien, Y.H.; Giugliani, R.; Kallish, S.; Kineen, S.; Lidove, O.; Niu, D.M.; et al. Consensus recommendations for the treatment and management of patients with Fabry disease on migalastat: A modified Delphi study. Front. Med. 2023, 10, 1220637. [Google Scholar] [CrossRef]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Mancia, G.; Kreutz, R.; Brunstrom, M.; Burnier, M.; Grassi, G.; Januszewicz, A.; Muiesan, M.L.; Tsioufis, K.; Agabiti-Rosei, E.; Algharably, E.A.E.; et al. 2023 ESH Guidelines for the management of arterial hypertension The Task Force for the management of arterial hypertension of the European Society of Hypertension: Endorsed by the International Society of Hypertension (ISH) and the European Renal Association (ERA). J. Hypertens. 2023, 41, 1874–2071. [Google Scholar] [CrossRef]
- Kleindorfer, D.O.; Towfighi, A.; Chaturvedi, S.; Cockroft, K.M.; Gutierrez, J.; Lombardi-Hill, D.; Kamel, H.; Kernan, W.N.; Kittner, S.J.; Leira, E.C.; et al. 2021 Guideline for the Prevention of Stroke in Patients with Stroke and Transient Ischemic Attack: A Guideline From the American Heart Association/American Stroke Association. Stroke 2021, 52, e364–e467. [Google Scholar] [CrossRef]
- Moon, J.S.; Kang, S.; Choi, J.H.; Lee, K.A.; Moon, J.H.; Chon, S.; Kim, D.J.; Kim, H.J.; Seo, J.A.; Kim, M.K.; et al. 2023 Clinical Practice Guidelines for Diabetes Management in Korea: Full Version Recommendation of the Korean Diabetes Association. Diabetes Metab. J. 2024, 48, 546–708. [Google Scholar] [CrossRef]
- Park, H.K.; Ko, S.B.; Jung, K.H.; Jang, M.U.; Kim, D.H.; Kim, J.T.; Choi, J.C.; Jeong, H.S.; Kim, C.; Heo, J.H.; et al. 2022 Update of the Korean Clinical Practice Guidelines for Stroke: Antithrombotic Therapy for Patients with Acute Ischemic Stroke or Transient Ischemic Attack. J. Stroke 2022, 24, 166–175. [Google Scholar] [CrossRef]
- Hughes, D.; Linhart, A.; Gurevich, A.; Kalampoki, V.; Jazukeviciene, D.; Feriozzi, S. Prompt Agalsidase Alfa Therapy Initiation is Associated with Improved Renal and Cardiovascular Outcomes in a Fabry Outcome Survey Analysis. Drug Des. Devel. Ther. 2021, 15, 3561–3572. [Google Scholar] [CrossRef]
- Shimohata, H.; Yamashita, M.; Yamada, K.; Hirayama, K.; Kobayashi, M. Treatment of Fabry Nephropathy: A Literature Review. Medicina 2023, 59, 1478. [Google Scholar] [CrossRef] [PubMed]
- Argoff, C.E.; Barton, N.W.; Brady, R.O.; Ziessman, H.A. Gastrointestinal symptoms and delayed gastric emptying in Fabry’s disease: Response to metoclopramide. Nucl. Med. Commun. 1998, 19, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Zar-Kessler, C.; Karaa, A.; Sims, K.B.; Clarke, V.; Kuo, B. Understanding the gastrointestinal manifestations of Fabry disease: Promoting prompt diagnosis. Therap. Adv. Gastroenterol. 2016, 9, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Mengel, E.; Kutschke, G.; Kim, K.S.; Birklein, F.; Krummenauer, F.; Beck, M. Use of gabapentin to reduce chronic neuropathic pain in Fabry disease. J. Inherit. Metab. Dis. 2003, 26, 413–414. [Google Scholar] [CrossRef]
- World Health Organization. World Report on Hearing; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Bauer, C.A. Tinnitus. N. Engl. J. Med. 2018, 378, 1224–1231. [Google Scholar] [CrossRef]
- Yazdanfard, P.D.W.; Effraimidis, G.; Madsen, C.V.; Nielsen, L.H.; Rasmussen, Å.K.; Petersen, J.H.; Sørensen, S.S.; Køber, L.; Fraga de Abreu, V.H.; Larsen, V.A.; et al. Hearing loss in fabry disease: A 16 year follow-up study of the Danish nationwide cohort. Mol. Genet. Metab. Rep. 2022, 31, 100841. [Google Scholar] [CrossRef]
- Chandrasekhar, S.S.; Tsai Do, B.S.; Schwartz, S.R.; Bontempo, L.J.; Faucett, E.A.; Finestone, S.A.; Hollingsworth, D.B.; Kelley, D.M.; Kmucha, S.T.; Moonis, G.; et al. Clinical Practice Guideline: Sudden Hearing Loss (Update). Otolaryngol. Head Neck Surg. 2019, 161, S1–S45. [Google Scholar] [CrossRef]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef]
- Schuller, Y.; Linthorst, G.E.; Hollak, C.E.; Van Schaik, I.N.; Biegstraaten, M. Pain management strategies for neuropathic pain in Fabry disease--a systematic review. BMC Neurol. 2016, 16, 25. [Google Scholar] [CrossRef]
- Chincholkar, M. Analgesic mechanisms of gabapentinoids and effects in experimental pain models: A narrative review. Br. J. Anaesth. 2018, 120, 1315–1334. [Google Scholar] [CrossRef]
- Obata, H. Analgesic Mechanisms of Antidepressants for Neuropathic Pain. Int. J. Mol. Sci. 2017, 18, 2483. [Google Scholar] [CrossRef] [PubMed]
- Politei, J.M.; Bouhassira, D.; Germain, D.P.; Goizet, C.; Guerrero-Sola, A.; Hilz, M.J.; Hutton, E.J.; Karaa, A.; Liguori, R.; Üçeyler, N.; et al. Pain in Fabry Disease: Practical Recommendations for Diagnosis and Treatment. CNS Neurosci. Ther. 2016, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Tremont-Lukats, I.W.; Megeff, C.; Backonja, M.M. Anticonvulsants for neuropathic pain syndromes: Mechanisms of action and place in therapy. Drugs 2000, 60, 1029–1052. [Google Scholar] [CrossRef] [PubMed]
- Vučković, S.; Srebro, D.; Vujović, K.S.; Vučetić, Č.; Prostran, M. Cannabinoids and Pain: New Insights From Old Molecules. Front. Pharmacol. 2018, 9, 1259. [Google Scholar] [CrossRef]
- O’Connor, A.B.; Dworkin, R.H. Treatment of neuropathic pain: An overview of recent guidelines. Am. J. Med. 2009, 122, S22–S32. [Google Scholar] [CrossRef]
- Rogawski, M.A.; Löscher, W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat. Med. 2004, 10, 685–692. [Google Scholar] [CrossRef]
- Jensen, T.S.; Gottrup, H.; Sindrup, S.H.; Bach, F.W. The clinical picture of neuropathic pain. Eur. J. Pharmacol. 2001, 429, 1–11. [Google Scholar] [CrossRef]
- Lockman, L.A.; Hunninghake, D.B.; Krivit, W.; Desnick, R.J. Relief of pain of Fabry’s disease by diphenylhydantoin. Neurology 1973, 23, 871–875. [Google Scholar] [CrossRef]
- Dworkin, R.H.; O’Connor, A.B.; Backonja, M.; Farrar, J.T.; Finnerup, N.B.; Jensen, T.S.; Kalso, E.A.; Loeser, J.D.; Miaskowski, C.; Nurmikko, T.J.; et al. Pharmacologic management of neuropathic pain: Evidence-based recommendations. Pain 2007, 132, 237–251. [Google Scholar] [CrossRef]
- Niesters, M.; Martini, C.; Dahan, A. Ketamine for chronic pain: Risks and benefits. Br. J. Clin. Pharmacol. 2014, 77, 357–367. [Google Scholar] [CrossRef]
- Teasell, R.W.; Arnold, J.M. Alpha-1 adrenoceptor hyperresponsiveness in three neuropathic pain states: Complex regional pain syndrome 1, diabetic peripheral neuropathic pain and central pain states following spinal cord injury. Pain Res. Manag. 2004, 9, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Paim-Marques, L.; de Oliveira, R.J.; Appenzeller, S. Multidisciplinary Management of Fabry Disease: Current Perspectives. J. Multidiscip. Healthc. 2022, 15, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Juan, P.; Hernan, A.; Beatriz, S.A.; Gustavo, C.; Antonio, M.; Eduardo, T.; Raul, D.; Margarita, L.; Mariana, B.; Daniela, G.; et al. Fabry disease: Multidisciplinary evaluation after 10 years of treatment with agalsidase Beta. JIMD Rep. 2014, 16, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Liu, J.; Chen, J.; Zhou, S.; Wang, Y.; Yu, L.; Sun, L.; Wang, L.; Wu, B.; Liu, F.; et al. Multidisciplinary approach to screening and management of children with Fabry disease: Practice at a Tertiary Children’s Hospital in China. Orphanet J. Rare Dis. 2021, 16, 509. [Google Scholar] [CrossRef]
- Zamorano, J.; Serra, V.; Pérez de Isla, L.; Feltes, G.; Calli, A.; Barbado, F.J.; Torras, J.; Hernandez, S.; Herrera, J.; Herrero, J.A.; et al. Usefulness of tissue Doppler on early detection of cardiac disease in Fabry patients and potential role of enzyme replacement therapy (ERT) for avoiding progression of disease. Eur. J. Echocardiogr. 2011, 12, 671–677. [Google Scholar] [CrossRef]
- Frustaci, A.; Verardo, R.; Galea, N.; Alfarano, M.; Magnocavallo, M.; Marchitelli, L.; Sansone, L.; Belli, M.; Cristina, M.; Frustaci, E.; et al. Long-Term Clinical-Pathologic Results of Enzyme Replacement Therapy in Prehypertrophic Fabry Disease Cardiomyopathy. J. Am. Heart Assoc. 2024, 13, e032734. [Google Scholar] [CrossRef]
- van der Veen, S.J.; Korver, S.; Hirsch, A.; Hollak, C.E.M.; Wijburg, F.A.; Brands, M.M.; Tondel, C.; van Kuilenburg, A.B.P.; Langeveld, M. Early start of enzyme replacement therapy in pediatric male patients with classical Fabry disease is associated with attenuated disease progression. Mol. Genet. Metab. 2022, 135, 163–169. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).