

Cardiotonic Steroids as a Potential Novel Approach for Immunomodulation in Inflammatory Bowel Disease

 , ,
, , {kind=link}
{kind=link}
Abstract
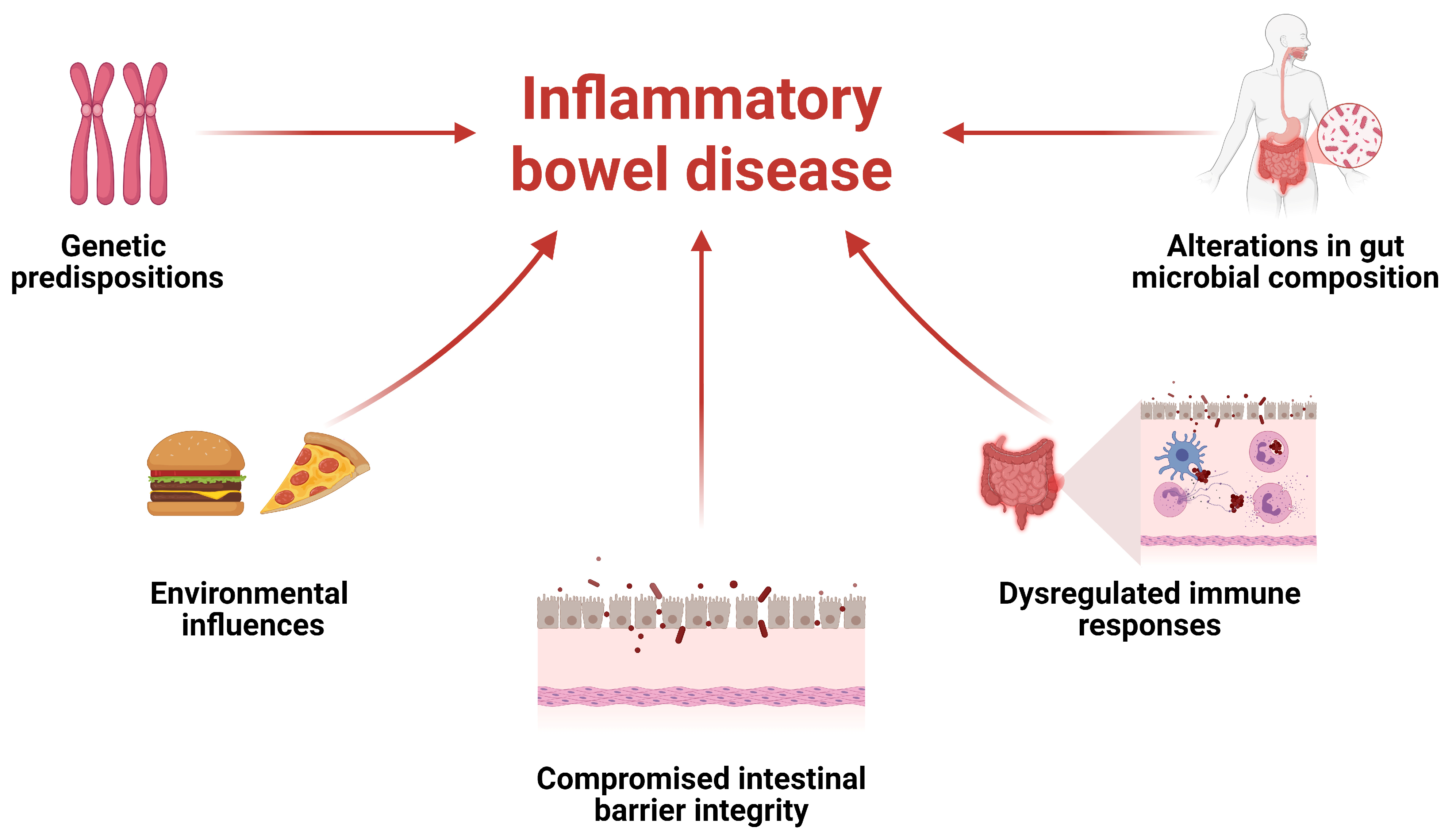
1. Introduction
2. Immunological Insights into IBD
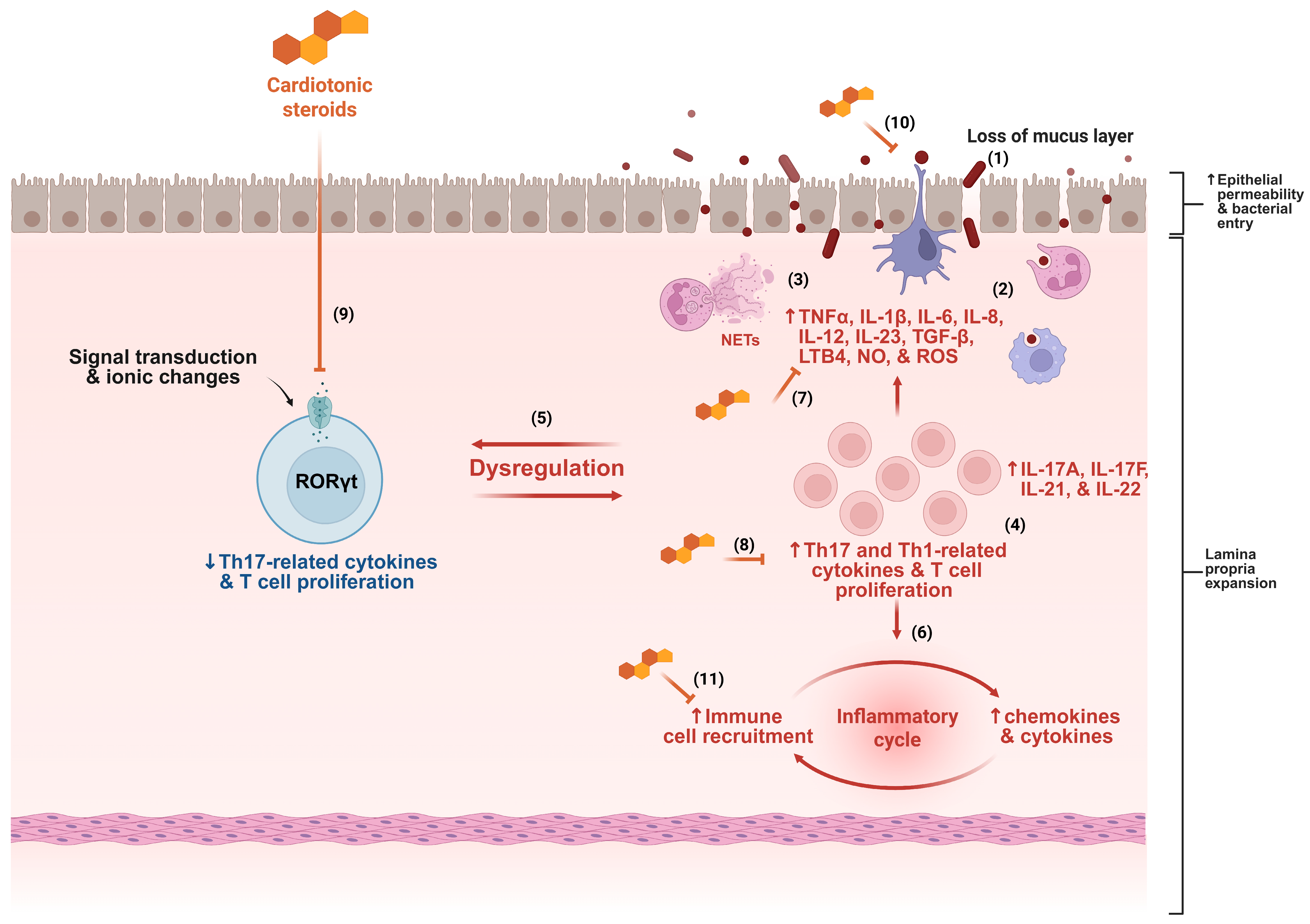
3. Cardiotonic Steroids as Potential New Therapeutics in IBD Treatment
4. The Gut Microbiota Plays an Important Role in Cardiotonic Steroid Bioavailability
5. Final Considerations and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farrell, D.; Artom, M.; Czuber-Dochan, W.; Jelsness-Jorgensen, L.P.; Norton, C.; Savage, E. Interventions for fatigue in inflammatory bowel disease. Cochrane Database Syst. Rev. 2020, 4, CD012005. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Diez-Martin, E.; Hernandez-Suarez, L.; Munoz-Villafranca, C.; Martin-Souto, L.; Astigarraga, E.; Ramirez-Garcia, A.; Barreda-Gomez, G. Inflammatory bowel disease: A comprehensive analysis of molecular bases, predictive biomarkers, diagnostic methods, and therapeutic options. Int. J. Mol. Sci. 2024, 25, 7062. [Google Scholar] [CrossRef] [PubMed]
- Roda, G.; Chien Ng, S.; Kotze, P.G.; Argollo, M.; Panaccione, R.; Spinelli, A.; Kaser, A.; Peyrin-Biroulet, L.; Danese, S. Crohn’s disease. Nat. Rev. Dis. Primers 2020, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Siegmund, B.; Le Berre, C.; Wei, S.C.; Ferrante, M.; Shen, B.; Bernstein, C.N.; Danese, S.; Peyrin-Biroulet, L.; Hibi, T. Ulcerative colitis. Nat. Rev. Dis. Primers 2020, 6, 74. [Google Scholar] [CrossRef]
- Caron, B.; Honap, S.; Peyrin-Biroulet, L. Epidemiology of inflammatory bowel disease across the ages in the era of advanced therapies. J. Crohns Colitis 2024, 18, ii3–ii15. [Google Scholar] [CrossRef]
- Kaplan, G.G.; Windsor, J.W. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 56–66. [Google Scholar] [CrossRef]
- M’Koma, A.E. Inflammatory bowel disease: Clinical diagnosis and surgical treatment-overview. Medicina 2022, 58, 567. [Google Scholar] [CrossRef]
- Van Assche, G.; Dignass, A.; Bokemeyer, B.; Danese, S.; Gionchetti, P.; Moser, G.; Beaugerie, L.; Gomollon, F.; Hauser, W.; Herrlinger, K.; et al. Second European evidence-based consensus on the diagnosis and management of ulcerative colitis part 3: Special situations. J. Crohns Colitis 2013, 7, 1–33. [Google Scholar] [CrossRef]
- Rogler, G.; Singh, A.; Kavanaugh, A.; Rubin, D.T. Extraintestinal manifestations of inflammatory bowel disease: Current concepts, treatment, and implications for disease management. Gastroenterology 2021, 161, 1118–1132. [Google Scholar] [CrossRef]
- Chang, J.T. Pathophysiology of inflammatory bowel diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.C.; Itzkowitz, S.H. Colorectal cancer in inflammatory bowel disease: Mechanisms and management. Gastroenterology 2022, 162, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.K.; Thabane, M.; Steinhart, A.H.; Newman, J.R.; Anand, A.; Irvine, E.J. Rectal 5-aminosalicylic acid for induction of remission in ulcerative colitis. Cochrane Database Syst. Rev. 2010, 1, 1–86. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.C.; Achkar, J.P.; Khan, K.J.; Kane, S.V.; Talley, N.J.; Marshall, J.K.; Moayyedi, P. Efficacy of 5-aminosalicylates in ulcerative colitis: Systematic review and meta-analysis. Am. J. Gastroenterol. 2011, 106, 601–616. [Google Scholar] [CrossRef]
- Travis, S.P.; Stange, E.F.; Lemann, M.; Oresland, T.; Bemelman, W.A.; Chowers, Y.; Colombel, J.F.; D’Haens, G.; Ghosh, S.; Marteau, P.; et al. European evidence-based Consensus on the management of ulcerative colitis: Current management. J. Crohns Colitis 2008, 2, 24–62. [Google Scholar] [CrossRef]
- Cohen, R.D.; Woseth, D.M.; Thisted, R.A.; Hanauer, S.B. A meta-analysis and overview of the literature on treatment options for left-sided ulcerative colitis and ulcerative proctitis. Am. J. Gastroenterol. 2000, 95, 1263–1276. [Google Scholar] [CrossRef]
- Sairenji, T.; Collins, K.L.; Evans, D.V. An update on inflammatory bowel disease. Prim. Care 2017, 44, 673–692. [Google Scholar] [CrossRef]
- Faubion, W.A., Jr.; Loftus, E.V., Jr.; Harmsen, W.S.; Zinsmeister, A.R.; Sandborn, W.J. The natural history of corticosteroid therapy for inflammatory bowel disease: A population-based study. Gastroenterology 2001, 121, 255–260. [Google Scholar] [CrossRef]
- Akiho, H.; Yokoyama, A.; Abe, S.; Nakazono, Y.; Murakami, M.; Otsuka, Y.; Fukawa, K.; Esaki, M.; Niina, Y.; Ogino, H. Promising biological therapies for ulcerative colitis: A review of the literature. World J. Gastrointest. Pathophysiol. 2015, 6, 219–227. [Google Scholar] [CrossRef]
- Ford, A.C.; Sandborn, W.J.; Khan, K.J.; Hanauer, S.B.; Talley, N.J.; Moayyedi, P. Efficacy of biological therapies in inflammatory bowel disease: Systematic review and meta-analysis. Am. J. Gastroenterol. 2011, 106, 644–659. [Google Scholar] [CrossRef]
- Clark, M.; Colombel, J.F.; Feagan, B.C.; Fedorak, R.N.; Hanauer, S.B.; Kamm, M.A.; Mayer, L.; Regueiro, C.; Rutgeerts, P.; Sandborn, W.J.; et al. American gastroenterological association consensus development conference on the use of biologics in the treatment of inflammatory bowel disease, June 21–23, 2006. Gastroenterology 2007, 133, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Axelrad, J.E.; Lichtiger, S.; Yajnik, V. Inflammatory bowel disease and cancer: The role of inflammation, immunosuppression, and cancer treatment. World J. Gastroenterol. 2016, 22, 4794–4801. [Google Scholar] [CrossRef] [PubMed]
- Vieujean, S.; Jairath, V.; Peyrin-Biroulet, L.; Dubinsky, M.; Iacucci, M.; Magro, F.; Danese, S. Understanding the therapeutic toolkit for inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2025, 22, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Bruner, L.P.; White, A.M.; Proksell, S. Inflammatory bowel disease. Prim. Care 2023, 50, 411–427. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Haneishi, Y.; Furuya, Y.; Hasegawa, M.; Picarelli, A.; Rossi, M.; Miyamoto, J. Inflammatory bowel diseases and gut microbiota. Int. J. Mol. Sci. 2023, 24, 3817. [Google Scholar] [CrossRef]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sanchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of inflammatory bowel disease: Innate immune system. Int. J. Mol. Sci. 2023, 24, 1526. [Google Scholar] [CrossRef]
- Zhou, G.; Yu, L.; Fang, L.; Yang, W.; Yu, T.; Miao, Y.; Chen, M.; Wu, K.; Chen, F.; Cong, Y.; et al. CD177+ neutrophils as functionally activated neutrophils negatively regulate IBD. Gut 2018, 67, 1052–1063. [Google Scholar] [CrossRef]
- Kang, L.; Fang, X.; Song, Y.H.; He, Z.X.; Wang, Z.J.; Wang, S.L.; Li, Z.S.; Bai, Y. Neutrophil-epithelial crosstalk during intestinal inflammation. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 1257–1267. [Google Scholar] [CrossRef]
- Drury, B.; Hardisty, G.; Gray, R.D.; Ho, G.T. Neutrophil extracellular traps in inflammatory bowel disease: Pathogenic mechanisms and clinical translation. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 321–333. [Google Scholar] [CrossRef]
- Chen, H.; Wu, X.; Xu, C.; Lin, J.; Liu, Z. Dichotomous roles of neutrophils in modulating pathogenic and repair processes of inflammatory bowel diseases. Precis. Clin. Med. 2021, 4, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Maronek, M.; Gromova, B.; Liptak, R.; Konecna, B.; Pastorek, M.; Cechova, B.; Harsanyova, M.; Budis, J.; Smolak, D.; Radvanszky, J.; et al. Extracellular DNA correlates with intestinal inflammation in chemically induced colitis in mice. Cells 2021, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.X.; Liu, Z.J. Potential roles of neutrophils in regulating intestinal mucosal inflammation of inflammatory bowel disease. J. Dig. Dis. 2017, 18, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, P.; Tsakmaki, A.; Pantazi, E.; Li, K.; Cozzetto, D.; Digby-Bell, J.; Yang, F.; Lo, J.W.; Alberts, E.; Sa, A.C.C.; et al. Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy. Nat. Commun. 2022, 13, 5820. [Google Scholar] [CrossRef]
- Biasi, F.; Leonarduzzi, G.; Oteiza, P.I.; Poli, G. Inflammatory bowel disease: Mechanisms, redox considerations, and therapeutic targets. Antioxid. Redox Signal. 2013, 19, 1711–1747. [Google Scholar] [CrossRef]
- Hansberry, D.R.; Shah, K.; Agarwal, P.; Agarwal, N. Fecal myeloperoxidase as a biomarker for inflammatory bowel disease. Cureus 2017, 9, e1004. [Google Scholar] [CrossRef]
- Seo, D.H.; Che, X.; Kim, S.; Kim, D.H.; Ma, H.W.; Kim, J.H.; Kim, T.I.; Kim, W.H.; Kim, S.W.; Cheon, J.H. Triggering receptor expressed on myeloid cells-1 agonist regulates intestinal inflammation via Cd177+ neutrophils. Front. Immunol. 2021, 12, 650864. [Google Scholar] [CrossRef]
- Bain, C.C.; Schridde, A. Origin, differentiation, and function of intestinal macrophages. Front. Immunol. 2018, 9, 2733. [Google Scholar] [CrossRef]
- Sun, R.; Abraham, C. IL23 promotes antimicrobial pathways in human macrophages, which are reduced with the IBD-protective IL23R R381Q variant. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 673–697. [Google Scholar] [CrossRef]
- Soufli, I.; Toumi, R.; Rafa, H.; Touil-Boukoffa, C. Overview of cytokines and nitric oxide involvement in immuno-pathogenesis of inflammatory bowel diseases. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 353–360. [Google Scholar] [CrossRef]
- El Sayed, S.; Patik, I.; Redhu, N.S.; Glickman, J.N.; Karagiannis, K.; El Naenaeey, E.S.Y.; Elmowalid, G.A.; Abd El Wahab, A.M.; Snapper, S.B.; Horwitz, B.H. CCR2 promotes monocyte recruitment and intestinal inflammation in mice lacking the interleukin-10 receptor. Sci. Rep. 2022, 12, 452. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Wang, B.Y.; Wang, T.T.; Wang, F.F.; Guo, Y.X.; Hua, R.X.; Shang, H.W.; Lu, X.; Xu, J.D. Functions of dendritic cells and its association with intestinal diseases. Cells 2021, 10, 583. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.G.; Vaida, A.; Thompson, L.M.; Ikuomola, F.I.; Caamano, J.H.; Burkitt, M.D.; Miyajima, F.; Williams, J.M.; Campbell, B.J.; Pritchard, D.M.; et al. NF-κB2 signalling in enteroids modulates enterocyte responses to secreted factors from bone marrow-derived dendritic cells. Cell Death Dis. 2019, 10, 896. [Google Scholar] [CrossRef]
- Shi, G.; Li, D.; Ren, J.; Li, X.; Wang, T.; Dou, H.; Hou, Y. mTOR inhibitor INK128 attenuates dextran sodium sulfate-induced colitis by promotion of MDSCs on Treg cell expansion. J. Cell. Physiol. 2019, 234, 1618–1629. [Google Scholar] [CrossRef]
- Xie, Y.; Zhao, Y.; Shi, L.; Li, W.; Chen, K.; Li, M.; Chen, X.; Zhang, H.; Li, T.; Matsuzawa-Ishimoto, Y.; et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J. Clin. Investig. 2020, 130, 2111–2128. [Google Scholar] [CrossRef]
- Ohtani, M.; Hoshii, T.; Fujii, H.; Koyasu, S.; Hirao, A.; Matsuda, S. Cutting edge: mTORC1 in intestinal CD11c+ CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL-10 production. J. Immunol. 2012, 188, 4736–4740. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, L.; Jia, A.; Chen, X.; Yang, Q.; Wang, Y.; Wang, Y.; Liu, R.; Cao, Y.; He, Y.; et al. Glucocorticoids promote the onset of acute experimental colitis and cancer by upregulating mTOR signaling in intestinal epithelial cells. Cancers 2020, 12, 945. [Google Scholar] [CrossRef]
- Franke, A.; Balschun, T.; Karlsen, T.H.; Hedderich, J.; May, S.; Lu, T.; Schuldt, D.; Nikolaus, S.; Rosenstiel, P.; Krawczak, M.; et al. Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat. Genet. 2008, 40, 713–715. [Google Scholar] [CrossRef]
- Anderson, C.A.; Massey, D.C.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Fisher, S.A.; Gwilliam, R.; Jacob, J.; Nimmo, E.R.; Drummond, H.; et al. Investigation of Crohn’s disease risk loci in ulcerative colitis further defines their molecular relationship. Gastroenterology 2009, 136, 523–529.e3. [Google Scholar] [CrossRef]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Brand, S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: New immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut 2009, 58, 1152–1167. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Read, S.; Asseman, C.; Malmstrom, V.; Mottet, C.; Stephens, L.A.; Stepankova, R.; Tlaskalova, H.; Powrie, F. Control of intestinal inflammation by regulatory T cells. Immunol. Rev. 2001, 182, 190–200. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef]
- Monteleone, I.; Pallone, F.; Monteleone, G. Th17-related cytokines: New players in the control of chronic intestinal inflammation. BMC Med. 2011, 9, 122. [Google Scholar] [CrossRef]
- O’Connor, W., Jr.; Kamanaka, M.; Booth, C.J.; Town, T.; Nakae, S.; Iwakura, Y.; Kolls, J.K.; Flavell, R.A. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 2009, 10, 603–609. [Google Scholar] [CrossRef]
- Wedebye Schmidt, E.G.; Larsen, H.L.; Kristensen, N.N.; Poulsen, S.S.; Lynge Pedersen, A.M.; Claesson, M.H.; Pedersen, A.E. TH17 cell induction and effects of IL-17A and IL-17F blockade in experimental colitis. Inflamm. Bowel Dis. 2013, 19, 1567–1576. [Google Scholar] [CrossRef]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis--results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef]
- Herrlinger, K.R.; Diculescu, M.; Fellermann, K.; Hartmann, H.; Howaldt, S.; Nikolov, R.; Petrov, A.; Reindl, W.; Otte, J.M.; Stoynov, S.; et al. Efficacy, safety and tolerability of vidofludimus in patients with inflammatory bowel disease: The ENTRANCE study. J. Crohns Colitis 2013, 7, 636–643. [Google Scholar] [CrossRef]
- Uhlig, H.H.; Powrie, F. Translating immunology into therapeutic concepts for inflammatory bowel disease. Annu. Rev. Immunol. 2018, 36, 755–781. [Google Scholar] [CrossRef] [PubMed]
- Plichta, D.R.; Graham, D.B.; Subramanian, S.; Xavier, R.J. Therapeutic opportunities in inflammatory bowel disease: Mechanistic dissection of host-microbiome relationships. Cell 2019, 178, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zhang, E.; Fang, R.H.; Gao, W.; Zhang, L. Capsulated cellular nanosponges for the treatment of experimental inflammatory bowel disease. ACS Nano 2023, 17, 15893–15904. [Google Scholar] [CrossRef] [PubMed]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef]
- Xu, Y.; Marck, P.; Huang, M.; Xie, J.X.; Wang, T.; Shapiro, J.I.; Cai, L.; Feng, F.; Xie, Z. Biased effect of cardiotonic steroids on Na/K-ATPase-mediated signal transduction. Mol. Pharmacol. 2021, 99, 217–225. [Google Scholar] [CrossRef]
- Hauptman, P.J.; Kelly, R.A. Digitalis. Circulation 1999, 99, 1265–1270. [Google Scholar] [CrossRef]
- Leite, J.A.; Alves, A.K.; Galvao, J.G.; Teixeira, M.P.; Cavalcante-Silva, L.H.; Scavone, C.; Morrot, A.; Rumjanek, V.M.; Rodrigues-Mascarenhas, S. Ouabain modulates zymosan-induced peritonitis in mice. Mediat. Inflamm. 2015, 2015, 265798. [Google Scholar] [CrossRef]
- Galvao, J.; Cavalcante-Silva, L.H.A.; de Almeida Lima, E.; Carvalho, D.C.M.; Alves, A.F.; Mascarenhas, S.R. Ouabain modulates airway remodeling caused by Th2-high asthma in mice. Int. Immunopharmacol. 2022, 109, 108808. [Google Scholar] [CrossRef]
- Cavalcante-Silva, L.H.A.; Carvalho, D.C.M.; de Almeida Lima, E.; Rodrigues-Mascarenhas, S. Ouabain inhibits p38 activation in mice neutrophils. Inflammopharmacology 2021, 29, 1829–1833. [Google Scholar] [CrossRef]
- Carvalho, D.C.M.; Cavalcante-Silva, L.H.A.; Lima, E.A.; Galvao, J.; Alves, A.K.A.; Feijo, P.R.O.; Quintas, L.E.M.; Rodrigues-Mascarenhas, S. Marinobufagenin inhibits neutrophil migration and proinflammatory cytokines. J. Immunol. Res. 2019, 2019, 1094520. [Google Scholar] [CrossRef]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef] [PubMed]
- Fender, J.; Klocker, J.; Boivin-Jahns, V.; Ravens, U.; Jahns, R.; Lorenz, K. “Cardiac glycosides”-quo vaditis?-past, present, and future? Naunyn Schmiedebergs Arch. Pharmacol. 2024, 397, 9521–9531. [Google Scholar] [CrossRef] [PubMed]
- Whayne, T.F., Jr. Clinical use of digitalis: A state of the art review. Am. J. Cardiovasc. Drugs 2018, 18, 427–440. [Google Scholar] [CrossRef]
- Xiao, S.; Yosef, N.; Yang, J.; Wang, Y.; Zhou, L.; Zhu, C.; Wu, C.; Baloglu, E.; Schmidt, D.; Ramesh, R.; et al. Small-molecule RORγt antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity 2014, 40, 477–489. [Google Scholar] [CrossRef]
- Huh, J.R.; Leung, M.W.; Huang, P.; Ryan, D.A.; Krout, M.R.; Malapaka, R.R.; Chow, J.; Manel, N.; Ciofani, M.; Kim, S.V.; et al. Digoxin and its derivatives suppress Th17 cell differentiation by antagonizing RORγt activity. Nature 2011, 472, 486–490. [Google Scholar] [CrossRef]
- Lee, J.; Baek, S.; Lee, J.; Lee, J.; Lee, D.G.; Park, M.K.; Cho, M.L.; Park, S.H.; Kwok, S.K. Digoxin ameliorates autoimmune arthritis via suppression of Th17 differentiation. Int. Immunopharmacol. 2015, 26, 103–111. [Google Scholar] [CrossRef]
- Tani, S.; Takano, R.; Tamura, S.; Oishi, S.; Iwaizumi, M.; Hamaya, Y.; Takagaki, K.; Nagata, T.; Seto, S.; Horii, T.; et al. Digoxin attenuates murine experimental colitis by downregulating Th17-related cytokines. Inflamm. Bowel Dis. 2017, 23, 728–738. [Google Scholar] [CrossRef]
- da Silva, J.M.C.; Azevedo, A.D.N.; Barbosa, R.; Teixeira, M.P.; Vianna, T.A.G.; Fittipaldi, J.; Cabral, V.R.; Paiva, L.S. Ouabain decreases regulatory T cell number in mice by reducing IL-2 secretion. Neuroimmunomodulation 2019, 26, 188–197. [Google Scholar] [CrossRef]
- Jacob, P.L.; Leite, J.A.; Alves, A.K.; Rodrigues, Y.K.; Amorim, F.M.; Neris, P.L.; Oliveira, M.R.; Rodrigues-Mascarenhas, S. Immunomodulatory activity of ouabain in Leishmania leishmania amazonensis-infected Swiss mice. Parasitol. Res. 2013, 112, 1313–1321. [Google Scholar] [CrossRef]
- Rawat, K.; Shrivastava, A. Neutrophils as emerging protagonists and targets in chronic inflammatory diseases. Inflamm. Res. 2022, 71, 1477–1488. [Google Scholar] [CrossRef]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.A.; Medeiros, A.B.A.; Soares, M.M.; Lima, E.A.; Oliveira, G.; Leite, M.; Machado, M.V.; Villar, J.; Barbosa, L.A.; Scavone, C.; et al. Evaluation of anti-inflammatory activity of the new cardiotonic steroid gamma-benzylidene digoxin 8 (BD-8) in mice. Cells 2024, 13, 1568. [Google Scholar] [CrossRef] [PubMed]
- Ponce, A.; Larre, I.; Castillo, A.; Garcia-Villegas, R.; Romero, A.; Flores-Maldonado, C.; Martinez-Rendon, J.; Contreras, R.G.; Cereijido, M. Ouabain increases gap junctional communication in epithelial cells. Cell. Physiol. Biochem. 2014, 34, 2081–2090. [Google Scholar] [CrossRef]
- Larre, I.; Lazaro, A.; Contreras, R.G.; Balda, M.S.; Matter, K.; Flores-Maldonado, C.; Ponce, A.; Flores-Benitez, D.; Rincon-Heredia, R.; Padilla-Benavides, T.; et al. Ouabain modulates epithelial cell tight junction. Proc. Natl. Acad. Sci. USA 2010, 107, 11387–11392. [Google Scholar] [CrossRef]
- Kinoshita, P.F.; Yshii, L.M.; Vasconcelos, A.R.; Orellana, A.M.; Lima Lde, S.; Davel, A.P.; Rossoni, L.V.; Kawamoto, E.M.; Scavone, C. Signaling function of Na,K-ATPase induced by ouabain against LPS as an inflammation model in hippocampus. J. Neuroinflammation 2014, 11, 218. [Google Scholar] [CrossRef]
- Mazala-de-Oliveira, T.; de Figueiredo, C.S.; de Rezende Correa, G.; da Silva, M.S.; Miranda, R.L.; de Azevedo, M.A.; Cossenza, M.; Dos Santos, A.A.; Giestal-de-Araujo, E. Ouabain-Na+/K+-ATPase signaling regulates retinal neuroinflammation and ROS production preventing neuronal death by an autophagy-dependent mechanism following optic nerve axotomy in vitro. Neurochem. Res. 2022, 47, 723–738. [Google Scholar] [CrossRef]
- Galvão, J.G.F.M.; Cavalcante-Silva, L.H.A.; Carvalho, D.C.M.; Ferreira, L.K.D.P.; Monteiro, T.M.; Alves, A.F.; Ferreira, L.A.M.P.; Gadelha, F.A.A.F.; Piuvezam, M.R.; Rodrigues-Mascarenhas, S. Ouabain attenuates ovalbumin-induced airway inflammation. Inflamm. Res. 2017, 66, 1117–1130. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Hamlyn, J.M. Ouabain, endogenous ouabain and ouabain-like factors: The Na+ pump/ouabain receptor, its linkage to NCX, and its myriad functions. Cell Calcium 2020, 86, 102159. [Google Scholar] [CrossRef]
- Ezike, T.C.; Okpala, U.S.; Onoja, U.L.; Nwike, C.P.; Ezeako, E.C.; Okpara, O.J.; Okoroafor, C.C.; Eze, S.C.; Kalu, O.L.; Odoh, E.C.; et al. Advances in drug delivery systems, challenges and future directions. Heliyon 2023, 9, e17488. [Google Scholar] [CrossRef]
- Snelson, M.; RMuralitharan, R.; Liu, C.F.; Markó, L.; Forslund, S.K.; Marques, F.Z.; Tang, W.W. Gut-heart axis: The role of gut microbiota and metabolites in heart failure. Circ. Res. 2025, 136, 1382–1406. [Google Scholar] [CrossRef]
- Brennan, C.A.; Garrett, W.S. Gut microbiota, inflammation, and colorectal cancer. Annu. Rev. Microbiol. 2016, 70, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Han, Y.; Huang, W.; Jin, M.; Gao, Z. The influence of the gut microbiota on the bioavailability of oral drugs. Acta Pharm. Sin. B 2021, 11, 1789–1812. [Google Scholar] [CrossRef]
- Saha, J.R.; Butler, V.P., Jr.; Neu, H.C.; Lindenbaum, J. Digoxin-inactivating bacteria: Identification in human gut flora. Science 1983, 220, 325–327. [Google Scholar] [CrossRef]
- Lindenbaum, J.; Rund, D.G.; Butler, V.P., Jr.; Tse-Eng, D.; Saha, J.R. Inactivation of digoxin by the gut flora: Reversal by antibiotic therapy. N. Engl. J. Med. 1981, 305, 789–794. [Google Scholar] [CrossRef]
- Haiser, H.J.; Gootenberg, D.B.; Chatman, K.; Sirasani, G.; Balskus, E.P.; Turnbaugh, P.J. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 2013, 341, 295–298. [Google Scholar] [CrossRef]
- Haiser, H.J.; Seim, K.L.; Balskus, E.P.; Turnbaugh, P.J. Mechanistic insight into digoxin inactivation by Eggerthella lenta augments our understanding of its pharmacokinetics. Gut Microbes 2014, 5, 233–238. [Google Scholar] [CrossRef]
- Kumar, K.; Jaiswal, S.K.; Dhoke, G.V.; Srivastava, G.N.; Sharma, A.K.; Sharma, V.K. Mechanistic and structural insight into promiscuity based metabolism of cardiac drug digoxin by gut microbial enzyme. J. Cell. Biochem. 2018, 119, 5287–5296. [Google Scholar] [CrossRef]
- Sperry, J.F.; Wilkins, T.D. Arginine, a growth-limiting factor for Eubacterium lentum. J. Bacteriol. 1976, 127, 780–784. [Google Scholar] [CrossRef]
- Doestzada, M.; Vila, A.V.; Zhernakova, A.; Koonen, D.P.Y.; Weersma, R.K.; Touw, D.J.; Kuipers, F.; Wijmenga, C.; Fu, J. Pharmacomicrobiomics: A novel route towards personalized medicine? Protein Cell 2018, 9, 432–445. [Google Scholar] [CrossRef]
- Alexander, M.; Ang, Q.Y.; Nayak, R.R.; Bustion, A.E.; Sandy, M.; Zhang, B.; Upadhyay, V.; Pollard, K.S.; Lynch, S.V.; Turnbaugh, P.J. Human gut bacterial metabolism drives Th17 activation and colitis. Cell Host Microbe 2022, 30, 17–30. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavalcante-Silva, L.H.A.; Sales-Neto, J.M.d.; Soares, M.M.; Ferreira, D.A.; Medeiros, A.B.A.; Rodrigues-Mascarenhas, S. Cardiotonic Steroids as a Potential Novel Approach for Immunomodulation in Inflammatory Bowel Disease. J. Clin. Med. 2025, 14, 4132. https://doi.org/10.3390/jcm14124132
Cavalcante-Silva LHA, Sales-Neto JMd, Soares MM, Ferreira DA, Medeiros ABA, Rodrigues-Mascarenhas S. Cardiotonic Steroids as a Potential Novel Approach for Immunomodulation in Inflammatory Bowel Disease. Journal of Clinical Medicine. 2025; 14(12):4132. https://doi.org/10.3390/jcm14124132
Chicago/Turabian StyleCavalcante-Silva, Luiz Henrique Agra, José Marreiro de Sales-Neto, Mariana Mendonça Soares, Davi Azevedo Ferreira, Anna Beatriz Araujo Medeiros, and Sandra Rodrigues-Mascarenhas. 2025. "Cardiotonic Steroids as a Potential Novel Approach for Immunomodulation in Inflammatory Bowel Disease" Journal of Clinical Medicine 14, no. 12: 4132. https://doi.org/10.3390/jcm14124132
APA StyleCavalcante-Silva, L. H. A., Sales-Neto, J. M. d., Soares, M. M., Ferreira, D. A., Medeiros, A. B. A., & Rodrigues-Mascarenhas, S. (2025). Cardiotonic Steroids as a Potential Novel Approach for Immunomodulation in Inflammatory Bowel Disease. Journal of Clinical Medicine, 14(12), 4132. https://doi.org/10.3390/jcm14124132