
Decoding SCN2A Variants: Bridging Genetics and Phenotypes in Autism Spectrum Disorder

 , ,
, , 

Abstract
1. Introduction
2. Methods/Materials
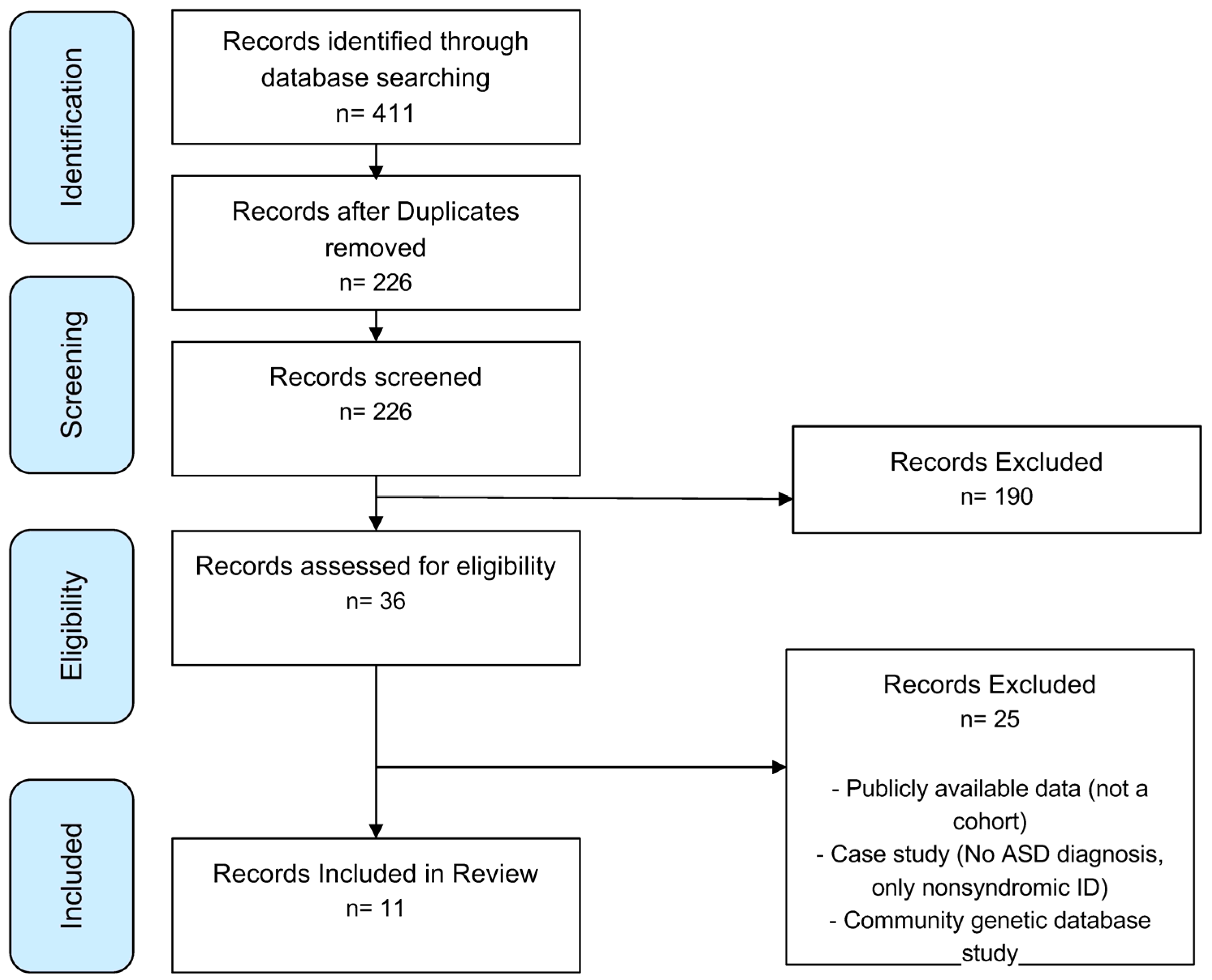
2.1. Search Strategy
2.2. Study Selection
2.3. Data Extraction
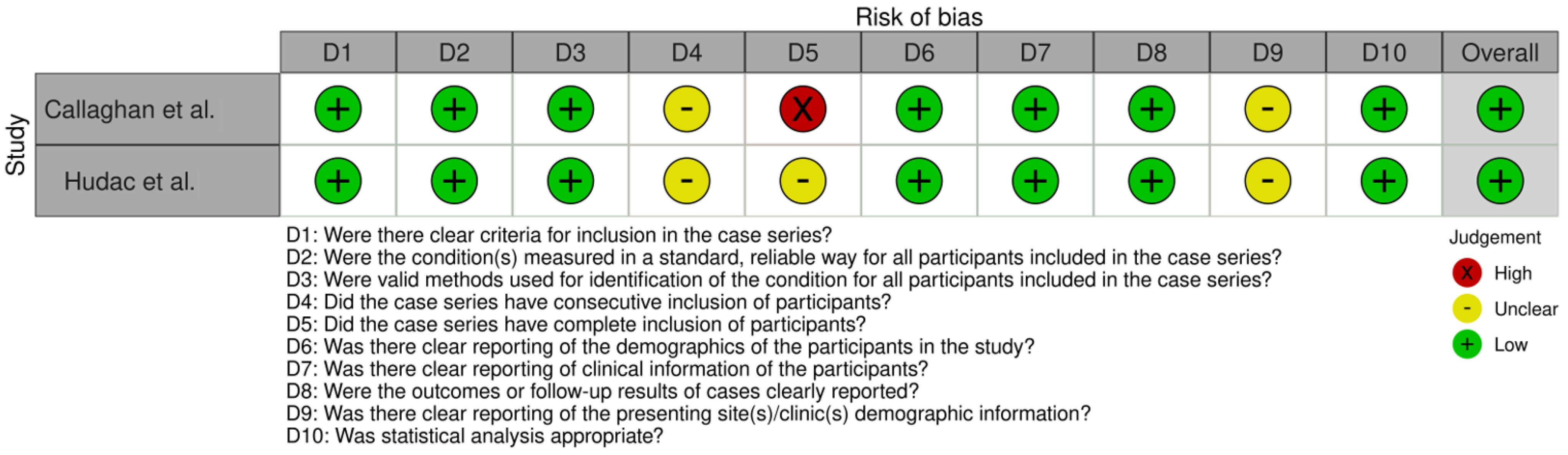
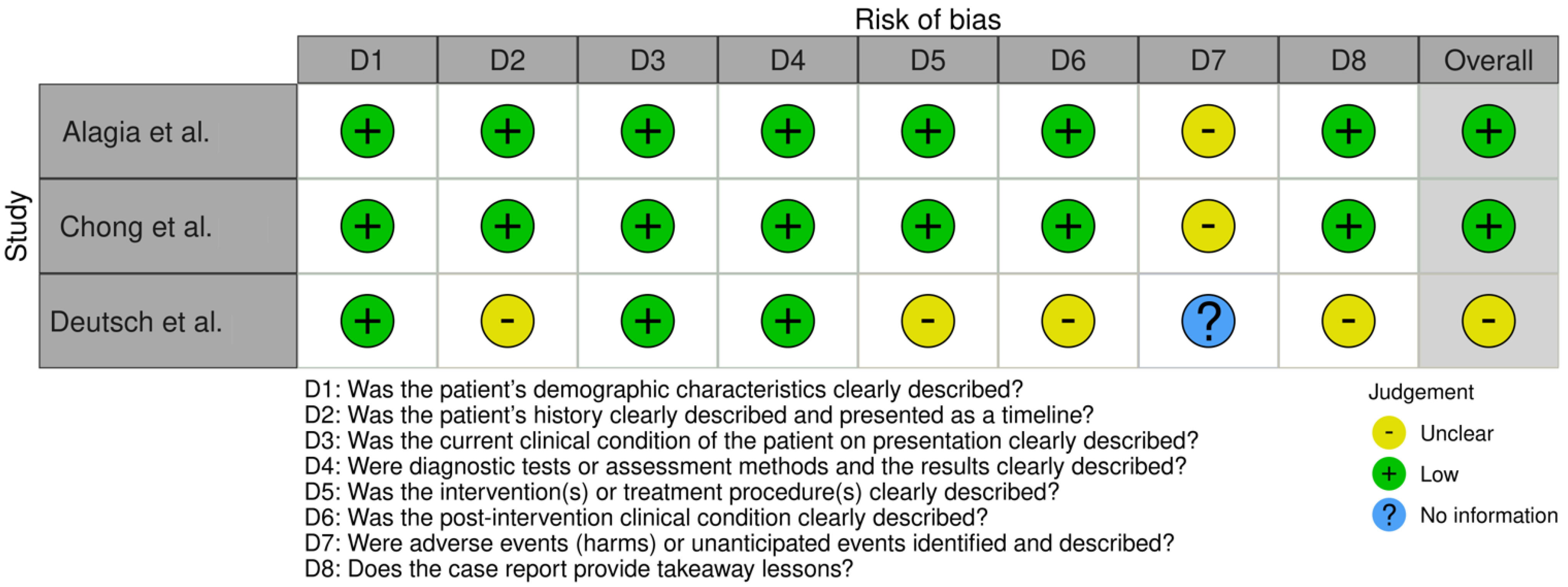
2.4. Risk of Bias and Quality Assessment
2.5. Data Synthesis
3. Results


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Observations | N (Total) | N (ASD) | N (ASD + SCN2a) | ASD Diagnosis Tool | Ethnicity | Race | M | F | Age (Range) | Consanguinity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Alagia et al. [59] | Includes dystonic movements | 1 | 1 | 1 | NR | African | Black | 1 | 14 | NR | |
| Arnett et al. [63] | Subset includes SCN2A cases | 65 | 45 | 10 | ADI-R | NR | NR | 4 | 6 | 5–21 | NR |
| Callaghan et al. [58] | Includes detailed sequencing info | 119 | 119 | 2 | DSM-IV-TR, ADI-R, ADOS-G | NR | NR | 2 | 13–23 | NR | |
| Chong et al. [60] | SCN2A and SCN3A deletions | 1 | 1 | 1 | M-CHAT-R | NR | NR | 1 | 1.75 | NR | |
| Deutsch et al. [61] | Case report, detailed phenotype | 1 | 1 | 1 | Clinical diagnosis | NR | NR | 1 | 29 | NR | |
| Hudac et al. [57] | Explores sensory phenotypes | 39 | 24 | 24 | NR | 33 White, 5 Hispanic | Mixed (Asian, African) | 19 | 20 | 3–22 | NR |
| Krupp et al. [64] | Focuses on mosaic mutations | 22 | 14 | 7 | DSM-V | White, African American, Asian | NR | 14 | 8 | 3–15 | NR |
| Richardson et al. [65] | Detailed phenotypic spectrum | 22 | 3 | 3 | Not specified | White, Asian, Middle Eastern | NR | 10 | 12 | 2–52 | NR |
| Tran et al. [62] | Vietnamese cohort | 100 | 100 | 1 | DSM-V | Vietnamese | NR | 0 | 1 | 3.5 | NR |
| Wolff et al. [49] | Large cohort with heterogeneity | 71 | 23 | 10 | NR | NR | NR | NR | NR | NR | NR |
| Zhang et al. [56] | Pipeline analysis for SCN2A mutations | 354 | 354 | 3 | DSM-V | NR | NR | 279 | 75 | NR | NR |
3.1. Patient Population and Diagnosis
3.2. Phenotypic Data
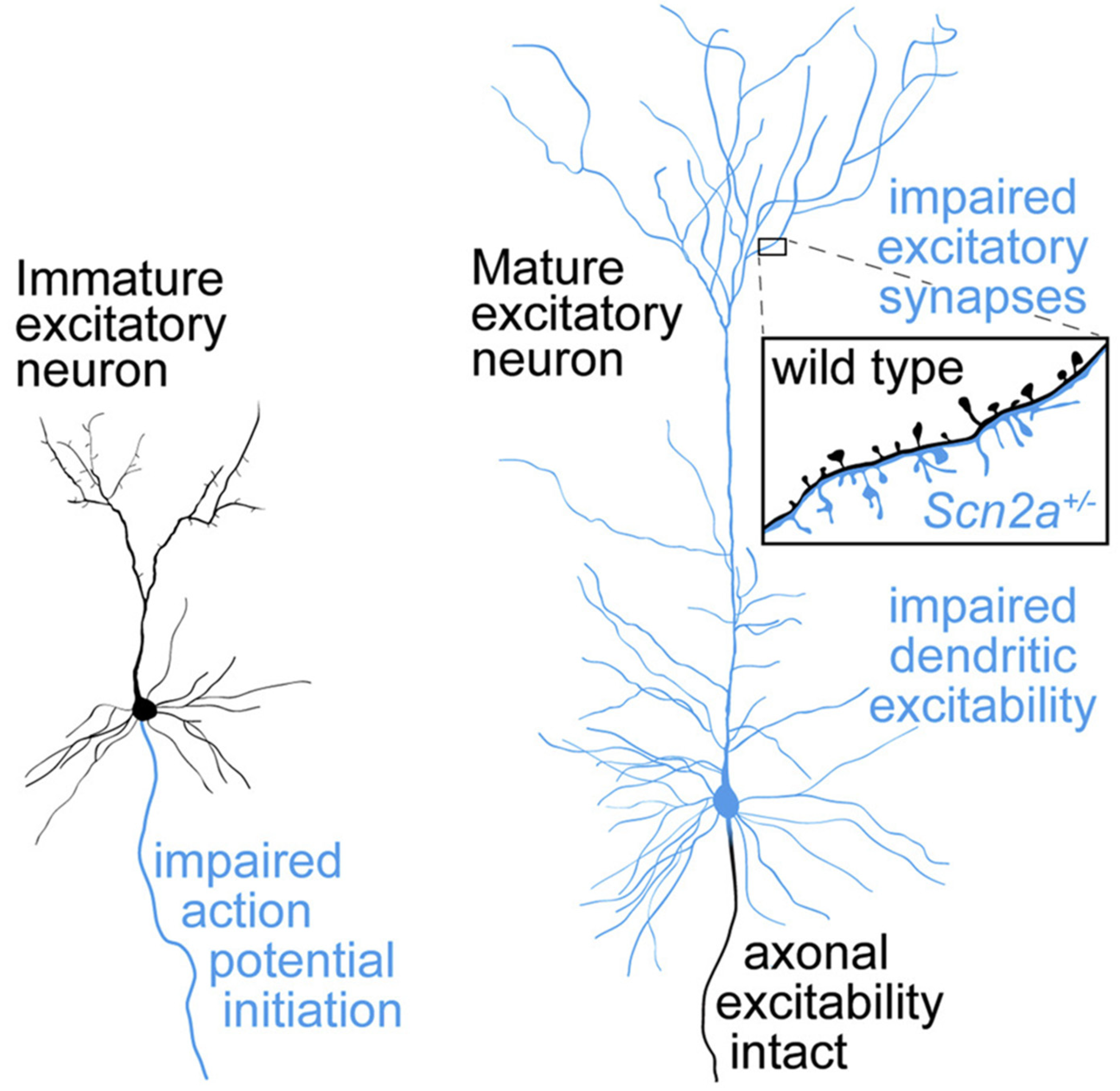
3.3. Genotypic Data
4. Discussion
Analysis of Genotypic and Phenotypic Trends
5. Limitations
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Psychiatric, A. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, Text Revision (DSM-5-TR (TM)); American Psychiatric Association Publishing: Washington, DC, USA, 2022; Volume 1. [Google Scholar]
- Pender, R.; Fearon, P.; Pourcain, B.S.; Heron, J.; Mandy, W. Developmental trajectories of autistic social traits in the general population. Psychol. Med. 2023, 53, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Rylaarsdam, L.E.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef] [PubMed]
- Hodges, H.; Fealko, C.; Soares, N. Autism spectrum disorder: Definition, epidemiology, causes, and clinical evaluation. Transl. Pediatr. 2020, 9 (Suppl. 1), S55–S65. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- Ben-Sasson, A.; Hen, L.; Fluss, R.; Cermak, S.A.; Engel-Yeger, B.; Gal, E. A meta-analysis of sensory modulation symptoms in individuals with autism spectrum disorders. J. Autism Dev. Disord. 2009, 39, 1–11. [Google Scholar] [CrossRef]
- Comparan-Meza, M.; de la Cruz, I.V.; Jauregui-Huerta, F.; Gonzalez-Castañeda, R.E.; Gonzalez-Perez, O.; Galvez-Contreras, A.Y. Biopsychological correlates of repetitive and restricted behaviors in autism spectrum disorders. Brain Behav. 2021, 11, e2341. [Google Scholar] [CrossRef]
- Chaxiong, P.; Burrows, C.; Botteron, K.N.; Dager, S.R.; Estes, A.M.; Hazlett, H.C.; Schultz, R.T.; Zwaigenbaum, L.; Piven, J.; Wolff, J. Relations of Restricted and Repetitive Behaviors to Social Skills in Toddlers with Autism. J. Autism Dev. Disord. 2022, 52, 1423–1434. [Google Scholar] [CrossRef]
- Turner, M. Annotation: Repetitive behaviour in autism: A review of psychological research. J. Child Psychol. Psychiatry 1999, 40, 839–849. [Google Scholar] [CrossRef]
- Davies, C.; Moosa, M.; McKenna, K.; Mittal, J.; Memis, I.; Mittal, R.; Eshraghi, A.A. Quality of Life, Neurosensory Disorders and Co-Occurring Medical Conditions in Individuals on the Spectrum, with a Special Focus on Females Diagnosed with Autism: A Systematic Review. J. Clin. Med. 2023, 12, 927. [Google Scholar] [CrossRef]
- Sato, M.; Nakai, N.; Fujima, S.; Choe, K.Y.; Takumi, T. Social circuits and their dysfunction in autism spectrum disorder. Mol. Psychiatry 2023, 28, 3194–3206. [Google Scholar] [CrossRef]
- McKenna, K.; Prasad, S.; Cooper, J.; King, A.M.; Shahzeidi, S.; Mittal, J.; Zalta, M.; Mittal, R.; Eshraghi, A.A. Incidence of Otolaryngological Manifestations in Individuals with Autism Spectrum Disorder: A Special Focus on Auditory Disorders. Audiol. Res. 2024, 14, 35–61. [Google Scholar] [CrossRef] [PubMed]
- Léna, I.; Mantegazza, M. NaV1.2 haploinsufficiency in Scn2a knock-out mice causes an autistic-like phenotype attenuated with age. Sci. Rep. 2019, 9, 12886. [Google Scholar] [CrossRef] [PubMed]
- Pediatrics, A.A.O.; Zwaigenbaum, L.; Bauman, M.L.; Stone, W.L.; Yirmiya, N.; Estes, A.; Hansen, R.L.; McPartland, J.C.; Natowicz, M.R.; Choueiri, R.; et al. Early Identification of Autism Spectrum Disorder: Recommendations for Practice and Research. Pediatrics 2015, 136 (Suppl. 1), S10–S40. [Google Scholar]
- Denisova, K.; Lin, Z. The importance of low IQ to early diagnosis of autism. Autism Res. 2023, 16, 122–142. [Google Scholar] [CrossRef]
- Charman, T.; Pickles, A.; Simonoff, E.; Chandler, S.; Loucas, T.; Baird, G. IQ in children with autism spectrum disorders: Data from the Special Needs and Autism Project (SNAP). Psychol. Med. 2011, 41, 619–627. [Google Scholar] [CrossRef]
- Spratt, P.W.; Ben-Shalom, R.; Keeshen, C.M.; Burke, K.J., Jr.; Clarkson, R.L.; Sanders, S.J.; Bender, K.J. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron 2019, 103, 673–685.e5. [Google Scholar] [CrossRef]
- Domínguez-Lucio, S.; Compañ-Gabucio, L.M.; Torres-Collado, L.; de la Hera, M.G. Occupational Therapy Interventions Using New Technologies in Children and Adolescents with Autism Spectrum Disorder: A Scoping Review. J. Autism Dev. Disord. 2023, 53, 332–358. [Google Scholar] [CrossRef]
- Özkan, E.; Çelik, S.B.; Yaran, M.; Bumin, G. Joint Attention–Based Occupational Therapy Intervention in Preschoolers with Autism Spectrum Disorder: A Randomized Controlled Trial. Am. J. Occup. Ther. 2023, 77, 7702205090. [Google Scholar] [CrossRef]
- Case-Smith, J.; Arbesman, M. Evidence-based review of interventions for autism used in or of relevance to occupational therapy. Am. J. Occup. Ther. 2008, 62, 416–429. [Google Scholar] [CrossRef]
- Eckes, T.; Buhlmann, U.; Holling, H.-D.; Möllmann, A. Comprehensive ABA-based interventions in the treatment of children with autism spectrum disorder—A meta-analysis. BMC Psychiatry 2023, 23, 133. [Google Scholar] [CrossRef]
- Karhu, E.; Zukerman, R.; Eshraghi, R.S.; Mittal, J.; Deth, R.C.; Castejon, A.M.; Trivedi, M.; Mittal, R.; Eshraghi, A.A. Nutritional interventions for autism spectrum disorder. Nutr. Rev. 2020, 78, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, A.; Griff, J.; Langlie, J.; Bencie, N.; Cromar, Z.; Mittal, J.; Memis, I.; Wallace, S.; Marcillo, A.; Mittal, R. Recent advancements in noninvasive brain modulation for individuals with autism spectrum disorder. Neural Regen. Res. 2023, 18, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Gevezova, M.; Sbirkov, Y.; Sarafian, V.; Plaimas, K.; Suratanee, A.; Maes, M. Autistic spectrum disorder (ASD)—Gene, molecular and pathway signatures linking systemic inflammation, mitochondrial dysfunction, transsynaptic signalling, and neurodevelopment. Brain Behav. Immun. Health 2023, 30, 100646. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.-C.; Lin, L.-S.; Long, S.; Ke, X.-Y.; Fukunaga, K.; Lu, Y.-M.; Han, F. Signalling pathways in autism spectrum disorder: Mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2022, 7, 229. [Google Scholar] [CrossRef]
- Memis, I.; Mittal, R.; Furar, E.; White, I.; Eshraghi, R.S.; Mittal, J.; Eshraghi, A.A. Altered Blood Brain Barrier Permeability and Oxidative Stress in Cntnap2 Knockout Rat Model. J. Clin. Med. 2022, 11, 2725. [Google Scholar] [CrossRef]
- Eshraghi, A.; Langlie, J.; Mittal, R.; Finberg, A.; Bencie, N.; Mittal, J.; Omidian, H.; Omidi, Y. Unraveling pathological mechanisms in neurological disorders: The impact of cell-based and organoid models. Neural Regen. Res. 2022, 17, 2131–2140. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Liu, G.; Kay, S.I.S.; Eshraghi, R.S.; Mittal, J.; Moshiree, B.; Mittal, R. Epigenetics and Autism Spectrum Disorder: Is There a Correlation? Front. Cell Neurosci. 2018, 12, 78. [Google Scholar] [CrossRef]
- Eshraghi, R.S.; Deth, R.C.; Mittal, R.; Aranke, M.; Kay, S.-I.S.; Moshiree, B.; Eshraghi, A.A. Early Disruption of the Microbiome Leading to Decreased Antioxidant Capacity and Epigenetic Changes: Implications for the Rise in Autism. Front. Cell. Neurosci. 2018, 12, 256. [Google Scholar] [CrossRef]
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and epigenetics of autism spectrum disorder—Current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar] [CrossRef]
- Zhuang, H.; Liang, Z.; Ma, G.; Qureshi, A.; Ran, X.; Feng, C.; Liu, X.; Yan, X.; Shen, L. Autism spectrum disorder: Pathogenesis, biomarker, and intervention therapy. MedComm 2024, 5, e497. [Google Scholar] [CrossRef]
- Rynkiewicz, A.; Janas-Kozik, M.; Słopień, A. Girls and women with autism. Psychiatr. Polska 2019, 53, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Milner, V.; McIntosh, H.; Colvert, E.; Happé, F. A Qualitative Exploration of the Female Experience of Autism Spectrum Disorder (ASD). J. Autism Dev. Disord. 2019, 49, 2389–2402. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Kweon, H.; Kang, R.; Kim, D.; Kim, K.; Kang, M.; Kim, S.Y.; Hwang, S.N.; Kim, J.Y.; Yang, E.; et al. Scn2a Haploinsufficiency in Mice Suppresses Hippocampal Neuronal Excitability, Excitatory Synaptic Drive, and Long-Term Potentiation, and Spatial Learning and Memory. Front. Mol. Neurosci. 2019, 12, 145. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, N.; Li, C.; Zhang, Z.; Teng, H.; Wang, Y.; Zhao, T.; Shi, L.; Zhang, K.; Xia, K.; et al. Genetic evidence of gender difference in autism spectrum disorder supports the female-protective effect. Transl. Psychiatry 2020, 10, 4. [Google Scholar] [CrossRef]
- Zhou, X.; Feliciano, P.; Shu, C.; Wang, T.; Astrovskaya, I.; Hall, J.B.; Obiajulu, J.U.; Wright, J.R.; Murali, S.C.; Xu, S.X.; et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat. Genet. 2022, 54, 1305–1319. [Google Scholar] [CrossRef]
- Schust, L.F.; Burke, J.; SanInocencio, C.; Bryan, B.A.; Ho, K.S.; Egan, S.M. A patient organization perspective: Charting the course to a cure for SCN2A-related disorders. Ther. Adv. Rare Dis. 2024, 5, 26330040241292645. [Google Scholar] [CrossRef]
- Seiffert, S.; Pendziwiat, M.; Bierhals, T.; Goel, H.; Schwarz, N.; van der Ven, A.; Boßelmann, C.M.; Lemke, J.; Syrbe, S.; Willemsen, M.H.; et al. Modulating effects of FGF12 variants on Na(V)1.2 and Na(V)1.6 being associated with developmental and epileptic encephalopathy and Autism spectrum disorder: A case series. EBioMedicine 2022, 83, 104234. [Google Scholar]
- Kruth, K.A.; Grisolano, T.M.; Ahern, C.A.; Williams, A.J. SCN2A channelopathies in the autism spectrum of neuropsychiatric disorders: A role for pluripotent stem cells? Mol. Autism 2020, 11, 23. [Google Scholar] [CrossRef]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef]
- Sanders, S.J.; Campbell, A.J.; Cottrell, J.R.; Moller, R.S.; Wagner, F.F.; Auldridge, A.L.; Bernier, R.A.; Catterall, W.A.; Chung, W.K.; Empfield, J.R.; et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 2018, 41, 442–456. [Google Scholar] [CrossRef]
- Nelson, A.D.; Catalfio, A.M.; Gupta, J.P.; Min, L.; Caballero-Florán, R.N.; Dean, K.P.; Elvira, C.C.; Derderian, K.D.; Kyoung, H.; Sahagun, A.; et al. Physical and functional convergence of the autism risk genes Scn2a and Ank2 in neocortical pyramidal cell dendrites. Neuron 2024, 112, 1133–1149.e6. [Google Scholar] [CrossRef] [PubMed]
- Ferri, S.L.; Abel, T.; Brodkin, E.S. Sex Differences in Autism Spectrum Disorder: A Review. Curr. Psychiatry Rep. 2018, 20, 9. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.L.; Bilder, D.A.; Zahorodny, W.; Pettygrove, S.; Durkin, M.S.; Fitzgerald, R.T.; Rice, C.; Kurzius-Spencer, M.; Baio, J.; Yeargin-Allsopp, M. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill. Summ. 2018, 65, 1–23. [Google Scholar] [CrossRef]
- Campbell, I.M.; Shaw, C.A.; Stankiewicz, P.; Lupski, J.R. Somatic mosaicism: Implications for disease and transmission genetics. Trends Genet. 2015, 31, 382–392. [Google Scholar] [CrossRef]
- Ogiwara, I.; Miyamoto, H.; Tatsukawa, T.; Yamagata, T.; Nakayama, T.; Atapour, N.; Miura, E.; Mazaki, E.; Ernst, S.J.; Cao, D.; et al. Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun. Biol. 2018, 1, 96. [Google Scholar] [CrossRef]
- Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Rusina, E.; Simonti, M.; Duprat, F.; Cestèle, S.; Mantegazza, M. Voltage-gated sodium channels in genetic epilepsy: Up and down of excitability. J. Neurochem. 2024, 168, 3872–3890. [Google Scholar] [CrossRef]
- Wolff, M.; Johannesen, K.M.; Hedrich, U.B.S.; Masnada, S.; Rubboli, G.; Gardella, E.; Lesca, G.; Ville, D.; Milh, M.; Villard, L.; et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017, 140, 1316–1336. [Google Scholar] [CrossRef]
- Ben-Shalom, R.; Keeshen, C.M.; Berrios, K.N.; An, J.Y.; Sanders, S.J.; Bender, K.J. Opposing Effects on Na V 1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biol. Psychiatry 2017, 82, 224–232. [Google Scholar] [CrossRef]
- Zeng, Q.; Yang, Y.; Duan, J.; Niu, X.; Chen, Y.; Wang, D.; Zhang, J.; Chen, J.; Yang, X.; Li, J.; et al. SCN2A-Related Epilepsy: The Phenotypic Spectrum, Treatment and Prognosis. Front. Mol. Neurosci. 2022, 15, 809951. [Google Scholar] [CrossRef]
- Munn, Z.; Barker, T.H.; Moola, S.; Tufanaru, C.; Stern, C.; McArthur, A.; Stephenson, M.; Aromataris, E. Methodological quality of case series studies: An introduction to the JBI critical appraisal tool. JBI Evid. Synth. 2020, 18, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Ekert, J.; Goyal, A.; Young, J.S.; Hervey-Jumper, S.L.; Berger, M.S. Interventional neurorehabilitation for glioma patients: A systematic review. Neurooncol. Pract. 2024, 11, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Barker, T.H.; Stone, J.C.; Sears, K.; Klugar, M.; Tufanaru, C.; Leonardi-Bee, J.; Aromataris, E.; Munn, Z. The revised JBI critical appraisal tool for the assessment of risk of bias for randomized controlled trials. JBI Évid. Synth. 2023, 21, 494–506. [Google Scholar] [CrossRef]
- Porritt, K.; Gomersall, J.; Lockwood, C. JBI’s Systematic Reviews: Study selection and critical appraisal. Am. J. Nurs. 2014, 114, 47–52. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Guo, R.; Xu, W.; Liu, X.; Zhao, C.; Guo, Q.; Xu, W.; Ni, X.; Hao, C.; et al. Genetic diagnostic yields of 354 Chinese ASD children with rare mutations by a pipeline of genomic tests. Front. Genet. 2023, 14, 1108440. [Google Scholar] [CrossRef]
- Hudac, C.M.; Friedman, N.R.; Ward, V.R.; Estreicher, R.E.; Dorsey, G.C.; Bernier, R.A.; Kurtz-Nelson, E.C.; Eichler, E.E.; Neuhaus, E. Characterizing Sensory Phenotypes of Subgroups with a Known Genetic Etiology Pertaining to Diagnoses of Autism Spectrum Disorder and Intellectual Disability. J. Autism Dev. Disord. 2023, 54, 2386–2401. [Google Scholar] [CrossRef]
- Callaghan, D.B.; Rogic, S.; Tan, P.P.C.; Calli, K.; Qiao, Y.; Baldwin, R.; Jacobson, M.; Belmadani, M.; Holmes, N.; Yu, C.; et al. Whole genome sequencing and variant discovery in the ASPIRE autism spectrum disorder cohort. Clin. Genet. 2019, 96, 199–206. [Google Scholar] [CrossRef]
- Alagia, M.; Fecarotta, S.; Romano, A.; Parrini, E.; Auricchio, G.; Miano, M.G.; Terrone, G. A Novel Splicing SCN2A Mutation in an Adolescent With Low-Functioning Autism, Acute Dystonic Movement Disorder, and Late-Onset Generalized Epilepsy. Pediatr. Neurol. 2023, 138, 58–61. [Google Scholar] [CrossRef]
- Chong, P.F.; Saitsu, H.; Sakai, Y.; Imagi, T.; Nakamura, R.; Matsukura, M.; Matsumoto, N.; Kira, R. Deletions of SCN2A and SCN3A genes in a patient with West syndrome and autistic spectrum disorder. Seizure 2018, 60, 91–93. [Google Scholar] [CrossRef]
- Deutsch, S.I.; Burket, J.A. A De Novo Missense Variant of SCN2A: Implications and Limitations for Understanding Clinical Phenotype and Treatment Recommendations. Clin. Neuropharmacol. 2021, 44, 138–140. [Google Scholar] [CrossRef]
- Tran, K.T.; Le, V.S.; Bui, H.T.P.; Do, D.H.; Nguyen, H.T.; Dao, L.T.M.; Nguyen, T.H.; Vu, D.M.; Ha, L.T.; Le, H.T.T.; et al. Genetic landscape of autism spectrum disorder in Vietnamese children. Sci. Rep. 2020, 10, 5034. [Google Scholar] [CrossRef] [PubMed]
- Arnett, A.B.; Beighley, J.S.; Kurtz-Nelson, E.C.; Hoekzema, K.; Wang, T.; Bernier, R.A.; Eichler, E.E. Developmental Predictors of Cognitive and Adaptive Outcomes in Genetic Subtypes of Autism Spectrum Disorder. Autism Res. 2020, 13, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Krupp, D.R.; Barnard, R.A.; Duffourd, Y.; Evans, S.A.; Mulqueen, R.M.; Bernier, R.; Rivière, J.-B.; Fombonne, E.; O’roak, B.J. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am. J. Hum. Genet. 2017, 101, 369–390. [Google Scholar] [CrossRef]
- Richardson, R.; Baralle, D.; Bennett, C.; Briggs, T.; Bijlsma, E.K.; Clayton-Smith, J.; Constantinou, P.; Foulds, N.; Jarvis, J.; Jewell, R.; et al. Further delineation of phenotypic spectrum of SCN2A-related disorder. Am. J. Med. Genet. A 2022, 188, 867–877. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, X.; Eaton, M.; Wu, J.; Ma, Z.; Lai, S.; Park, A.; Ahmad, T.S.; Que, Z.; Lee, J.H.; et al. Severe deficiency of the voltage-gated sodium channel Na(V)1. 2 elevates neuronal excitability in adult mice. Cell Rep. 2021, 36, 109495. [Google Scholar]
- Zhang, J.; Chen, X.; Eaton, M.; Wu, J.; Ma, Z.; Lai, S.; Park, A.; Ahmad, T.S.; Que, Z.; Lee, J.H.; et al. Paradoxical hyperexcitability from Na(V)1. 2 sodium channel loss in neocortical pyramidal cells. Cell Rep. 2021, 36, 109483. [Google Scholar]
- Gould, E.; Kim, J.H. SCN2A contributes to oligodendroglia excitability and development in the mammalian brain. Cell Rep. 2021, 36, 109653. [Google Scholar] [CrossRef]
- Scala, M.; Efthymiou, S.; Sultan, T.; De Waele, J.; Panciroli, M.; Salpietro, V.; Maroofian, R.; Striano, P.; Van Petegem, F.; Houlden, H.; et al. Homozygous SCN1B variants causing early infantile epileptic encephalopathy 52 affect voltage-gated sodium channel function. Epilepsia 2021, 62, e82–e87. [Google Scholar] [CrossRef]
- Thompson, C.H.; Potet, F.; Abramova, T.V.; DeKeyser, J.-M.; Ghabra, N.F.; Vanoye, C.G.; Millichap, J.J.; George, A.L. Epilepsy-associated SCN2A (NaV1.2) variants exhibit diverse and complex functional properties. J. Gen. Physiol. 2023, 155, e202313375. [Google Scholar] [CrossRef]
- Werling, D.M.; Geschwind, D.H. Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 2013, 26, 146–153. [Google Scholar] [CrossRef]
- Fombonne, E. Epidemiology of pervasive developmental disorders. Pediatr. Res. 2009, 65, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, B.; Wu, C.; Wang, J.; Sun, M. Autism Spectrum Disorder: Neurodevelopmental Risk Factors, Biological Mechanism, and Precision Therapy. Int. J. Mol. Sci. 2023, 24, 1819. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, J.D.; Marrus, N.; Maloney, S.E.; Yip, B.; Sandin, S.; Turner, T.N.; Selmanovic, D.; Kroll, K.L.; Gutmann, D.H.; Constantino, J.N.; et al. Can the “female protective effect” liability threshold model explain sex differences in autism spectrum disorder? Neuron 2022, 110, 3243–3262. [Google Scholar] [CrossRef]
- Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D’Gama, A.M.; Kim, S.N.; Hill, R.S.; Goldberg, A.P.; et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 2017, 20, 1217–1224. [Google Scholar] [CrossRef]
- D’gama, A.M. Somatic Mosaicism and Autism Spectrum Disorder. Genes 2021, 12, 1699. [Google Scholar] [CrossRef]
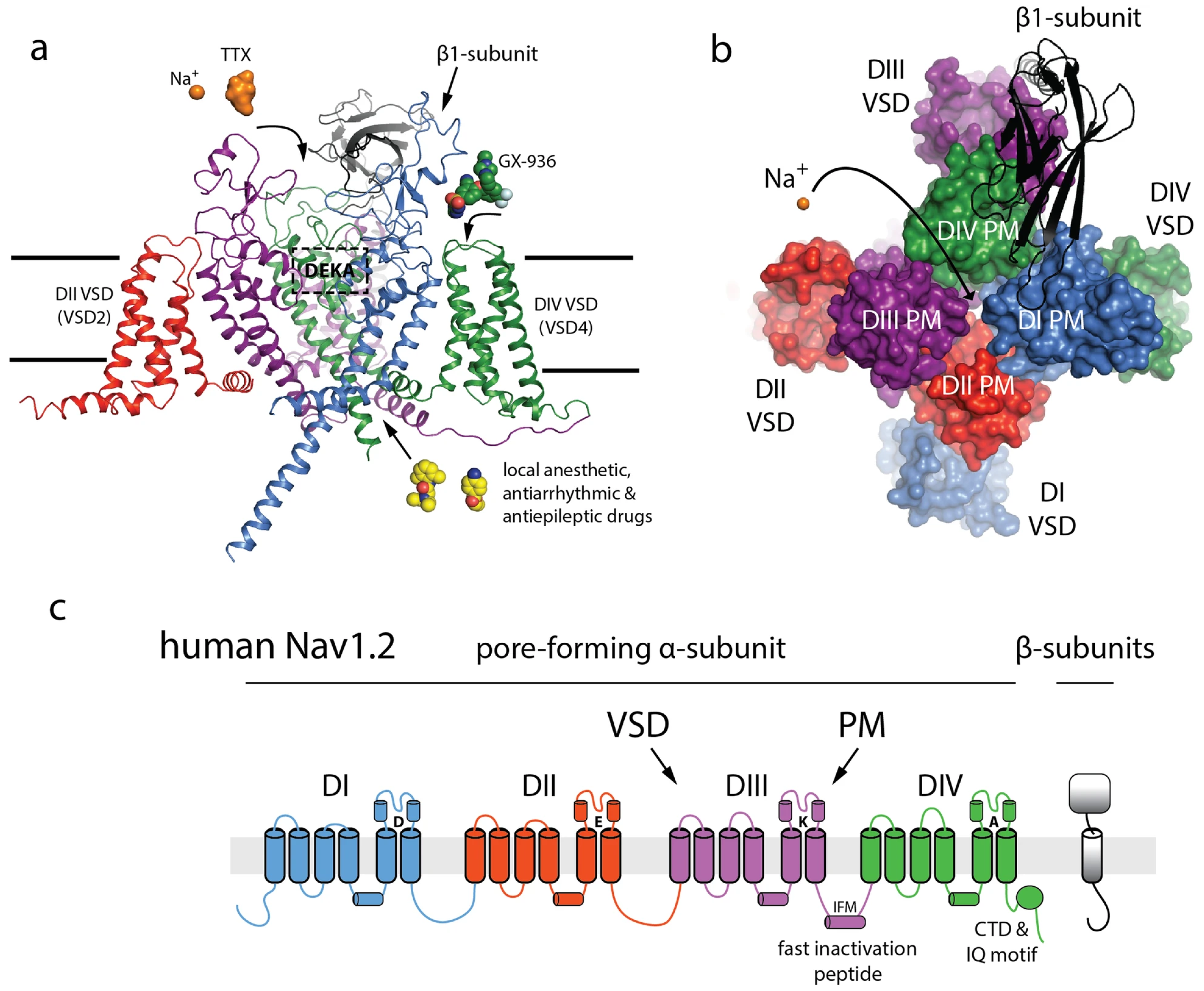
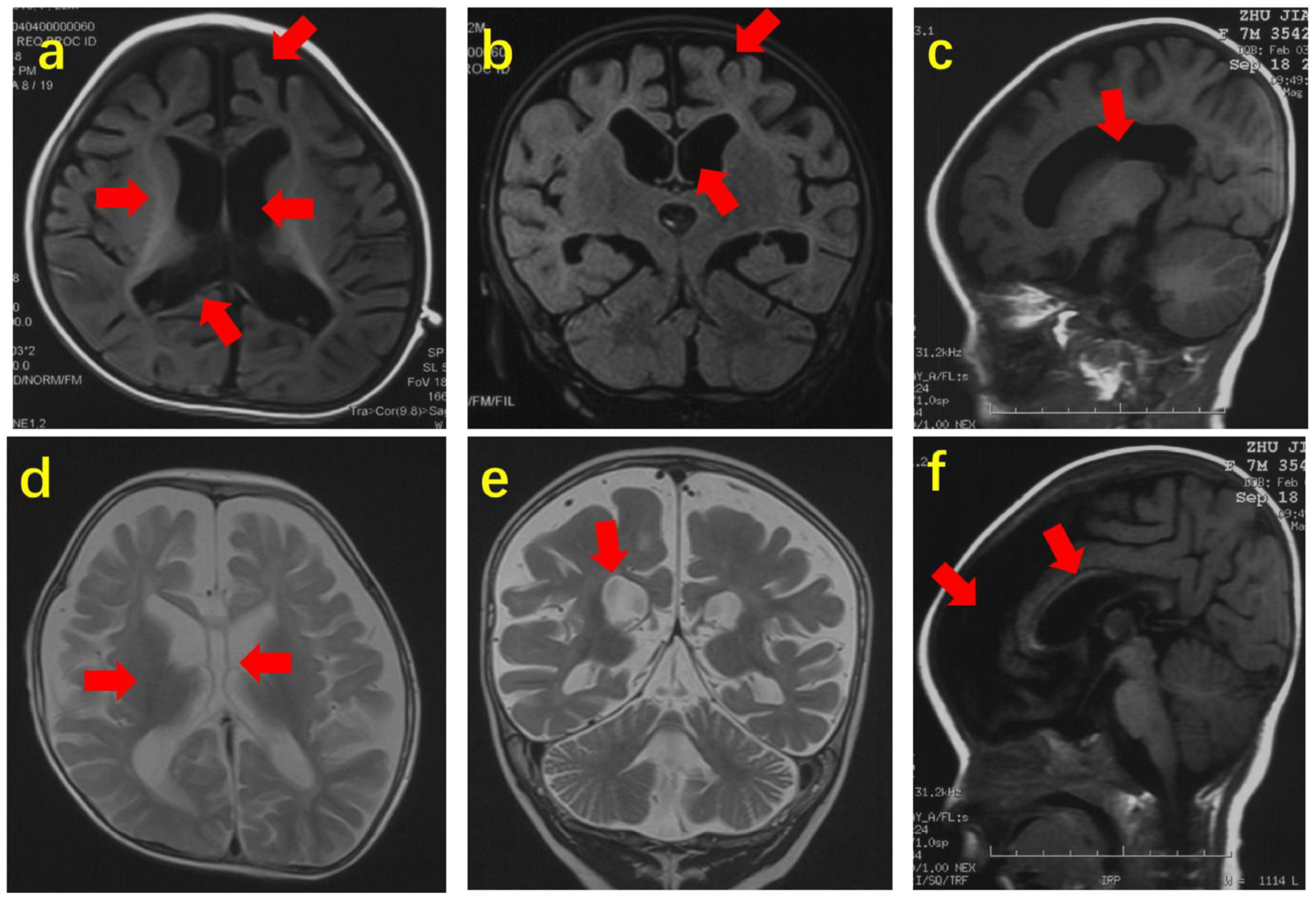
| Reference | Seizures | Epilepsy | Psychiatric | Repetitive Actions | Verbal | Nonverbal | Speech Delay | Developmental Delay (DD) | ID | Physical Abnormalities | Movement Disorders | Dysphagia | Walking | Crawling | Unsteady/Ataxic Gait |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Alagia et al. [59] | Yes | Yes | Anxiety | Pelvis rocking | 1 | 1 | Severe | Yes | NR | Facial dysmorphism | Dystonia | NR | NR | NR | Yes |
| Arnett et al. [63] | No | No | NR | Echolalia | 5 | 2 | Moderate | No | NR | None | None | NR | Yes | Yes | NR |
| Callaghan et al. [58] | Yes | No | Hyperactivity | None | 3 | NR | Mild | Yes | NR | Microcephaly | Hypotonia | NR | No | NR | Yes |
| Chong et al. [60] | Yes | Yes | NR | Stereotypic rocking | 1 | NR | Severe | Yes | NR | Dysmorphic features | None | NR | Yes | Yes | Yes |
| Deutsch et al. [61] | No | No | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR |
| Hudac et al. [57] | Yes | NR | NR | Echolalia | 2 | 5 | Severe | Yes | NR | Microcephaly | Hypotonia | NR | Yes | NR | Yes |
| Krupp et al. [64] | No | NR | Behavioral issues | NR | NR | NR | NR | Yes | NR | None | None | NR | No | No | NR |
| Richardson et al. [65] | Yes | NR | Aggression | Stereotypy | 5 | 6 | Severe | Yes | NR | Dysmorphic features | NR | NR | Yes | NR | Yes |
| Tran et al. [62] | No | No | Hyperactivity | None | NR | NR | Mild | No | NR | None | None | NR | NR | NR | NR |
| Wolff et al. [49] | Yes | Yes | NR | NR | NR | NR | Severe | Yes | NR | Microcephaly | Ataxia | NR | NR | NR | Yes |
| Zhang et al. [56] | No | None | None | None | NR | NR | None | No | NR | None | None | NR | NR | NR | NR |
| Reference | Mutation Type | Homozygous/ Heterozygous | Maternal Inheritance Paternal Inheritance | Unknown Origin | Mosaic | De Novo | Exon | Intron | GoF | LoF |
|---|---|---|---|---|---|---|---|---|---|---|
| Alagia et al. [59] | 1 (Splicing) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Arnett et al. [63] | 2 (Frameshift), 6 (Missense), 1 (Nonsense), 1 (Splicing), 10 (SNV) | 10 Heterozygous | 0 | 3 | 0 | 7 | 6 | 0 | 0 | 0 |
| Callaghan et al. [58] | 1 (Missense), 1 (Splicing), 2 (SNV) | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 |
| Chong et al. [60] | 1 (Deletion) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Deutsch et al. [61] | 1 (Missense), 1 (SNV) | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Hudac et al. [57] | 3 (Frameshift), 18 (Missense), 5 (Nonsense) 1 (Splicing), 1 (Duplication), 1 (Stop/Gain) | 0 | 2 (Paternal) | 7 | 0 | 26 | 0 | 0 | 0 | 0 |
| Krupp et al. [64] | 1 (Deletion), 2 (Frameshift), 5 (Missense), 2 (Nonsense), 1 (Splicing), 1 (Stop/Gain Included in Nonsense), 9 (SNV) | 9 Heterozygous | 1 (Maternal) | 0 | 2 | 7 | 11 | 0 | 0 | Implicated in frameshift and nonsense mutations |
| Richardson et al. [65] | 2 (Deletion), 2 (Frameshift), 14 (Missense), 5 (Nonsense), 3 (Splicing) | 0 | 1 (Maternal) 1 (Paternal) | 2 | 2 | 18 | 0 | 0 | 0 | 6 |
| Tran et al. [62] | 1 (Frameshift) | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 1 |
| Wolff et al. [49] | 5 (Frameshift), 13 (Missense), 2 (Nonsense), 2 (Splicing) | NR | 0 | 0 | 0 | 22 | 20 | 2 | NR | NR |
| Zhang et al. [56] | 1 (Deletion), 1 (Frameshift), 1 (Nonsense), 1 (Splicing) | NR | 0 | 0 | 0 | 3 | 2 | 1 | NR | NR |
| Reference | Variant(s) | Notes |
|---|---|---|
| Alagia et al. [59] | c.2919+4delT | NM_001040142 |
| Arnett et al. [63] | c.34G>A, c.2464G>A, c.2645G>A, c.2877C>A, c.2932T>C, c.4264A>G, c.4896_4897insT, c.4996C>T, c.425delA, c.605+1G>T | Multiple variants |
| Callaghan et al. [58] | p.Arg102Gln, splice acceptor variant | Missense and splicing |
| Chong et al. [60] | 1.1 Mb deletion including SCN2A and SCN3A | Large deletion |
| Deutsch et al. [61] | p.A704K | Alanine to lysine |
| Hudac et al. [57] | c.34G>A, c.1289A>C, c.5339G>T, c.2932T>C, c.5318C>T, c.5272A>C, c.2877C>A, c.605+1G>T, c.3849+2T>C, c.4801G>T, c.2635G>A, c.4938_4939insGAT, c.4591C>T, c.823C>T, c.252C>A, c.1712G>A, c.632G>A, c.2464G>A | Multiple variants |
| Krupp et al. [64] | c.3370A>T, c.272A>G, c.1094C>T, c.3922C>T, c.1819C>T, c.5536C>T, c.3435_3436delCG, c.469delA, c.1384+1G>T | Multiple types |
| Richardson et al. [65] | c.2932T>C, c.2674G>A, c.5192G>A, c.4644G>C, c.640T>G, c.2774T>C, c.5318C>T, c.1184G>A, c.4543C>T, c.515T>G, c.4780T>A, c.4886G>A, c.5638G>A | Multiple variants |
| Tran et al. [62] | p.Leu78fs | De novo |
| Wolff et al. [49] | Q1811E, M1548V, E430A, E999K, R1319Q (x2), R1882P, A1652P, H930Q, P1622S, c.605+1G4T, V1528Cfs*7, C1170Vfs*15, R1235*, A1773V, K1933M, N503Kfs*19, K1387Sfs*4, R1435*, T1711Lfs*8, G1744E, c.386+2T>C | All de novo |
| Zhang et al. [56] | c.4550_4551del, c.605+1G>A, c.1570C>T | Subjects 3F, 3M, 4M |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DiStefano, N.; Cooper, J.N.; Elisha, D.H.; Zalta, M.; Mittal, J.; Cohen, D.; Monterrubio, A.; Hossain, R.; Sangadi, A.; Mittal, R.; et al. Decoding SCN2A Variants: Bridging Genetics and Phenotypes in Autism Spectrum Disorder. J. Clin. Med. 2025, 14, 3790. https://doi.org/10.3390/jcm14113790
DiStefano N, Cooper JN, Elisha DH, Zalta M, Mittal J, Cohen D, Monterrubio A, Hossain R, Sangadi A, Mittal R, et al. Decoding SCN2A Variants: Bridging Genetics and Phenotypes in Autism Spectrum Disorder. Journal of Clinical Medicine. 2025; 14(11):3790. https://doi.org/10.3390/jcm14113790
Chicago/Turabian StyleDiStefano, Nicholas, Jaimee N. Cooper, David H. Elisha, Max Zalta, Jeenu Mittal, David Cohen, Andrea Monterrubio, Ryan Hossain, Akhila Sangadi, Rahul Mittal, and et al. 2025. "Decoding SCN2A Variants: Bridging Genetics and Phenotypes in Autism Spectrum Disorder" Journal of Clinical Medicine 14, no. 11: 3790. https://doi.org/10.3390/jcm14113790
APA StyleDiStefano, N., Cooper, J. N., Elisha, D. H., Zalta, M., Mittal, J., Cohen, D., Monterrubio, A., Hossain, R., Sangadi, A., Mittal, R., & Eshraghi, A. A. (2025). Decoding SCN2A Variants: Bridging Genetics and Phenotypes in Autism Spectrum Disorder. Journal of Clinical Medicine, 14(11), 3790. https://doi.org/10.3390/jcm14113790