
Therapeutic Challenges and New Era in Fibrillary Glomerulonephritis with the Introduction of DNAJB9: Experience from a Tertiary Nephrology Center
 , and
, and
Abstract
1. Introduction
2. Methods
2.1. Patients
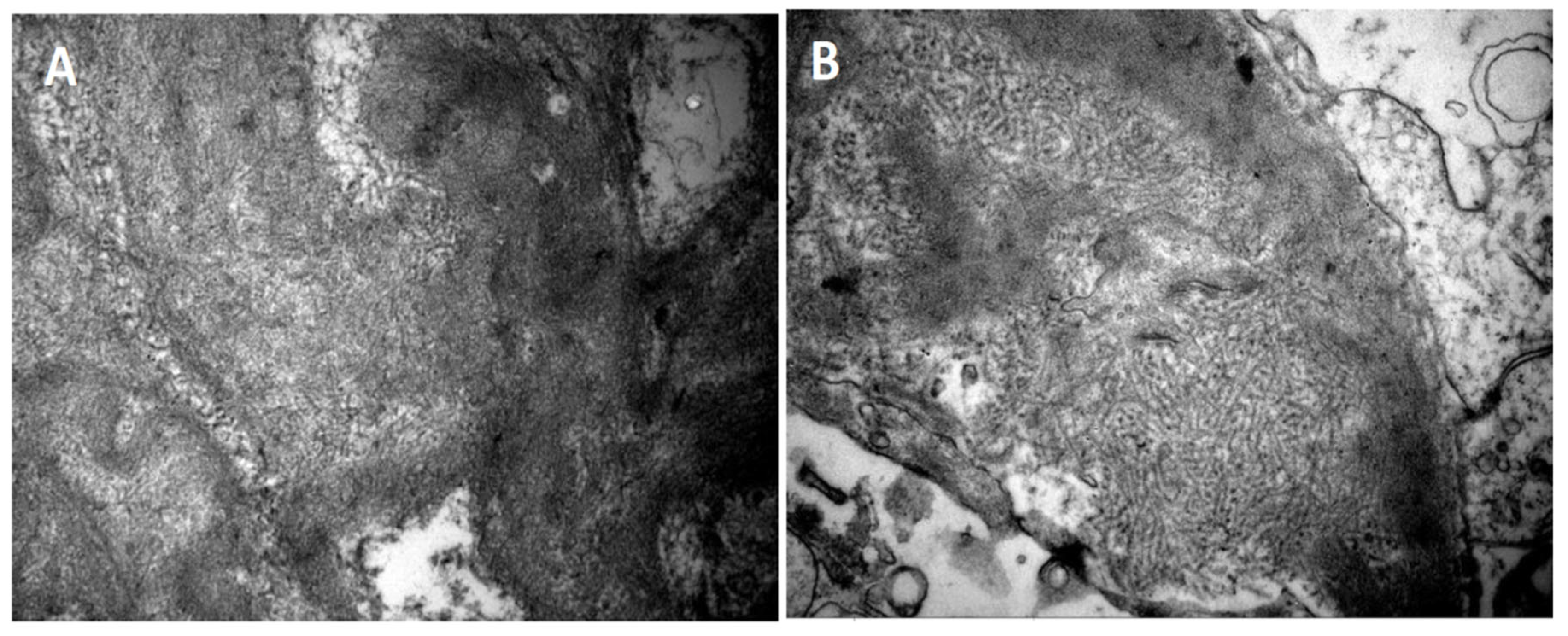
- Deposition of fibrils in the glomerulus by light electron microscopy. The fibrils must have the following characteristics:
- Random arrangement;
- Diameter of 14 to 24 nm;
- Deposition in the mesangium and the glomerular basement membrane.
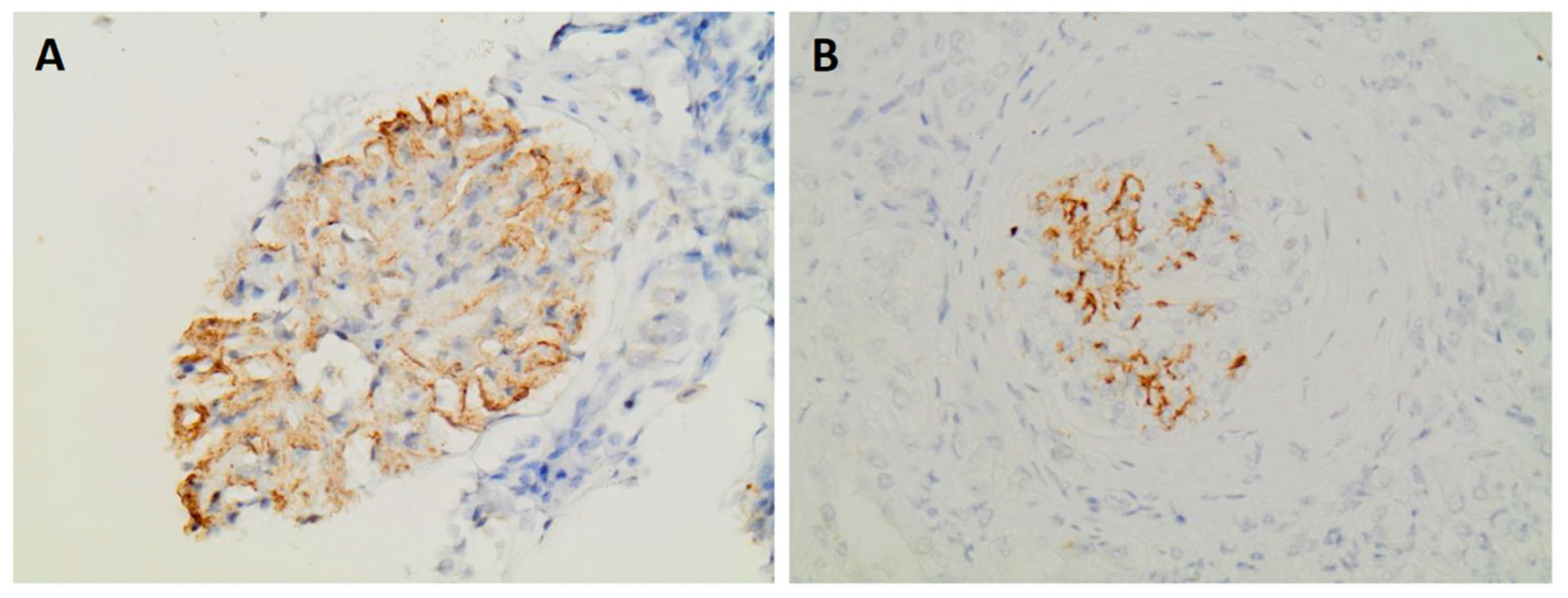
- Expression of DNAJB9 in immunohistochemistry
- Complete response (CR): proteinuria < 0.5 g/day and normal renal function (serum creatinine < 1.2 mg/dL);
- Partial response (PR): reduction in proteinuria by >50% from the peak recorded value, stable renal function;
- Persistent renal dysfunction (PRD): failure to meet the criteria for CR or PR or worsening renal function without progression to ESRD;
- ESRD: eGFR < 15 mL/min.
2.2. Statistics
3. Results
3.1. Demographic, Clinical Features and Laboratory Data
3.2. Pathology Findings
3.3. Treatment and Clinical Outcome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alpers, C.E.; Rennke, H.G.; Hopper, J.; Biava, C.G. Fibrillary glomerulonephritis: An entity with unusual immunofluorescence features. Kidney Int. 1987, 31, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.L.; Markowitz, G.S.; Valeri, A.M. Fibrillary and immunotactoid glomerulonephritis: Distinct entities with different clinical and pathologic features. Kidney Int. 2003, 63, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.L.; Markowitz, G.S. Fibrillary Glomerulonephritis: An Update. Kidney Int. Rep. 2019, 4, 917–922. [Google Scholar] [CrossRef]
- O’shaughnessy, M.M.; Hogan, S.L.; Poulton, C.J.; Falk, R.J.; Singh, H.K.; Nickeleit, V.; Jennette, J.C. Temporal and Demographic Trends in Glomerular Disease Epidemiology in the Southeastern United States, 1986–2015. Clin. J. Am. Soc. Nephrol. 2017, 12, 614–623. [Google Scholar] [CrossRef]
- Nasr, S.H.; Valeri, A.M.; Cornell, L.D.; Fidler, M.E.; Sethi, S.; Leung, N.; Fervenza, F.C. Fibrillary glomerulonephritis: A report of 66 cases from a single institution. Clin. J. Am. Soc. Nephrol. 2011, 6, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Normand, G.; Jolivot, A.; Rabeyrin, M.; Hervieu, V.; Valette, P.-J.; Scoazec, J.-Y.; Gougon, J.-M.; Juillard, L.; Dumortier, J. Paraneoplastic fibrillary glomerulonephritis associated with intrahepatic cholangiocarcinoma: When diagnosis of a rare kidney disease leads to successful hepatic cancer treatment. Clin. Res. Hepatol. Gastroenterol. 2017, 41, e8–e11. [Google Scholar] [CrossRef]
- Andeen, N.K.; Troxell, M.L.; Riazy, M.; Avasare, R.S.; Lapasia, J.; Jefferson, J.A.; Akilesh, S.; Najafian, B.; Nicosia, R.F.; Alpers, C.E.; et al. Fibrillary Glomerulonephritis: Clinicopathologic Features and Atypical Cases from a Multi-Institutional Cohort. Clin. J. Am. Soc. Nephrol. 2019, 6, 1741–1750. [Google Scholar] [CrossRef]
- Payan Schober, F.; Jobson, M.A.; Poulton, C.J.; Singh, H.K.; Nickeleit, V.; Falk, R.J.; Jennette, J.C.; Nachman, P.H.; Pendergraft, W.F., III. Clinical Features and Outcomes of a Racially Diverse Population with Fibrillary Glomerulonephritis. Am. J. Nephrol. 2017, 45, 248–256. [Google Scholar] [CrossRef]
- Zhang, L.; Carson, J.M.; Lucia, M.S. Fibrillary glomerulonephritis in an HIV patient without concurrent hepatitis C infection: Case report and review of the literature. Clin. Nephrol. 2018, 89, 381–386. [Google Scholar] [CrossRef]
- Javaugue, V.; Karras, A.; Glowacki, F.; McGregor, B.; Lacombe, C.; Goujon, J.-M.; Ragot, S.; Aucouturier, P.; Touchard, G.; Bridoux, F. Long-term kidney disease outcomes in fibrillary glomerulonephritis: A case series of 27 patients. Am. J. Kidney Dis. 2013, 62, 679–690. [Google Scholar] [CrossRef]
- Marinaki, S.; Tsiakas, S.; Liapis, G.; Skalioti, C.; Kapsia, E.; Lionaki, S.; Boletis, J. Clinicopathologic features and treatment outcomes of patients with fibrillary glomerulonephritis: A case series. Medicine 2021, 100, e26022. [Google Scholar] [CrossRef] [PubMed]
- Andeen, N.K.; Yang, H.-Y.; Dai, D.-F.; MacCoss, M.J.; Smith, K.D. DnaJ Homolog Subfamily B Member 9 Is a Putative Autoantigen in Fibrillary GN. J. Am. Soc. Nephrol. 2018, 29, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Alexander, M.P.; Vrana, J.A.; Theis, J.D.; Mills, J.R.; Negron, V.; Sethi, S.; Dispenzieri, A.; Highsmith, W.E.; Nasr, S.H.; et al. DnaJ Heat Shock Protein Family B Member 9 Is a Novel Biomarker for Fibrillary GN. J. Am. Soc. Nephrol. 2018, 29, 51–56. [Google Scholar] [CrossRef]
- Nasr, S.H.; Vrana, J.A.; Dasari, S.; Bridoux, F.; Fidler, M.E.; Kaaki, S.; Quellard, N.; Rinsant, A.; Goujon, J.M.; Sethi, S.; et al. DNAJB9 Is a Specific Immunohistochemical Marker for Fibrillary Glomerulonephritis. Kidney Int. Rep. 2018, 3, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Chen, D.; Liang, D.; Xu, F.; Zhang, M.; Yang, F.; Zhu, X.; Li, P.; Zeng, C. Clinicopathological characteristics and outcome of patients with fibrillary glomerulonephritis: DNAJB9 is a valuable histologic marker. J. Nephrol. 2021, 34, 883–892. [Google Scholar] [CrossRef]
- Gambella, A.; Pitino, C.; Barreca, A.; Nocifora, A.; Giarin, M.M.; Bertero, L.; Biancone, L.; Roccatello, D.; Papotti, M.; Cassoni, P. DNAJB9 Is a Reliable Immunohistochemical Marker of Fibrillary Glomerulonephritis: Evaluation of Diagnostic Efficacy in a Large Series of Kidney Biopsies. Biomedicines 2022, 27, 2102. [Google Scholar] [CrossRef]
- Daverkausen-Fischer, L.; Pröls, F. The function of the co-chaperone ERdj4 in diverse (patho-)physiological conditions. Cell. Mol. Life Sci. 2021, 79, 9. [Google Scholar] [CrossRef]
- KDIGO Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, S1–S276. [Google Scholar] [CrossRef]
- Said, S.M.; Leung, N.; Alexander, M.P.; Cornell, L.D.; Fidler, M.E.; Grande, J.P.; Herrera, L.H.; Sethi, S.; Zhang, P.; Nasr, S.H. DNAJB9-positive monotypic fibrillary glomerulonephritis is not associated with monoclonal gammopathy in the vast majority of patients. Kidney Int. 2020, 98, 498–504. [Google Scholar] [CrossRef]
- Da, Y.; Goh, G.H.; Lau, T.; Chng, W.J.; Soekojo, C.Y. Fibrillary glomerulonephritis and monoclonal gammopathy: Potential diagnostic challenges. Front. Oncol. 2022, 12, 880923. [Google Scholar] [CrossRef]
- Andeen, N.K.; Smith, K.D. Fibrillary glomerulonephritis: An update. The Authors Reply. Kidney Int. Rep. 2020, 5, 956–964. [Google Scholar]
- Pronovost, P.H.; Brady, H.R.; Gunning, M.E.; Espinoza, O.; Rennke, H.G. Clinical features, predictors of disease progression and results of renal transplantation in fibrillary/immunotactoid glomerulopathy. Nephrol. Dial. Transplant. 1996, 11, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Attieh, R.M.; Yang, Y.; Rosenstock, J.L. Updates on the Diagnosis and Management of Fibrillary Glomerulonephritis. Adv. Kidney Dis. Health 2024, 31, 374–383. [Google Scholar] [CrossRef]
- Erickson, S.B.; Zand, L.; Nasr, S.H.; Alexander, M.P.; Leung, N.; Drosou, M.E.; Fervenza, F.C. Treatment of fibrillary glomerulonephritis with rituximab: A 12-month pilot study. Nephrol. Dial. Transplant. 2021, 36, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Sabanis, N.; Liaveri, P.; Geladari, V.; Liapis, G.; Moustakas, G. DNAJB9 Fibrillary Glomerulonephritis with Membranous-like Pattern: A Case-Based Literature Review. Cureus 2023, 28, e47862. [Google Scholar] [CrossRef]
- Behnke, J.; Mann, M.J.; Scruggs, F.L. Members of the Hsp70 Family Recognize distinct types of sequences to execute ER quality control. Mol. Cell 2016, 1, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Dasari, S.; Lieske, J.C.; Benson, L.M.; Vanderboom, P.M.; Holtz-Heppelmann, C.J.; Giesen, C.D.; Snyder, M.R.; Erickson, S.B.; Fervenza, F.C.; et al. Serum levels of DNAJB9 are elevated in fibrillary glomerulonephritis patients. Kidney Int. 2019, 95, 1269–1272. [Google Scholar] [CrossRef]
- Hvala, A.; Kobenter, T.; Ferluga, D. Fingerprint and other organised deposits in lupus nephritis. Wien. Klin. Wochenschr. 2000, 112, 711–715. [Google Scholar]
- Menon, S.; Zeng, X.; Valentini, R. Fibrillary glomerulonephritis and renal failure in a child with systemic lupus erythematosus. Pediatr. Nephrol. 2009, 24, 1577–1581. [Google Scholar] [CrossRef]
- Bhat, Z.Y.; Zeng, X.; Hingorani, J. Fibrillary glomerulonephritis in a patient with systemic lupus erythematosus: A rare association. Int. Urol. Nephrol. 2013, 45, 281–284. [Google Scholar] [CrossRef]
- Whelband, M.C.; Willingham, T.; Thirunavukkarasu, S. Fibrillar glomerulonephritis in a patient with systemic lupus erythematosus with no evidence of lupus nephritis. BMJ Case Rep. 2023, 16, e253388. [Google Scholar] [CrossRef] [PubMed]
- Stehlé, T.; Joly, D.; Vanhille, P.; Boffa, J.-J.; Rémy, P.; Mesnard, L.; Hoffmann, M.; Grimbert, P.; Choukroun, G.; Vrtovsnik, F.; et al. Clinicopathological study of glomerular diseases associated with sarcoidosis: A multicenter study. Orphanet J. Rare Dis. 2013, 30, 65. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Patients | Gender | Age | Serum Creatinine [mg/dL] | Proteinuria [gr/24 h] | Hematuria | Hypertension | Comorbidities |
|---|---|---|---|---|---|---|---|
| 1 | Male | 45 | 1.7 | 3.70 | − | + | - |
| 2 | Female | 58 | 1.4 | 3.50 | − | + | DM2, rheumatologic disease |
| 3 | Female | 31 | 0.9 | 3.00 | + | + | Hypothyroidism, tonsillectomy |
| 4 | Female | 69 | 1.7 | 4.50 | + | + | DM2, hypothyroidism, AF, CKD, smoking |
| 5 | Male | 34 | 0.8 | 1.33 | + | − | Smoking |
| 6 | Female | 60 | 2.4 | 3.78 | + | − | Thyroid nodules, dyslipidemia, SLE |
| 7 | Male | 63 | 1.2 | 11.68 | + | + | Obesity, smoking |
| 8 | Male | 61 | 1.6 | 8.00 | + | + | Colon polyps |
| 9 | Female | 56 | 0.8 | 1.80 | − | − | Hypothyroidism, dyslipidemia, sarcoidosis |
| 10 | Male | 74 | 4.5 | 5.60 | + | + | Hypothyroidism, CKD |
| 11 | Female | 57 | 3.6 | 4.00 | + | − | Lung cancer, dyslipidemia |
| Patients | Histological Pattern | Glomerulosclerosis | Tubular Atrophy/Interstitial Fibrosis | Crescents | DNAJB9 | Immunofluorescence |
|---|---|---|---|---|---|---|
| 1 | Diffuse sclerosing | 75% | 35% | − | NA | IgG 1+, IgM 1+, IgA 1+, C3 3+, C1q trace, κ negative, λ 1–2 + |
| 2 | Membranoproliferative | 66% | 25–30% | − | NA | IgG 1–2+, IgM 1–2+, IgA trace, C3 1–2+, C1q trace, κ λ negative |
| 3 | Mesangial proliferative | 30% | 25% | − | + | IgG 2+, IgM 2–3+, IgA trace, C3 3–4+, C1q, κ, λ trace |
| 4 | Mesangial proliferative | 63% | 35% | − | + | IgG 1–2 +, IgM trace, ΙgA negative, C3 1–2+, C1q negative, λ 1–2+, κ negative |
| 5 | Mesangial proliferative | 18% | 20% | 1 cellular | NA | IgG 2–3+, IgM trace, ΙgA 2–3+, C3 2+, C1q 2+, λ 3+, κ 2+ |
| 6 | Mesangial proliferative + membranoproliferative | 18% | 25% | − | NA | IgG 1–2+, IgM 1–2+ IgA negative, C3 3+, C1q, κ, λ trace |
| 7 | Membranous | 60% | 25% | − | + | IgG 1–2+, IgM 1+, IgA trace, C3 2–3+, C1q trace, κ trace, λ 2+ |
| 8 | Mesangial proliferative | 40% | 20% | − | NA | IgG 1+, IgM trace – 1+, IgA negative, C3 2–3+, C1q 1+, κ 1+, λ 2–3+ |
| 9 | Mesangial proliferative | 6% | 20% | − | NA | IgG 2–3+, IgM trace, IgA 1+, C3 2+, C1q trace, κ 1–2+, λ 3+ |
| 10 | Crescentic | 27% | 35% | 4 [2 cellular, 2 fibrocellular] | + | IgG 1+, IgM trace, IgA negative, C3 trace, C1q negative, κ, λ trace |
| 11 | Crescentic | 8% | 25% | 8 [5 cellular, 3 fibrocellular] | + | IgG, IgA negative, IgM trace, C3 1–2+, C1q trace, κ, λ negative |
| Patients | Follow up Period [Months] | Immunosuppression | sCr [mg/dL] Diagnosis | sCr [mg/dL] Last Measurement | Upr [g/24 h] Diagnosis | Upr [g/24 h] Last Measurement | Therapeutic Response |
|---|---|---|---|---|---|---|---|
| 1 | 24 | GC + RTX | 1.7 | 1.7 | 3.7 | 0.8 | PR |
| 2 | 24 | CYC + RTX | 1.4 | 1.5 | 3.5 | 0.5 | PR |
| 3 | 6 | GC + RTX | 0.9 | 0.9 | 3 | 1.2 | PR |
| 4 | 12 | GC + RTX | 1.7 | 1.4 | 4.5 | 2.7 | PR |
| 5 | 48 | GC + RTX | 0.8 | 1 | 1.3 | 0.1 | CR |
| 6 | 36 | GC + CYC + MMF | 2.4 | 1.7 | 3.7 | 0.05 | PR |
| 7 | 12 | GC + RTX | 1.2 | 1.2 | 11.6 | 1.9 | PR |
| 8 | 60 | GC + RTX | 1.6 | 2.2 | 8 | 7.8 | PRD |
| 9 | 72 | GC + RTX + MMF | 0.8 | 0.7 | 1.8 | 2.5 | PRD |
| 10 | 6 | GC + CYC + RTX | 4.5 | 3.3 | 5.6 | 1.6 | PR |
| 11 | 2 | GC + CYC + RTX | 3.8 | 1.6 | 4 | 1.5 | PR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poulli, T.; Liaveri, P.; Liapis, G.; Daoudaki, M.; Fouza, A.; Stangou, M.; Moustakas, G. Therapeutic Challenges and New Era in Fibrillary Glomerulonephritis with the Introduction of DNAJB9: Experience from a Tertiary Nephrology Center. J. Clin. Med. 2025, 14, 3709. https://doi.org/10.3390/jcm14113709
Poulli T, Liaveri P, Liapis G, Daoudaki M, Fouza A, Stangou M, Moustakas G. Therapeutic Challenges and New Era in Fibrillary Glomerulonephritis with the Introduction of DNAJB9: Experience from a Tertiary Nephrology Center. Journal of Clinical Medicine. 2025; 14(11):3709. https://doi.org/10.3390/jcm14113709
Chicago/Turabian StylePoulli, Tsielestina, Paraskevi Liaveri, George Liapis, Maria Daoudaki, Ariadni Fouza, Maria Stangou, and George Moustakas. 2025. "Therapeutic Challenges and New Era in Fibrillary Glomerulonephritis with the Introduction of DNAJB9: Experience from a Tertiary Nephrology Center" Journal of Clinical Medicine 14, no. 11: 3709. https://doi.org/10.3390/jcm14113709
APA StylePoulli, T., Liaveri, P., Liapis, G., Daoudaki, M., Fouza, A., Stangou, M., & Moustakas, G. (2025). Therapeutic Challenges and New Era in Fibrillary Glomerulonephritis with the Introduction of DNAJB9: Experience from a Tertiary Nephrology Center. Journal of Clinical Medicine, 14(11), 3709. https://doi.org/10.3390/jcm14113709