
State of the Art on Inherited Retinal Dystrophies: Management and Molecular Genetics

 , , , , and
, , , , and 
Abstract

1. Introduction
- Loss of function, when the gene product is either not produced or is nonfunctional;
- Gain of function, where a change in the gene product becomes toxic to the cell;
- Dominant-negative effect, when the mutant gene product interferes with the normal function of the protein.
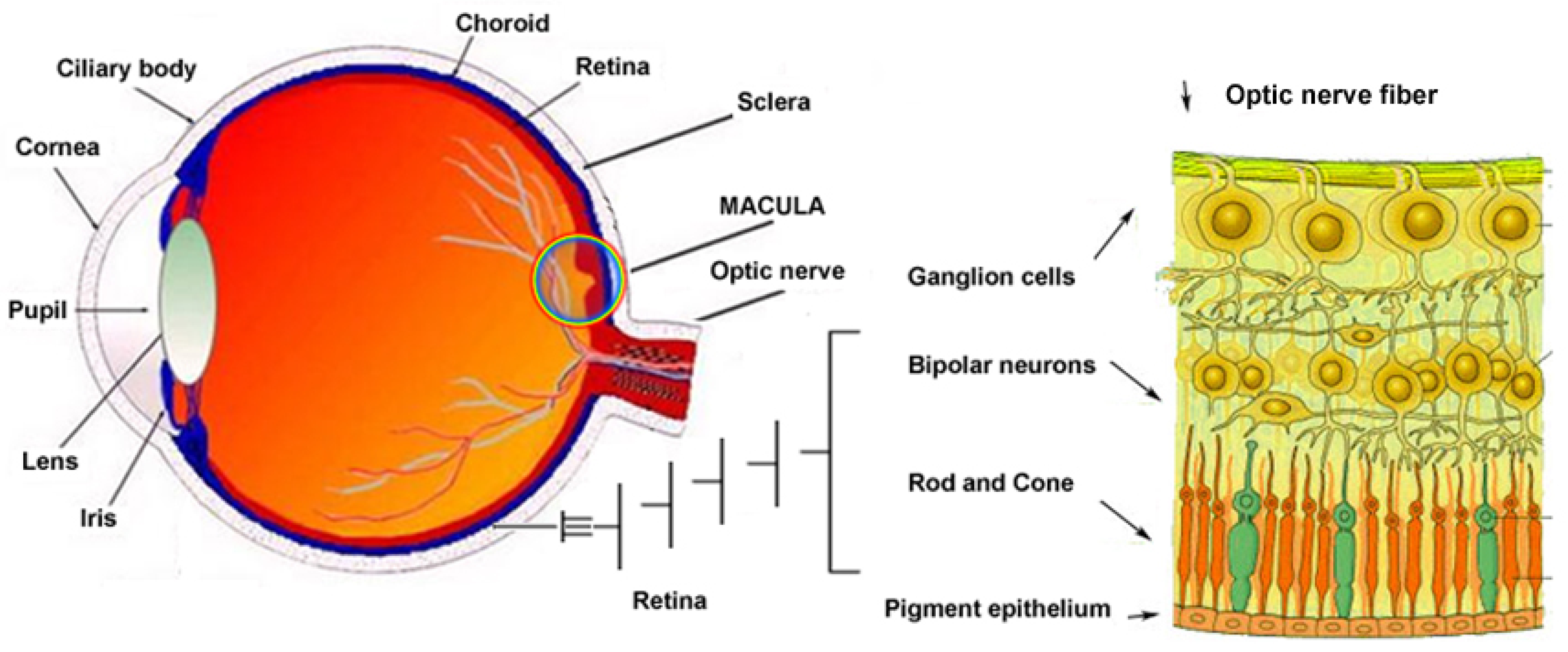
2. Brief Overview of the Key Genes Involved in IRDs (Figure 2 and Figure 3)
2.1. Leber Congenital Amaurosis (LCA)
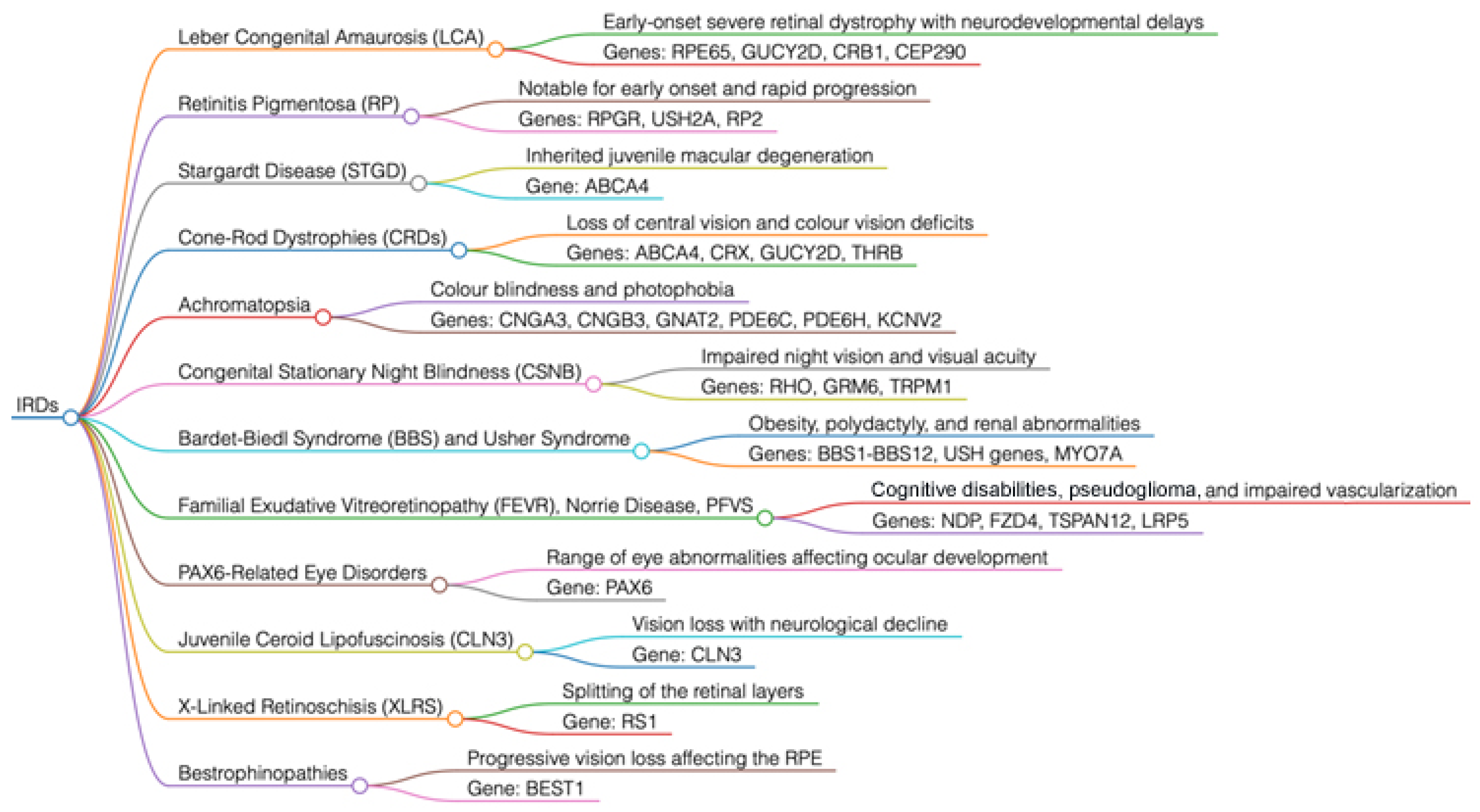

2.2. Retinitis Pigmentosa (RP)
2.3. X-Linked Retinoschisis (XLRS)
2.4. Stargardt Disease (STGD)
2.5. Cone and Rod Dystrophies (CRDs)
2.6. Achromatopsia
2.7. Congenital Stationary Night Blindness (CSNB)
2.8. Bestrophinopathies
3. Key Genes Involved in Syndromic Forms of IRDs (Figure 2)
3.1. Bardet–Biedl Syndrome (BBS)
3.2. Usher Syndrome
- ○
- MYO7A (USH1B)
- ○
- USH1C (Harmonin)
- ○
- CDH23 (Cadherin-23)
- ○
- PCDH15 (Protocadherin-15)
- ○
- SANS (USH1G)
- ○
- USH2A (Usherin)
- ○
- GPR98 (VLGR1)
- ○
- DFNB31 (Whirlin)
- ○
- CLRN1
- ○
- HARS
3.3. Familial Exudative Vitreoretinopathy (FEVR), Norrie Disease, and Pseudoglioma
3.4. Ocular Disorders Associated with PAX6
3.5. Juvenile Neuronal Ceroid Lipofuscinosis (JNCL) or Batten Disease
3.6. Mitochondrial Eye Diseases
3.7. Screening of High Myopia Syndromic Monogenic (msHM)
4. Clinical Practice Implications, Criteria, and Diagnostic Techniques Applied to IRDs
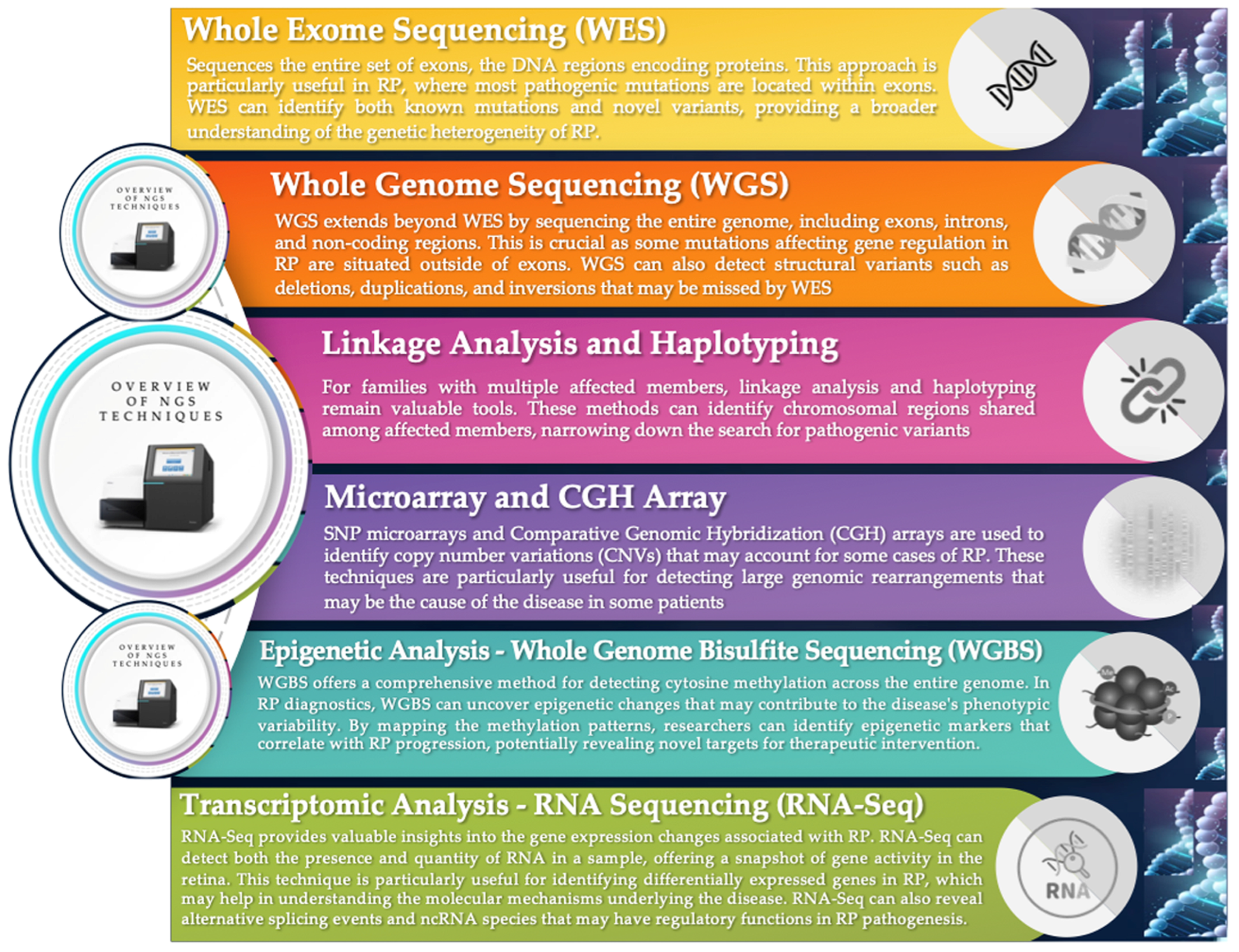
5. Genetic Analysis Technologies: From WES to Advanced Genomic Profiling
6. Effective Patient Communication: Genetic Counseling, Interpretation of Reports, and Management Strategies for IRDs
- ❖
- Pathogenic (P)
- ❖
- Likely pathogenic (LP)
- ❖
- Uncertain significance (VUS)
- ❖
- Likely benign (LB)
- ❖
- Benign (B)
7. Focus on Gene Therapy for IRDs: Genome Editing, Ethical and Regulatory Issues
Clinical Results and Long-Term Effects of the Luxturna Gene Therapy
8. Future Directions in Research and Therapy for IRDs
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAVs | Adeno-associated viruses |
| ACMG | American College of Medical Genetics and Genomics |
| AD | Autosomal dominant |
| ADOA | Autosomic dominant optic atrophy Kjer type |
| AI | Artificial intelligence |
| AR | Autosomal recessive |
| B | Benign |
| BBS | Bardet–Biedl syndrome |
| CGH | Comparative genomic hybridization |
| CRDs | Cone and rod dystrophies |
| CSNB | Congenital stationary night blindness |
| ERG | Electroretinogram |
| ETDRS | Early treatment diabetic retinopathy study grid |
| FA | Fluorescein angiography |
| FAF | Fundus autofluorescence |
| FDA | Food and Drug Administration |
| FEVR | Familial exudative vitreoretinopathy |
| GWASs | Genome-wide association studies |
| ICG | Indocyanine green angiography |
| IRDs | Inherited retinal dystrophies |
| JNCL | Juvenile neuronal ceroid lipofuscinosis |
| LB | Likely benign |
| LCA | Leber congenital amaurosis |
| LHON | Leber hereditary optic neuropathy |
| LP | Likely pathogenic |
| MAGIC | Myopia Associated Genetics and Intervention Consortium |
| ML | Machine learning |
| msHM | High myopia syndromic monogenic |
| mtDNA | Mitochondrial and nuclear genes |
| NGS | Next-generation sequencing |
| OCT | Optical coherence tomography |
| OCTA | Optical coherence tomography angiography |
| P | Pathogenic |
| PGD | Preimplantation genetic diagnosis |
| RD | Retinal detachment |
| RNA-Seq | RNA sequencing |
| RP | Retinitis pigmentosa |
| RPE | Retinal pigment epithelial |
| STGD | Stargardt disease |
| VF | Visual field |
| VUS | Uncertain significance |
| WES | Whole exome sequencing |
| WGBS | Whole genome bisulfite sequencing |
| WGS | Whole genome sequencing |
| XLRS | X-linked retinoschisis |
References
- Hu, M.L.; Edwards, T.L.; O’Hare, F.; Hickey, D.G.; Wang, J.H.; Liu, Z.; Ayton, L.N. Gene therapy for inherited retinal diseases: Progress and possibilities. Clin. Exp. Optom. 2021, 104, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Tsui, I.; Song, B.J.; Lin, C.S.; Tsang, S.H. A Practical approach to retinal dystrophies. Adv. Exp. Med. Biol. 2018, 1085, 245–259. [Google Scholar] [CrossRef]
- Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal inflammation, cell death and inherited retinal dystrophies. Int. J. Mol. Sci. 2021, 22, 2096. [Google Scholar] [CrossRef]
- Vázquez-Domínguez, I.; Garanto, A.; Collin, R.W.J. Molecular therapies for inherited retinal diseases-current standing, opportunities and Challenges. Genes 2019, 10, 654. [Google Scholar] [CrossRef]
- Lam, B.L.; Leroy, B.P.; Black, G.; Ong, T.; Yoon, D.; Trzupek, K. Genetic testing and diagnosis of inherited retinal diseases. Orphanet J. Rare Dis. 2021, 16, 514. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, M.; Robson, A.G.; Fujinami, K.; de Guimarães, T.A.; Fujinami-Yokokawa, Y.; Varela, M.D.; Pontikos, N.; Kalitzeos, A.; Mahroo, O.A.; Webster, A.R.; et al. Phenotyping and genotyping inherited retinal diseases: Molecular genetics, clinical and imaging features, and therapeutics of macular dystrophies, cone and cone-rod dystrophies, rod-cone dystrophies, Leber congenital amaurosis, and cone dysfunction syndromes. Prog. Retin. Eye Res. 2024, 100, 101244. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Okan, I.C.T.; Uyanik, G.; Tschopp, M.; Agca, C. Precise gene editing technologies in retinal applications. Adv. Exp. Med. Biol. 2025, 1468, 119–123. [Google Scholar] [CrossRef]
- Schneider, N.; Sundaresan, Y.; Gopalakrishnan, P.; Beryozkin, A.; Hanany, M.; Levanon, E.Y.; Banin, E.; Ben-Aroya, S.; Sharon, D. Inherited retinal diseases: Linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin. Eye Res. 2022, 89, 101029. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Abdalla, E.M.; Nabil, K.M.; D’Angelo, R.; Sidoti, A. New omics-derived perspectives on retinal dystrophies: Could ion channels-encoding or related genes act as modifier of pathological phenotype? Int. J. Mol. Sci. 2020, 22, 70. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Alibrandi, S.; Vadalà, M.; Giglia, G.; Sidoti, A.; D’Angelo, R. N-retinylidene-N-retinylethanolamine adduct induces expression of chronic inflammation cytokines in retinal pigment epithelium cells. Exp. Eye Res. 2021, 209, 108641. [Google Scholar] [CrossRef]
- Huang, C.H.; Yang, C.M.; Yang, C.H.; Hou, Y.C.; Chen, T.C. Leber’s congenital amaurosis: Current concepts of genotype-phenotype correlations. Genes 2021, 12, 1261. [Google Scholar] [CrossRef] [PubMed]
- Sather, R., 3rd; Ihinger, J.; Simmons, M.; Lobo, G.P.; Montezuma, S.R. The clinical findings, pathogenic variants, and gene therapy qualifications found in a Leber congenital amaurosis phenotypic spectrum patient cohort. Int. J. Mol. Sci. 2024, 25, 1253. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, L.; Xie, Y.; Liu, W.; Zhang, S.; Liu, L.; Lin, P.; Li, N. Clinical and genetic studies for a cohort of patients with Leber congenital amaurosis. Graefes Arch. Clin. Exp. Ophthalmol. 2024, 262, 3029–3038. [Google Scholar] [CrossRef] [PubMed]
- Miraldi Utz, V.; Coussa, R.G.; Antaki, F.; Traboulsi, E.I. Gene therapy for RPE65-related retinal disease. Ophthalmic Genet. 2018, 39, 671–677. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Bowne, S.J. Mutations in CRB1, GUCY2D, and other genes in Leber congenital amaurosis: Clinical features and molecular mechanisms. Human Genet. 2019, 138, 813–828. [Google Scholar]
- Wang, J.; Li, S.; Jiang, Y.; Wang, Y.; Ouyang, J.; Yi, Z.; Sun, W.; Jia, X.; Xiao, X.; Wang, P.; et al. Pathogenic variants in CEP290 or IQCB1 cause earlier-onset retinopathy in Senior-Loken syndrome compared to those in INVS, NPHP3, or NPHP4. Am. J. Ophthalmol. 2023, 252, 188–204. [Google Scholar] [CrossRef]
- Battu, R.; Ratra, D.; Gopal, L. Newer therapeutic options for inherited retinal diseases: Gene and cell replacement therapy. Indian. J. Ophthalmol. 2022, 70, 2316–2325. [Google Scholar] [CrossRef]
- Takkar, B.; Bansal, P.; Venkatesh, P. Leber’s Congenital Amaurosis and Gene Therapy. Indian. J. Pediatr. 2018, 85, 237–242. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef]
- Pungor, K.; Lee, J.; Denee, T.; Kambarov, Y.; Nissinen, R.; Ampeh, K.; Pellegrini, M.; Parmeggiani, F. Impacts of X-linked retinitis pigmentosa and patient pathways in European countries: Results from the cross-sectional EXPLORE XLRP-1 Physician Survey. Advances in therapy 2024, 41, 3378–3395. [Google Scholar] [CrossRef]
- Karali, M.; Testa, F.; Di Iorio, V.; Torella, A.; Zeuli, R.; Scarpato, M.; Romano, F.; Onore, M.E.; Pizzo, M.; Melillo, P.; et al. Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy. Sci. Rep. 2022, 12, 20815. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, S.E.; Song, E.; Raoof, N.; van Bysterveldt, K.; Oliver, V.F.; Hong, S.C.; Al-Taie, R.; Wilson, G.; Vincent, A.L. Genotypic and phenotypic characterisation of RP2- and RPGR-associated X-linked inherited retinal dystrophy, including female manifestations. Clin. Exp. Ophthalmol. 2023, 51, 300–312. [Google Scholar] [CrossRef]
- Maurya, R.; Vikal, A.; Narang, R.K.; Patel, P.; Kurmi, B.D. Recent advancements and applications of ophthalmic gene therapy strategies: A breakthrough in ocular therapeutics. Exp. Eye Res. 2024, 245, 109983. [Google Scholar] [CrossRef]
- Ku, C.A.; Wei, L.W.; Sieving, P.A. X-Linked retinoschisis. Cold Spring Harb. Perspect. Med. 2023, 13, a041288. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S. Current clinical applications of in vivo gene therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef]
- Heymann, J.B.; Vijayasarathy, C.; Fariss, R.N.; Sieving, P.A. Advances in understanding the molecular structure of retinoschisin while questions remain of biological function. Prog. Retin. Eye Res. 2023, 95, 101147. [Google Scholar] [CrossRef] [PubMed]
- Tanna, P.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2017, 101, 25–30. [Google Scholar] [CrossRef]
- Fujinami, K.; Waheed, N.; Laich, Y.; Yang, P.; Fujinami-Yokokawa, Y.; Higgins, J.J.; Lu, J.T.; Curtiss, D.; Clary, C.; Michaelides, M. Stargardt macular dystrophy and therapeutic approaches. Br. J. Ophthalmol. 2024, 108, 495–505. [Google Scholar] [CrossRef]
- Sahel, J.A.; Marazova, K.; Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb. Perspect. Med. 2014, 5, a017111. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Rinaldi, C.; Aragona, P.; Briuglia, S.; D’Ascola, A.; D’Angelo, R.; Sidoti, A. Stargardt phenotype associated with two ELOVL4 promoter variants and ELOVL4 downregulation: New Possible Perspective to Etiopathogenesis? Invest. Ophthalmol. Vis. Sci. 2018, 59, 843–857. [Google Scholar] [CrossRef]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Nassisi, M.; Smirnov, V.M.; Solis Hernandez, C.; Mohand-Saïd, S.; Condroyer, C.; Antonio, A.; Kühlewein, L.; Kempf, M.; Kohl, S.; Wissinger, B.; et al. CNGB1-related rod-cone dystrophy: A mutation review and update. Hum. Mutat. 2021, 42, 641–666. [Google Scholar] [CrossRef]
- Fernández-Suárez, E.; González-del Pozo, M.; García-Núñez, A.; Méndez-Vidal, C.; Martín-Sánchez, M.; Mejías-Carrasco, J.M.; Ramos-Jiménez, M.; Morillo-Sánchez, M.J.; Rodríguez-de la Rúa, E.; Borrego, S.; et al. Expanding the phenotype of THRB: A range of macular dystrophies as the major clinical manifestations in patients with a dominant splicing variant. Front. Cell Dev. Biol. 2023, 11. [Google Scholar] [CrossRef]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef] [PubMed]
- Hirji, N.; Aboshiha, J.; Georgiou, M.; Bainbridge, J.; Michaelides, M. Achromatopsia: Clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet. 2018, 39, 149–157. [Google Scholar] [CrossRef]
- Pascual-Camps, I.; Barranco-Gonzalez, H.; Aviñó-Martínez, J.; Silva, E.; Harto-Castaño, M. Diagnosis and treatment options for achromatopsia: A review of the literature. J. Pediatr. Ophthalmol. Strabismus. 2018, 55, 85–92. [Google Scholar] [CrossRef]
- Katta, M.; de Guimaraes, T.A.C.; Fujinami-Yokokawa, Y.; Fujinami, K.; Georgiou, M.; Mahroo, O.A.; Webster, A.R.; Michaelides, M. Congenital stationary night blindness: Structure, function and genotype-phenotype correlations in a cohort of 122 patients. Ophthalmol. Retina. 2024, 8, 932–941. [Google Scholar] [CrossRef]
- Stefaniuk-Szmukier, M.; Bieniek, A.; Ropka-Molik, K.; Bellone, R.R. Genetic testing as a tool for diagnosis of congenital stationary night blindness (CSNB) in white spotted breeds in Poland. J. Equine Vet. Sci. 2025, 147, 105405. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.; Wongchaisuwat, N.; Lamborn, A.; Schmidt, R.; Everett, L.; Yang, P.; Pennesi, M.E. Gene therapy in bestrophinopathies: Insights from preclinical studies in preparation for clinical trials. Saudi J. Ophthalmol. 2023, 37, 287–295. [Google Scholar] [CrossRef]
- Miyagi, M.; Takeuchi, J.; Koyanagi, Y.; Mizobuchi, K.; Hayashi, T.; Ito, Y.; Terasaki, H.; Nishiguchi, K.M.; Ueno, S. Clinical findings in eyes with BEST1-related retinopathy complicated by choroidal neovascularization. Graefes Arch. Clin. Exp. Ophthalmol. 2022, 260, 1125–1137. [Google Scholar] [CrossRef]
- Shoemaker, A. Bardet-Biedl syndrome: A clinical overview focusing on diagnosis, outcomes and best-practice management. Diabetes Obes. Metab. 2024, 26 (Suppl. 2), 25–33. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.R.; Krentz, A.D.; Berg, R.L.; Richardson, J.G.; Pomeroy, J.; Hebbring, S.J.; Haws, R.M. Kidney failure in Bardet-Biedl syndrome. Clin. Genet. 2022, 101, 429–441. [Google Scholar] [CrossRef]
- Tomlinson, J.W. Bardet-Biedl syndrome: A focus on genetics, mechanisms and metabolic dysfunction. Diabetes Obes. Metab. 2024, 26 (Suppl. 2), 13–24. [Google Scholar] [CrossRef]
- Satariano, M.; Ghose, S.; Raina, R. The pathophysiology of inherited renal cystic diseases. Genes 2024, 15, 91. [Google Scholar] [CrossRef]
- Niederlova, V.; Modrak, M.; Tsyklauri, O.; Huranova, M.; Stepanek, O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum. Mutat. 2019, 40, 2068–2087. [Google Scholar] [CrossRef]
- Delmaghani, S.; El-Amraoui, A. The genetic and phenotypic landscapes of Usher syndrome: From disease mechanisms to a new classification. Hum. Genet. 2022, 141, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Riahi, Z.; Chantot-Bastaraud, S.; Smagghe, L.; Letexier, M.; Marcaillou, C.; Lefèvre, G.M.; Hardelin, J.P.; El-Amraoui, A.; Singh-Estivalet, A.; et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur. J. Hum. Genet. 2016, 24, 1730–1738. [Google Scholar] [CrossRef] [PubMed]
- Toms, M.; Pagarkar, W.; Moosajee, M. Usher syndrome: Clinical features, molecular genetics and advancing therapeutics. Ther. Adv. Ophthalmol. 2020, 12, 2515841420952194. [Google Scholar] [CrossRef]
- Fuster-García, C.; García-Bohórquez, B.; Rodríguez-Muñoz, A.; Aller, E.; Jaijo, T.; Millán, J.M.; García-García, G. Usher syndrome: Genetics of a human ciliopathy. Int. J. Mol. Sci. 2021, 22, 6723. [Google Scholar] [CrossRef]
- Le Quesne Stabej, P.; Saihan, Z.; Rangesh, N.; Steele-Stallard, H.B.; Ambrose, J.; Coffey, A.; Emmerson, J.; Haralambous, E.; Hughes, Y.; Steel, K.P.; et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J. Med. Genet. 2012, 49, 27–36. [Google Scholar] [CrossRef]
- Fuster-García, C.; García-García, G.; Jaijo, T.; Fornés, N.; Ayuso, C.; Fernández-Burriel, M.; Sánchez-De la Morena, A.; Aller, E.; Millán, J.M. High-throughput sequencing for the molecular diagnosis of Usher syndrome reveals 42 novel mutations and consolidates CEP250 as Usher-like disease causative. Sci. Rep. 2018, 8, 17113. [Google Scholar] [CrossRef]
- Tauqeer, Z.; Yonekawa, Y. Familial exudative vitreoretinopathy: Pathophysiology, diagnosis, and management. Asia-Pacific J. Ophthalmol. 2018, 7, 176–182. [Google Scholar] [CrossRef]
- Dickinson, J.L.; Sale, M.M.; Passmore, A.; FitzGerald, L.M.; Wheatley, C.M.; Burdon, K.P.; Craig, J.E.; Tengtrisorn, S.; Carden, S.M.; Maclean, H.; et al. Mutations in the NDP gene: Contribution to Norrie disease, familial exudative vitreoretinopathy and retinopathy of prematurity. Clin. Exp. Ophthalmol. 2006, 34, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Wawrzynski, J.; Patel, A.; Badran, A.; Dowell, I.; Henderson, R.; Sowden, J.C. Spectrum of mutations in NDP resulting in ocular disease; a systematic review. Front. Genet. 2022, 13, 884722. [Google Scholar] [CrossRef]
- Deml, B.; Reis, L.M.; Lemyre, E.; Clark, R.D.; Kariminejad, A.; Semina, E.V. Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur. J. Hum. Genet. 2016, 24, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Bardakjian, T.M.; Schneider, A. The genetics of anophthalmia and microphthalmia. Curr. Opin. Ophthalmol. 2011, 22, 309–313. [Google Scholar] [CrossRef]
- Singh, R.B.; Gupta, P.; Kartik, A.; Farooqui, N.; Singhal, S.; Shergill, S.; Singh, K.P.; Agarwal, A. Ocular manifestations of neuronal ceroid lipofuscinoses. Semin. Ophthalmol. 2021, 36, 582–595. [Google Scholar] [CrossRef]
- Mole, S.E. The neuronal ceroid lipofuscinoses. In Jasper’s Basic Mechanisms of the Epilepsies, 5th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Vezzani, A., Delgado-Escueta, A.V., Eds.; Oxford University Press: New York, NY, USA, 2024; pp. 1067–1084. [Google Scholar]
- Chen, B.S.; Harvey, J.P.; Gilhooley, M.J.; Jurkute, N.; Yu-Wai-Man, P. Mitochondria and the eye-manifestations of mitochondrial diseases and their management. Eye 2023, 37, 2416–2425. [Google Scholar] [CrossRef]
- Finsterer, J.; Mancuso, M.; Pareyson, D.; Burgunder, J.M.; Klopstock, T. Mitochondrial disorders of the retinal ganglion cells and the optic nerve. Mitochondrion 2018, 42, 1–10. [Google Scholar] [CrossRef]
- Donato, L.; Morda, D.; Scimone, C.; Alibrandi, S.; D’Angelo, R.; Sidoti, A. From powerhouse to regulator: The role of mitoepigenetics in mitochondrion-related cellular functions and human diseases. Free Rad. Biol. Med. 2024, 218, 105–119. [Google Scholar] [CrossRef]
- Chen, C.; An, G.; Yu, X.; Wang, S.; Lin, P.; Yuan, J.; Zhuang, Y.; Lu, X.; Bai, Y.; Zhang, G.; et al. Screening mutations of the monogenic syndromic high myopia by whole exome sequencing from MAGIC project. Investig. Ophthalmol. Vis. Sci. 2024, 65, 9. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Q. Insight into the molecular genetics of myopia. Mol. Vis. 2017, 23, 1048–1080. [Google Scholar] [PubMed]
- Muftuoglu, I.K.; Al-Sheikh, M.J.S.; Rasheed, M.A.; Singh, S.R.; Chhablani, J. Imaging in inherited retinal disorders. Eur. J. Ophthalmol. 2021, 31, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Khurram Butt, D.; Gurbaxani, A.; Kozak, I. Ultra-wide-field fundus autofluorescence for the detection of inherited retinal disease in difficult-to-examine children. J. Pediatr. Ophthalmol. Strabismus 2019, 56, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Merin, S.; Auerbach, E. Retinitis pigmentosa. Surv. Ophthalmol. 1976, 20, 303–346. [Google Scholar] [CrossRef]
- Daich Varela, M.; Esener, B.; Hashem, S.A.; Cabral de Guimaraes, T.A.; Georgiou, M.; Michaelides, M. Structural evaluation in inherited retinal diseases. Br. J. Ophthalmol. 2021, 105, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Iovino, C.; Iodice, C.M.; Pisani, D.; Damiano, L.; Di Iorio, V.; Testa, F.; Simonelli, F. Clinical applications of optical coherence tomography angiography in inherited retinal diseases: An up-to-date review of the literature. J. Clin. Med. 2023, 12, 3170. [Google Scholar] [CrossRef]
- Sujirakul, T.; Lin, M.K.; Duong, J.; Wei, Y.; Lopez-Pintado, S.; Tsang, S.H. Multimodal imaging of central retinal disease progression in a 2-year mean follow-up of retinitis pigmentosa. Am. J. Ophthalmol. 2015, 160, 786–798.e4. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef]
- Gregori, N.Z.; Hussain, R.M.; Kay, C.N.; Lam, B.L.; Dermer, H.; Davis, J.L. Current Concepts and Emerging Gene Therapies for Inherited Retinal Diseases. Int. Ophthalmol. Clin. 2019, 59, 83–110. [Google Scholar] [CrossRef]
- Stöhr, H.; Weber, B.H.F. Genetics and diagnostics of inherited retinal diseases in the era of whole genome sequencing. Med. Genet. 2025, 37, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.; Naeem, M.A.; Ali, M.H.; Assir, M.Z.; Khan, S.N.; Riazuddin, S.; Hejtmancik, J.F.; Riazuddin, S.A.; Ayyagari, R. Whole-Exome Sequencing Identifies Novel Variants that Co-segregates with Autosomal Recessive Retinal Degeneration in a Pakistani Pedigree. Adv. Exp. Med. Biol. 2018, 1074, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenbergh, C.; Van Schil, K.; Cannoodt, R.; Bauwens, M.; Van Laethem, T.; De Jaegere, S.; Steyaert, W.; Sante, T.; Menten, B.; Leroy, B.P.; et al. arrEYE: A customized platform for high-resolution copy number analysis of coding and noncoding regions of known and candidate retinal dystrophy genes and retinal noncoding RNAs. Genet. Med. 2017, 19, 457–466. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Vadalà, M.; Castellucci, M.; Bonfiglio, V.M.E.; Scalinci, S.Z.; Abate, G.; D’Angelo, R.; Sidoti, A. The genomic mosaic of mitochondrial dysfunction: Decoding nuclear and mitochondrial epigenetic contributions to maternally inherited diabetes and deafness pathogenesis. Heliyon 2024, 10, e34756. [Google Scholar] [CrossRef]
- Voigt, A.P.; Mullin, N.K.; Stone, E.M.; Tucker, B.A.; Scheetz, T.E.; Mullins, R.F. Single-cell RNA sequencing in vision research: Insights into human retinal health and disease. Prog. Retin. Eye Res. 2021, 83, 100934. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Alibrandi, S.; Pitruzzella, A.; Scalia, F.; D’Angelo, R.; Sidoti, A. Possible A2E Mutagenic Effects on RPE Mitochondrial DNA from Innovative RNA-Seq Bioinformatics Pipeline. Antioxidants 2020, 9, 1158. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa. Antioxidants 2020, 9, 318. [Google Scholar] [CrossRef]
- Yan, A.L.; Du, S.W.; Palczewski, K. Genome editing, a superior therapy for inherited retinal diseases. Vis. Res. 2023, 206, 108192. [Google Scholar] [CrossRef]
- Dockery, A.; Whelan, L.; Humphries, P.; Farrar, G.J. Next-Generation Sequencing Applications for Inherited Retinal Diseases. Int. J. Mol. Sci. 2021, 22, 5684. [Google Scholar] [CrossRef]
- Chen, D.; Xu, Y.; Fu, Y.; Wang, Y.; Liu, Y.; Ding, C.; Cai, B.; Pan, J.; Wang, J.; Li, R.; et al. Clinical application of next generation sequencing-based haplotype linkage analysis in the preimplantation genetic testing for germline mosaicisms. Orphanet J. Rare Dis. 2023, 18, 137. [Google Scholar] [CrossRef]
- Tanudisastro, H.A.; Deveson, I.W.; Dashnow, H.; MacArthur, D.G. Sequencing and characterizing short tandem repeats in the human genome. Nat. Rev. Genet. 2024, 25, 460–475. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Calzetti, G.; Schwarzwälder, K.; Ottonelli, G.; Kaminska, K.; Strauss, R.W.; Baere, E.; Leroy, B.P.; Audo, I.; Zeitz, C.; Cursiefen, C.; et al. Genetic Testing of Patients with Inherited Retinal Diseases in the European Countries: An International Survey by the European Vision Institute. Ophthalmic Res. 2024, 67, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Benati, D.; Patrizi, C.; Recchia, A. Gene editing prospects for treating inherited retinal diseases. J. Med. Genet. 2020, 57, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, N.; Mai, Q.; Zhou, C. The frontier of precision medicine: Application of single-cell multi-omics in preimplantation genetic diagnosis. Brief. Funct. Genomics. 2024, 23, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Gage, H.; Wetherill, L.; Anderson, K.; Conboy, E.; Haider, K. Motivations and expectations of parents seeking genetic testing for their children with ocular genetic disease. Ophthalmic Genet. 2023, 44, 371–378. [Google Scholar] [CrossRef]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef]
- Alsalloum, A.; Gornostal, E.; Mingaleva, N.; Pavlov, R.; Kuznetsova, E.; Antonova, E.; Nadzhafova, A.; Kolotova, D.; Kadyshev, V.; Mityaeva, O.; et al. A Comparative Analysis of Models for AAV-Mediated Gene Therapy for Inherited Retinal Diseases. Cells 2024, 13, 1706. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860, Erratum in Lancet 2017, 390, 848. [Google Scholar] [CrossRef]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S.; Chakraborty, C. CRISPR-Cas9: A preclinical and clinical perspective for the treatment of human diseases. Mol. Ther. 2021, 29, 571–586. [Google Scholar] [CrossRef]
- Hernández-Juárez, J.; Rodríguez-Uribe, G.; Borooah, S. Toward the Treatment of Inherited Diseases of the Retina Using CRISPR-Based Gene Editing. Front. Med. 2021, 8, 698521. [Google Scholar] [CrossRef]
- Zhao, Q.; Wei, L.; Chen, Y. From bench to bedside: Developing CRISPR/Cas-based therapy for ocular diseases. Pharmacol. Res. 2025, 213, 107638. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, I.S.; Radziwon, A.; St Laurent, C.D.; MacDonald, I.M. Choroideremia. Curr. Opin. Ophthalmol. 2017, 28, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Cehajic-Kapetanovic, J.; Bellini, M.P.; Taylor, L.J.; Yusuf, I.H.; Soomro, T.; da Cruz, L.; MacLaren, R.E. Regenerate Study Group. Gene Therapy for Choroideremia Using an Adeno-Associated Viral Vector Encoding Rab Escort Protein 1: The Regenerate Open-Label Trial; National Institute for Health and Care Research: Southampton, UK, 2024. [Google Scholar] [CrossRef]
- Mishra, A.; Sieving, P.A. X-linked Retinoschisis and Gene Therapy. Int. Ophthalmol. Clin. 2021, 61, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.S.C.; McCaughey, T.; Swann, O.; Pébay, A.; Hewitt, A.W. Genome engineering in ophthalmology: Application of CRISPR/Cas to the treatment of eye disease. Prog. Retin. Eye Res. 2016, 53, 1–20. [Google Scholar] [CrossRef]
- Bansal, M. Advances in retina genetics: Progress, potential, and challenges. Indian. J. Ophthalmol. 2025, 73 (Suppl. 1), S31–S36. [Google Scholar] [CrossRef]
- Bucher, K.; Rodríguez-Bocanegra, E.; Dauletbekov, D.; Fischer, M.D. Immune responses to retinal gene therapy using adeno-associated viral vectors—Implications for treatment success and safety. Prog. Retin. Eye Res. 2021, 83, 100915. [Google Scholar] [CrossRef]
- Britten-Jones, A.C.; Jin, R.; Gocuk, S.A.; Cichello, E.; O’Hare, F.; Hickey, D.G.; Edwards, T.L.; Ayton, L.N. The safety and efficacy of gene therapy treatment for monogenic retinal and optic nerve diseases: A systematic review. Genet. Med. 2022, 24, 521–534. [Google Scholar] [CrossRef]
- Turriff, A.E.; Cukras, C.A.; Brooks, B.P.; Huryn, L.A. Considerations in multi-gene panel testing in pediatric ophthalmology. J. AAPOS. 2019, 23, 163–165.e1. [Google Scholar] [CrossRef]
- Moreno Villares, J.M. Reflexión en torno al informe sobre consentimiento informado en pediatría del Comité de Bioética de la Academia Americana de Pediatría (2016) [Reflection on the report on the informed consent in paediatrics by the Bioethics Committee of the American Academy of Pediatrics (2016)]. An. Pediatría 2017, 86, 56–57. [Google Scholar] [CrossRef]
- Lorenz, B. Long-term experience with gene augmentation therapy in patients with inherited retinal disease associated with biallelic mutations in RPE65. Med. Genet. 2025, 37, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Russell, S.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Drack, A.V.; Simonelli, F.; Leroy, B.P.; Reape, K.Z.; High, K.A.; et al. Durability of voretigene neparvovec for Biallelic RPE65-mediated inherited retinal disease: Phase 3 results at 3 and 4 years. Ophthalmology 2021, 128, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Pechnikova, N.A.; Poimenidou, M.; Iliadis, I.; Zafeiriou-Chatziefraimidou, M. Pre-Clinical and Clinical Advances in Gene Therapy of X-Linked Retinitis Pigmentosa: Hope on the Horizon. J. Clin. Med. 2025, 14, 898. [Google Scholar] [CrossRef]
- Abdalla Elsayed, M.E.A.; Cehajic-Kepetanovic, J.; MacLaren, R.E. Gene therapy for choroideremia: Progress, potential and pitfalls. Expert Opin. Biol. Ther. 2025, 25, 257–263. [Google Scholar] [CrossRef]
- Pennesi, M.E.; Yang, P.; Birch, D.G.; Weng, C.Y.; Moore, A.T.; Iannaccone, A.; Comander, J.I.; Jayasundera, T.; Chulay, J.; XLRS-001 Study Group. Intravitreal Delivery of rAAV2tYF-CB-hRS1 Vector for Gene Augmentation Therapy in Patients with X-Linked Retinoschisis: 1-Year Clinical Results. Ophthalmol. Retin. 2022, 6, 1130–1144. [Google Scholar] [CrossRef]
- Jain, R.; Daigavane, S. Advances and challenges in gene therapy for inherited retinal dystrophies: A comprehensive review. Cureus 2024, 16, e69895. [Google Scholar] [CrossRef]
- Szabó, V.; Varsányi, B.; Barboni, M.; Takács, Á.; Knézy, K.; Molnár, M.J.; Nagy, Z.Z.; György, B.; Rivolta, C. Insights into eye genetics and recent advances in ocular gene therapy. Mol. Cell. Probes 2025, 79, 102008. [Google Scholar] [CrossRef]
- Jony, M.J.; Joshi, A.; Dash, A.; Shukla, S. Non-Viral Delivery Systems to Transport Nucleic Acids for Inherited Retinal Disorders. Pharmaceuticals 2025, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Fang, D.; Chen, L.; Diao, Y.Y.; Xie, T.; Zou, Z.H.; Zheng, H.Y.; Zhang, S.C. Research progress in optogenetics for the treatment of retinitis pigmentos. Zhonghua Yan Ke Za Zhi. 2025, 61, 66–70. [Google Scholar] [CrossRef]
- Issa, M.; Sukkarieh, G.; Gallardo, M.; Sarbout, I.; Bonnin, S.; Tadayoni, R.; Milea, D. Applications of artificial intelligence to inherited retinal diseases: A systematic review. Surv. Ophthalmol. 2025, 70, 255–264. [Google Scholar] [CrossRef]
- Inaba, A.; Yoshida, A.; Maeda, A.; Kawai, K.; Kosugi, S.; Takahashi, M. Perception of genetic testing among patients with inherited retinal disease: Benefits and challenges in a Japanese population. J. Genet. Couns. 2022, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.M.; Tham, Y.C.; Simunovic, M.P.; Chen, F.K.; Luu, C.D.; Chen, H.; Jin, Z.B.; Shen, R.J.; Li, S.; Sui, R.; et al. Rationale and protocol paper for the Asia Pacific Network for inherited eye diseases. Asia-Pacific J. Ophthalmol. 2024, 13, 100030. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; de Guimaraes, T.A.C.; Filho, A.G.; Fabozzi, L.; Pearson, R.A.; Michaelides, M. Stem cell-based therapies for retinal diseases: Focus on clinical trials and future prospects. Ophthalmic Genet. 2024, 1–14. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CT/Phase | Study Type | Gene | Disease | Drug | Cohort | Link |
|---|---|---|---|---|---|---|
| NCT05392179 Phase 2 | Open label non-randomized | Rhodopsin mutations including P23H | Retinitis pigmentosa | ADX-2191 serial IVT (methotrexate) | 8 | https://clinicaltrials.gov/study/NCT05392179?term=retinitis%20pigmentosa&rank=3#study-plan (accessed on 11 May 2025) |
| NCT03252847 Phase 1/2 | Open label multicenter, dose escalation trial | RPGR | X-linked retinitis pigmentosa | AAV5-RPGR vector SR | 49 | https://clinicaltrials.gov/study/NCT03252847?term=NCT03252847&rank=1 (accessed on 11 May 2025) |
| NCT02317887 Phase 1/2a |
Prospective, single-center dose escalation | RS1 | Retinoschisis X-linked |
AAV-RS1 vector IVT | 12 | https://clinicaltrials.gov/study/NCT02317887?tab=results (accessed on 11 May 2025) |
| NCT03140969 Phase 1/2 |
Open label, multiple dose escalation | CEP290 p.Cys998X mutation | Amaurosi congenita di Leber | QR-110 RNA antisense oligonucleotide targeting the c.2991+1655A>G IVT | 11 | https://clinicaltrials.gov/study/NCT03140969 (accessed on 11 May 2025) |
| NCT05417126 Phase 2a |
Multicenter open label | ABCA4, ELOVL4, PROM 1 | Stargardt disease |
vMCO-010 vector IVT | 6 | https://clinicaltrials.gov/study/NCT05417126?term=NCT05417126&rank=1 (accessed on 11 May 2025) |
| NCT05748873 Phase 1/2A |
Multicenter double masked randomized | RHO, PDE6A, PDE6B | Rod cone dystrophies |
SPVN06 vector SR | 33 | https://clinicaltrials.gov/study/NCT05748873?term=NCT05748873&rank=1 (accessed on 11 May 2025) |
| NCT03758404 Phase 1/2 |
Open label Multicenter | CNGA3 | Achromatopsia |
AAV-CNGA3 vector SR | 11 | https://clinicaltrials.gov/study/NCT03758404?term=NCT03758404&rank=1 (accessed on 11 May 2025) |
| Therapy | Disease Treated | Gene | AAV | Clinical Outcome |
| Luxturna | RPE65-related retinal Dystrophy | RPE65 | AAV2 | Improvement in low-light vision, long-term visual maintenance |
| Gene therapy for choroideremia | Choroideremia | CHM | AAV2-REP1 | Increased retinal sensitivity, improved visual acuity |
| Gene therapy for X-linked retinoschisis | X-linked retinoschisis | RS1 | AAV | Improvements in retinal morphology and visual acuity preliminary results |
| Clinical Results, Follow-Up Duration and Significant Improvement | ||||
| Study/Trial | Follow-Up | Visual Outcome | Improvement | Observations |
| Phase 3 Luxturna trial | 1–2 years | Improvement in low light vision | About 2 lines on the ETDRS* grid | Variability in improvements among patients, impact on quality of life |
| 3-year follow-up of Luxturna | 3 years | Maintenance or improvement in vision | Better adaptation to low light conditions | Stable long-term results for most patients |
| Choroideremia AAV2-REP1 study | 1 year | Increased retinal sensitivity | Improved sensitivity in the treated group | More noticeable improvements in patients with residual retinal function prior to treatment |
| Clinical Variable | Patients with Significant Improvements | Patients with Lesser Improvements | Factors Associated with Reduced Improvement |
| Age at treatment | Greater improvement in younger patients | Less improvement in older patients | Advanced age may affect response to treatment |
| Pre-existing retinal degeneration | Better results in patients with mild to moderate degeneration | Less pronounced results in patients with advanced degeneration | Presence of pre-existing retinal damage may limit treatment efficacy |
| Global Initiatives and International Cooperation | |||
| Initiative/Program | Primary Objective | Participants/Collaborators | Expected Benefits |
| Global registry for IRDs | Collection of global data on inherited retinal diseases | Hospitals, clinics, and international research institutes | Accelerating research, international data comparison, development of personalized therapies |
| Single-country IRD records to compare | Creation of a global registry for IRD patients | Patient organizations, universities, research centers | Improving clinical decisions and therapeutic protocols |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nebbioso, M.; Artico, M.; Gharbiya, M.; Mannocci, A.; Limoli, P.G.; Iannetta, D.; Donato, L. State of the Art on Inherited Retinal Dystrophies: Management and Molecular Genetics. J. Clin. Med. 2025, 14, 3526. https://doi.org/10.3390/jcm14103526
Nebbioso M, Artico M, Gharbiya M, Mannocci A, Limoli PG, Iannetta D, Donato L. State of the Art on Inherited Retinal Dystrophies: Management and Molecular Genetics. Journal of Clinical Medicine. 2025; 14(10):3526. https://doi.org/10.3390/jcm14103526
Chicago/Turabian StyleNebbioso, Marcella, Marco Artico, Magda Gharbiya, Alice Mannocci, Paolo Giuseppe Limoli, Danilo Iannetta, and Luigi Donato. 2025. "State of the Art on Inherited Retinal Dystrophies: Management and Molecular Genetics" Journal of Clinical Medicine 14, no. 10: 3526. https://doi.org/10.3390/jcm14103526
APA StyleNebbioso, M., Artico, M., Gharbiya, M., Mannocci, A., Limoli, P. G., Iannetta, D., & Donato, L. (2025). State of the Art on Inherited Retinal Dystrophies: Management and Molecular Genetics. Journal of Clinical Medicine, 14(10), 3526. https://doi.org/10.3390/jcm14103526