
Gaucher Disease or Acid Sphingomyelinase Deficiency? The Importance of Differential Diagnosis

 , , , , , ,
, , , , , ,
Abstract

1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Glucocerebrosidase and Acid Sphingomyelinase Activity Assays
2.3. DNA Extraction
2.4. GBA and SMPD1 Genetic Analyses
2.5. Biomarkers Assays
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, B.E.; Weinreb, N.J. Gaucher disease: A comprehensive review. Crit. Rev. Oncog. 2013, 18, 163–175. [Google Scholar] [CrossRef]
- Nalysnyk, L.; Rotella, P.; Simeone, J.C.; Hamed, A.; Weinreb, N. Gaucher disease epidemiology and natural history: A comprehensive review of the literature. Hematology 2017, 22, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Dandana, A.; Ben Khelifa, S.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology 2016, 83, 13–23. [Google Scholar] [CrossRef]
- Hughes, D.; Mikosch, P.; Belmatoug, N.; Carubbi, F.; Cox, T.; Goker-Alpan, O.; Kindmark, A.; Mistry, P.; Poll, L.; Weinreb, N.; et al. Gaucher Disease in Bone: From Pathophysiology to Practice. J. Bone Miner. Res. 2019, 34, 996–1013. [Google Scholar] [CrossRef] [PubMed]
- Baris, H.N.; Cohen, I.J.; Mistry, P.K. Gaucher disease: The metabolic defect, pathophysiology, phenotypes and natural history. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. 1), 72–81. [Google Scholar]
- Mignot, C.; Gelot, A.; De Villemeur, T.B. Gaucher disease. Handb. Clin. Neurol. 2013, 113, 1709–1715. [Google Scholar]
- Sidransky, E. Gaucher disease: Insights from a rare Mendelian disorder. Discov. Med. 2012, 14, 273–281. [Google Scholar]
- Hughes, D.A.; Pastores, G.M. GeneReviews; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Cappellini, M.D.; Motta, I.; Barbato, A.; Giuffrida, G.; Manna, R.; Carubbi, F.; Giona, F. Similarities and differences between Gaucher disease and acid sphingomyelinase deficiency: An algorithm to support the diagnosis. Eur. J. Intern. Med. 2023, 108, 81–84. [Google Scholar] [CrossRef]
- Brianna, M.Q.; Deschenes Nm Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar]
- Wasserstein, M.; Dionisi-Vici, C.; Giugliani, R.; Hwu, W.L.; Lidove, O.; Lukacs, Z.; Mengel, E.; Mistry, P.K.; Schuchman, E.H.; McGovern, M. Recommendations for clinical monitoring of patients with acid sphingomyelinase deficiency (ASMD). Mol. Genet. Metab. 2019, 126, 98–105. [Google Scholar] [CrossRef]
- McGovern, M.M.; Aron, A.; Brodie, S.E.; Desnick, R.J.; Wasserstein, M.P. Natural history of type A Niemann-Pick disease: Possible endpoints for therapeutic trials. Neurology 2006, 66, 228–232. [Google Scholar] [CrossRef] [PubMed]
- McGovern, M.M.; Avetisyan, R.; Sanson, B.J.; Lidove, O. Disease manifestations and burden of illness in patients with acid sphin-gomyelinase deficiency (ASMD). Orphanet J. Rare Dis. 2017, 12, 41. [Google Scholar] [CrossRef]
- McGovern, M.M.; Dionisi-Vici, C.; Giugliani, R.; Hwu, P.; Lidove, O.; Lukacs, Z.; Mengel, E.; Mistry, P.; Schuchman, E.; McGovern, M. Consensus recommendation on a diagnostic guideline for acid sphingomyelinase deficiency. Genet. Med. 2017, 19, 967–974.e16. [Google Scholar] [CrossRef]
- Mistry, P.K.; Cappellini, M.D.; Hayri Ozsan, E.L.; Mach Pascual, S.; Rosenbaum, H.; Solano, M.H.; Spigelman, Z.; Villarrubia, J.; Watman, N.P.; Massenkeil, G. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am. J. Hematol. 2011, 86, 110–115. [Google Scholar] [CrossRef]
- Deodato, F.; Boenzi, S.; Taurisano, R.; Semeraro, M.; Sacchetti, E.; Carrozzo, R.; Dionisi-Vici, C. The impact of biomarkers analysis in the diagnosis of Niemann-Pick C disease and acid sphingomyelinase deficiency. Clin. Chim. Acta. 2018, 486, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, M.; Dionisi Vici, C.; Burlina, A.; Venturelli, F.; Fiumara, A.; Fecarotta, S.; Donati, M.A.; Spada, M.; Concolino, D.; Pession, A. Screening for lysosomal diseases in a selected pediatric population: The case of Gaucher disease and acid sphingomyelinase deficiency. Orphanet J. Rare Dis. 2023, 18, 197. [Google Scholar] [CrossRef]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D.; Casentini, C. Gaucher and Niemann–Pick diseases—Enzymatic diagnosis in dried blood spots on filter paper: Retrospective diagnoses in newborn-screening cards. Clin. Chim. Acta 2002, 317, 191–197. [Google Scholar] [CrossRef]
- Dardis, A.; Michelakakis, H.; Rozenfeld, P.; Fumic, K.; Wagner, J.; Pavan, E.; Fuller, M.; Revel-Vilk, S.; Hughes, D.; Cox, T.; et al. Patient centered guidelines for the laboratory diagnosis of Gaucher disease type 1. Orphanet J. Rare Dis. 2022, 17, 442. [Google Scholar] [CrossRef]
- Tang, C.; Jia, X.; Tang, F.; Liu, S.; Jiang, X.; Zhao, X.; Sheng, H.; Peng, M.; Liu, L.; Huang, Y. Detection of glucosylsphingosine in dried blood spots for diagnosis of Gaucher disease by LC-MS/MS. Clin. Biochem. 2021, 87, 79–84. [Google Scholar] [CrossRef]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. [Google Scholar] [CrossRef]
- Polo, G.; Burlina, A.P.; Ranieri, E.; Colucci, F.; Rubert, L.; Pascarella, A.; Duro, G.; Tummolo, A.; Padoan, A.; Plebani, M.; et al. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: A comparative study. Clin. Chem. Lab. Med. 2019, 57, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef]
- Breilyn, M.S.; Zhang, W.; Yu, C.; Wasserstein, M.P. Plasma lyso-sphingomyelin levels are positively associated with clinical severity in acid sphingomyelinase deficiency. Mol. Genet. Metab. Rep. 2021, 28, 100780. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; Burlina, A.; Pereira, D.; Silva, C.; Herbst, Z.M.; Trapp, F.B.; Michelin-Tirelli, K.; Lopes, F.F.; Burin, M.G.; Brusius-Facchin, A.C.; et al. Quantification of lysosphingomyelin and lysosphingomyelin-509 for the screening of acid sphingomyelinase deficiency. Orphanet J. Rare Dis. 2022, 17, 407. [Google Scholar] [CrossRef]
- Diaz, G.A.; Jones, S.A.; Scarpa, M.; Mengel, K.E.; Giugliani, R.; Guffon, N.; Batsu, I.; Fraser, P.A.; Li, J.; Zhang, Q.; et al. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet. Med. 2022, 24, 2209. [Google Scholar] [CrossRef]
- Simonaro, C.M.; Desnick, R.J.; McGovern, M.M.; Wasserstein, M.P.; Schuchman, E.H. The demographics and distribution of type B Niemann-Pick disease: Novel mutations lead to new genotype/phenotype correlations. Am. J. Hum. Genet. 2002, 71, 1413–1419. [Google Scholar] [CrossRef]
- Ferlinz, K.; Hurwitz, R.; Weiler, M.; Suzuki, K.; Sandhoff, K.; Vanier, M.T. Molecular analysis of the acid sphingomyelinase deficiency in a family with an intermediate form of Niemann-Pick disease. Am. J. Hum. Genet. 1995, 56, 1343–1349. [Google Scholar]
- Irun, P.; Mallén, M.; Dominguez, C.; Rodriguez-Sureda, V.; Alvarez-Sala, L.A.; Arslan, N.; Bermejo, N.; Guerrero, C.; Perez de Soto, I.; Villalón, L.; et al. Identification of seven novel SMPD1 mutations causing Niemann-Pick disease types A and B. Clin. Genet. 2013, 84, 356–361. [Google Scholar] [CrossRef]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Stirnemann, J.; Vigan, M.; Hamroun, D.; Heraoui, D.; Rossi-Semerano, L.; Berger, M.G.; Rose, C.; Camou, F.; de Roux-Serratrice, C.; Grosbois, B.; et al. The French Gaucher’s disease registry: Clinical characteristics, complications and treatment of 562 patients. Orphanet J. Rare Dis. 2012, 7, 77. [Google Scholar] [CrossRef]
- Schuchman, E.H. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J. Inherit. Metab. Dis. 2007, 30, 654–663. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef]
- Motta, I.; Filocamo, M.; Poggiali, E.; Stroppiano, M.; Dragani, A.; Consonni, D.; Barcellini, W.; Gaidano, G.; Facchini, L.; Specchia, G.; et al. A multicentre observational study for early diagnosis of Gaucher disease in patients with Splenomegaly and/or Thrombocytopenia. Eur. J. Haematol. 2016, 96, 352–359. [Google Scholar] [CrossRef]
- Motta, I.; Consonni, D.; Stroppiano, M.; Benedetto, C.; Cassinerio, E.; Tappino, B.; Ranalli, P.; Borin, L.; Facchini, L.; Patriarca, A.; et al. Predicting the probability of Gaucher disease in subjects with splenomegaly and thrombocytopenia. Sci. Rep. 2021, 11, 2594. [Google Scholar] [CrossRef]
- Lei, K.; Zhao, Y.; Sun, L.; Liang, H.; Luo, R.; Sun, X.; Tao, Y.; Chen, L.; Zhang, L.; Li, A.; et al. A pilot screening of high-risk Gaucher disease children using dried blood spot methods in Shandong province of China. Orphanet J. Rare Dis. 2018, 13, 48. [Google Scholar] [CrossRef]
- Huang, Y.; Jia, X.; Tang, C.; Liu, S.; Sheng, H.; Zhao, X.; Zeng, C.; Liu, L. High risk screening for Gaucher disease in patients with splenomegaly and/or thrombocytopenia in China: 55 cases identified. Clin. Chim. Acta 2020, 506, 22–27. [Google Scholar] [CrossRef]
- Russell, S.A.; Sholzberg, M.; Mangel, J.; Keeney, M.; Hedley, B.; Bode, M.; Gob, A.; Lam, S.; Phua, C.; Hsia, C.C. Gaucher disease screening at a general adult hematology tertiary care centre: A prospective study. Int. J. Lab. Hematol. 2019, 41, e66–e69. [Google Scholar] [CrossRef]
- Petra Oliva, P.; Schwarz, M.; Mechtler, T.P.; Sansen, S.; Keutzer, J.; Prusa, A.R.; Streubel, B.; Kasper, D.C. Importance to include differential diagnostics for acid sphingomyelinase deficiency (ASMD) in patients suspected to have to Gaucher disease. Mol. Genet. Metab. 2023, 139, 107563. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, R.B.; Salazar, D.; Tanpaiboon, P. Laboratory diagnostic approaches in metabolic disorders. Ann. Transl. Med. 2018, 6, 470. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Diaz, G.A.; Lachmann, R.H.; Jouvin, M.H.; Nandy, I.; Ji, A.J.; Puga, A.C. Olipudase alfa for treatment of acid sphingomyelinase deficiency (ASMD): Safety and efficacy in adults treated for 30 months. J. Inherit. Metab. Dis. 2018, 41, 829–838. [Google Scholar] [CrossRef]
- Wasserstein, M.; Lachmann, R.; Hollak, C.; Arash-Kaps, L.; Barbato, A.; Gallagher, R.C.; Giugliani, R.; Guelbert, N.B.; Ikezoe, T.; Lidove, O.; et al. A randomized, placebo-controlled clinical trial evaluating olipudase alfa enzyme replacement therapy for chronic acid sphingomyelinase deficiency (ASMD) in adults: One-year results. Genet. Med. 2022, 24, 1425–1436. [Google Scholar] [CrossRef]
- Keam, S.J. Olipudase Alfa: First Approval. Drugs 2022, 82, 941–947. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GBA1 PCR Primers exon 1–intron 5 | 5′-3′ sequence |
| GBA_EX1FOR-PC | CTCCATGCAAATCTGTGTTC |
| GBA_EX5Rev-PC | GGCCTGAAAAAGCTAGAATG |
| GBA1 PCR Primers intron 5–exon 11 | 5′-3′ sequence |
| GBA_EX5FOR-PC | CCAGGATGATTGCGAACTC |
| GBA_EX11Rev-PC | TGCTGTGCCCTCTTTAGTC |
| GBA1 Sequencing Primers exon 1–intron 5 | 5′-3′ sequence |
| Ex1F seq GBA | AGATGAGAGGAAGCCAA |
| ex2R seq GBA | TGGTCTCAGTCACTCAAAAG |
| EX3F seq GBA | TCTTTTGAAACAGAGTCTT |
| EX4R_GBA | CAGAATGGGCAGAGTGAGAT |
| EX5F seq GBA | GGCCTCCCAAAGTGCTGG |
| GBA1 Sequencing Primers intron 5–exon 11 | 5′-3′ sequence |
| Ex6R seq bb | ATTGAGAGGCCCAAGGCT |
| Ex7R seq bb | CCCTAGAAAGGTTTCAAGCGA |
| EX8F_GBA | TCCAGGATCAGTTGCTCTTC |
| EX8R seq bb | AGTAAGAGGTCTGAGGTCTG |
| EX9F seq bb | TCTTACTAGTTTCACCAAAG |
| EX9R seq bb | AAGTTACGCACCCAATTGGG |
| EX9F seq bb2 | CCTTGCCCTGAACCCCGAA |
| SMPD1 PCR Sequencing Primers | 5′-3′ sequence |
| Ex1F SMPD1 | TGAGCGCGGATTCTGACA |
| Ex1R SMPD1 | AGCAAACTCAGTGATGGATT |
| Ex2F SMPD1 | GTTGGCCTGGTTCCTCTGCT |
| Ex2R SMPD1 | GTTCCCTTCTCCCTTCACTT |
| Ex3F SMPD1 | CAGCACAGGAGGACCAGGA |
| Ex4R SMPD1 | CAGCCTTCAGACACTCACCC |
| Ex5F SMPD1 | CATCTCACCATCCCTGTTGT |
| Ex5R SMPD1 | CTCCAACCTCCTTCCCCTAT |
| Ex6F SMPD1 | TCCCTGGAGTTACCCTTGCT |
| Ex6R SMPD1 | CTTGCCCTGCTTGCCTGGAA |
| Overall | Subjects | Male | Female |
|---|---|---|---|
| Subjects (%) | 627 | 367 (58.5) | 260 (41.5) |
| Average age [min–max] | 56.3 [1–90] | 55.4 [1–90] | 57.5 [1–86] |
| Hematology % | 70.8 | 69.1 | 73.5 |
| Internal Medicine % | 17.1 | 17.4 | 16.5 |
| Pediatrics % | 6.8 | 7.6 | 5.8 |
| Other % | 5.3 | 6.0 | 4.2 |
| GD patients (%) | 8 (1.3) | 5 (1.4) | 3 (1.1) |
| Niemann Pick A/B patients (%) | 3 (0.5) | 2 (0.5) | 1 (0.4) |
| Unaffected GD patients (%) | 619 (98.7) | 362 (98.6) | 257 (98.8) |
| Unaffected ASMD patients (%) | 624 (99.5) | 365 (99.4) | 259 (99.6) |
| Unaffected patients (%) | 616 (98.2) | 360 (98.1) | 256 (98.5) |
| Average GCase activity in the probands | 0.9 | 0.8 | 1.0 |
| Average ASM activity in the probands | 1.0 | 0.9 | 1.0 |
| Average GCase activity in unaffected patients | 8.3 | 8.6 | 7.9 |
| Average ASM activity in unaffected patients | 10.1 | 10.0 | 10.3 |
| Patient No. | Gender/Age | ASM Activity | GCase Activity | Lyso-Gb1 | GBA1 Mutations | Gaucher Disease | Splenomegaly | Thrombocytopenia | Anemia | Skeletal Pathology | Pulmonary Involvement | Other Signs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 23 | F/66 | normal | 0.4 | -- | G119R/N370S | Type I | + | + | - | - | - | - |
| 82 | M/53 | normal | 0.8 | -- | N370S/L444P | Type III | - | + | - | - | - | - |
| 199 | M/71 | normal | 0.3 | 346.0 | R131C/N370S | Type I | + | + | - | - | - | - |
| 206 | M/23 | normal | 0.5 | 643.0 | R131C/N370S | Type I | + | + | + | - | - | - |
| 284 | F/43 | normal | 1.7 | 332.0 | N370S/L444P | Type I | + | + | -- | - | - | - |
| 464 | M/77 | normal | 1.5 | 14.6 | R131C/L444P | Type I | + | + | - | - | - | - |
| 565 | M/63 | normal | 1.0 | 82.0 | N370S/N370S | Type I | + | + | -- | -- | -- | -- |
| 627 | F/12 | normal | 1.0 | 218.0 | Q286R/N370S | Type I | + | + | + | + | - | Growth disturbance and leukopenia. |
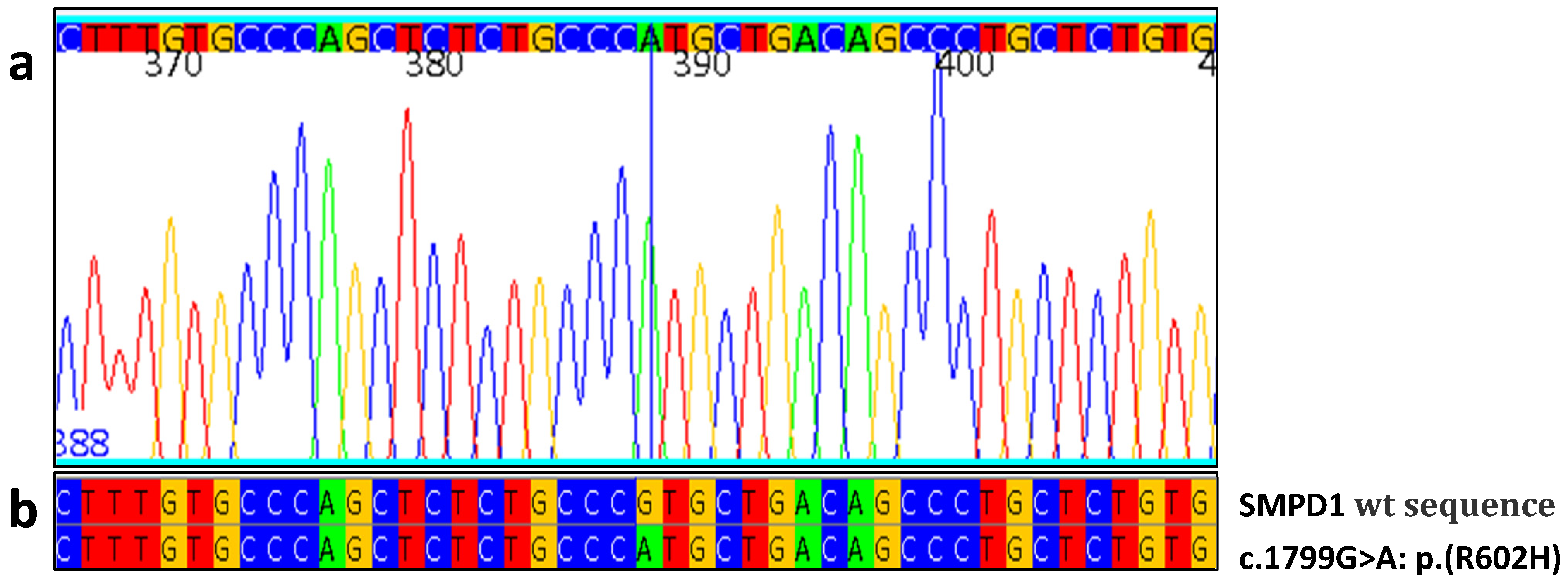
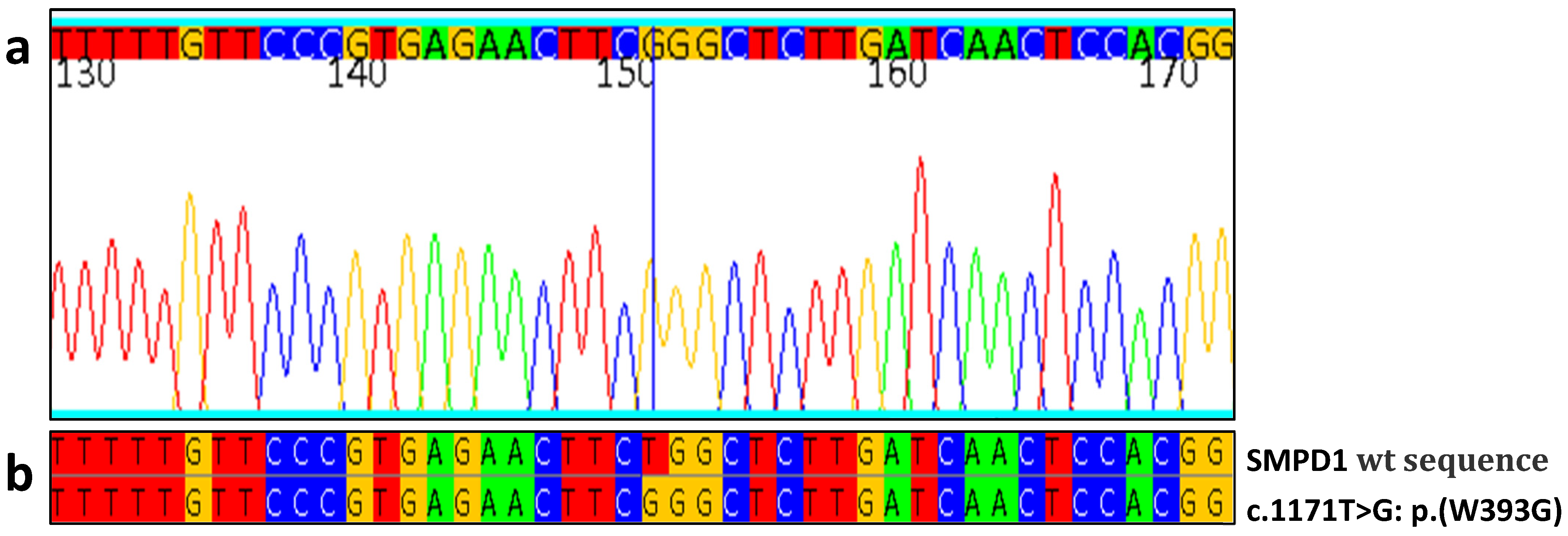
| Patient No | Gender/Age | GCase Activity | ASM Activity | Lyso-SM | Lyso-SM-509 | SMPD1 Mutations | Splenomegaly | Piastrynopenia | Anemia | Skeletal Pathology | Pulmonary Involvement | Other Signs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M/64 | 8,8 | 0.7 | -- | 4.7 | R600H/R600H | + | + | - | - | - | - |
| 11 | M/1 | 16.7 | 1.2 | -- | 4.3 | W393G/W393G | + | + | + | - | + | - |
| 210 | F/51 | 7.2 | 1.0 | 148.0 | 4.0 | c.1829_1831del/c.1829_1831del | + | + | + | - | - | Recurrent ear infections |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacomarra, M.; Colomba, P.; Francofonte, D.; Zora, M.; Caocci, G.; Diomede, D.; Giuffrida, G.; Fiori, L.; Montanari, C.; Sapuppo, A.; et al. Gaucher Disease or Acid Sphingomyelinase Deficiency? The Importance of Differential Diagnosis. J. Clin. Med. 2024, 13, 1487. https://doi.org/10.3390/jcm13051487
Giacomarra M, Colomba P, Francofonte D, Zora M, Caocci G, Diomede D, Giuffrida G, Fiori L, Montanari C, Sapuppo A, et al. Gaucher Disease or Acid Sphingomyelinase Deficiency? The Importance of Differential Diagnosis. Journal of Clinical Medicine. 2024; 13(5):1487. https://doi.org/10.3390/jcm13051487
Chicago/Turabian StyleGiacomarra, Miriam, Paolo Colomba, Daniele Francofonte, Marcomaria Zora, Giovanni Caocci, Daniela Diomede, Gaetano Giuffrida, Laura Fiori, Chiara Montanari, Annamaria Sapuppo, and et al. 2024. "Gaucher Disease or Acid Sphingomyelinase Deficiency? The Importance of Differential Diagnosis" Journal of Clinical Medicine 13, no. 5: 1487. https://doi.org/10.3390/jcm13051487
APA StyleGiacomarra, M., Colomba, P., Francofonte, D., Zora, M., Caocci, G., Diomede, D., Giuffrida, G., Fiori, L., Montanari, C., Sapuppo, A., Scortechini, A. R., Vitturi, N., Duro, G., & Zizzo, C. (2024). Gaucher Disease or Acid Sphingomyelinase Deficiency? The Importance of Differential Diagnosis. Journal of Clinical Medicine, 13(5), 1487. https://doi.org/10.3390/jcm13051487