
The Role of Tau Pathology in Alzheimer’s Disease and Down Syndrome


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
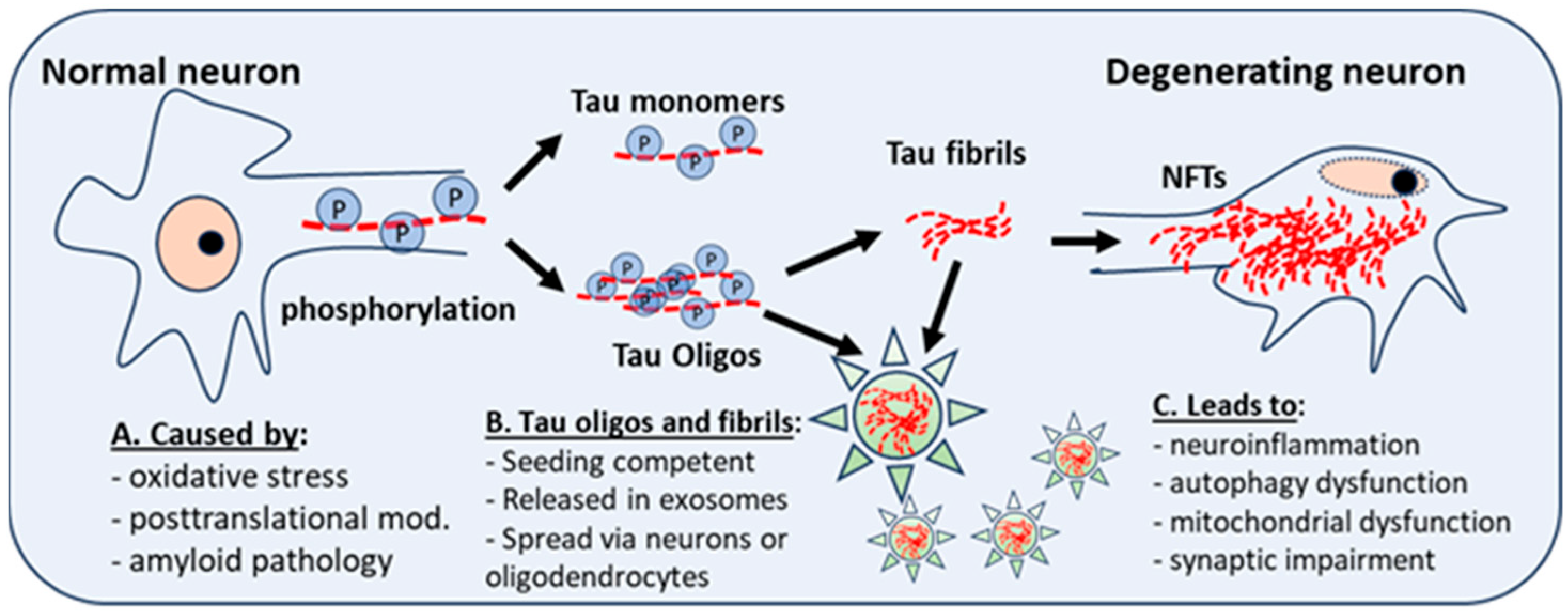
2. Microtubule-Associated Protein Tau (MAPT) Structure and Function
2.1. Posttranslational Modifications of Tau
2.2. The Role of RNA and DNA in Tau Aggregation
2.3. Local Tau Folding Patterns Confer Seed Competence
3. Tauopathy Seeding and Its Spread in the DS Brain
3.1. P-Tau Spreading in Neurotransmitter Systems with AD
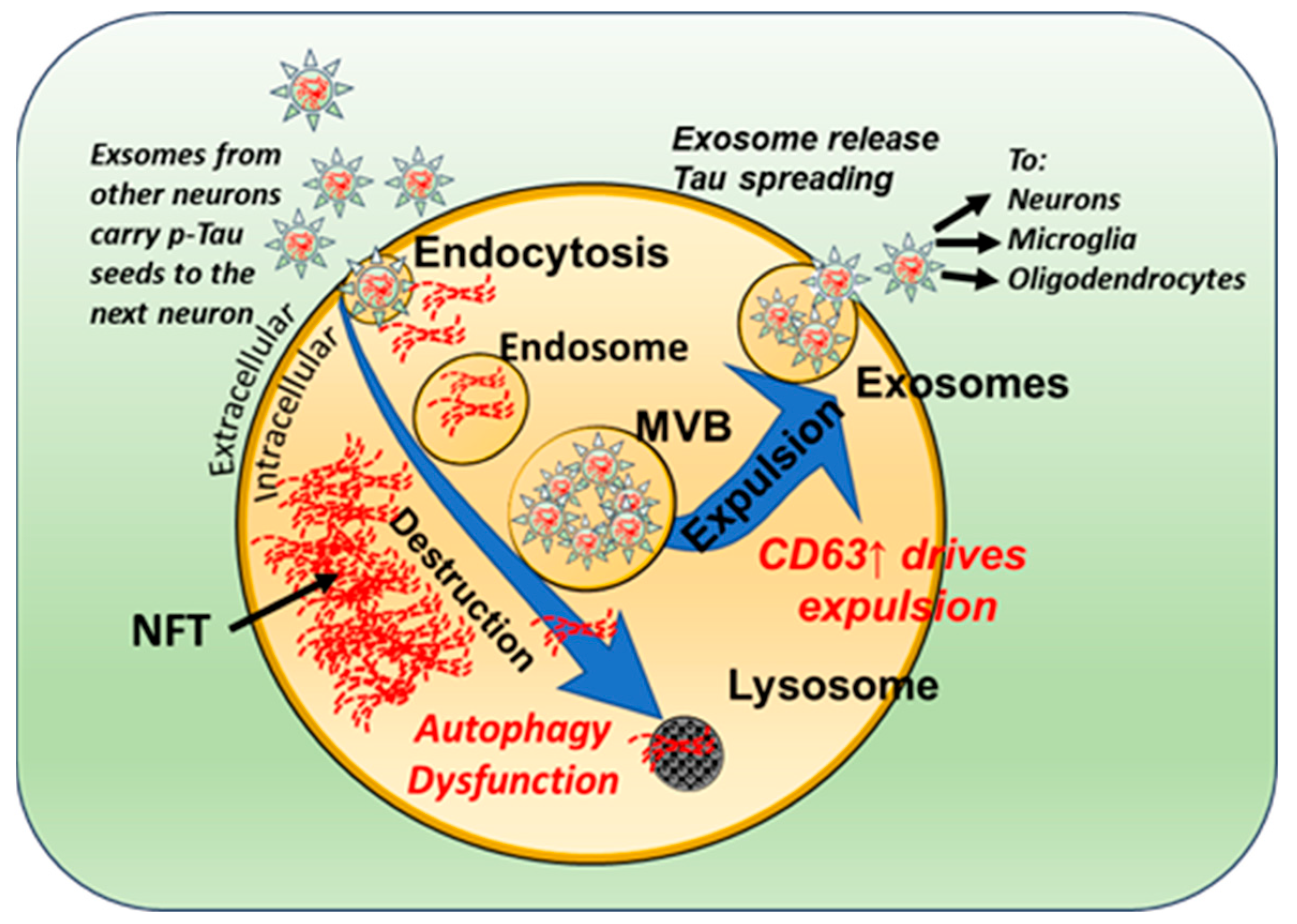
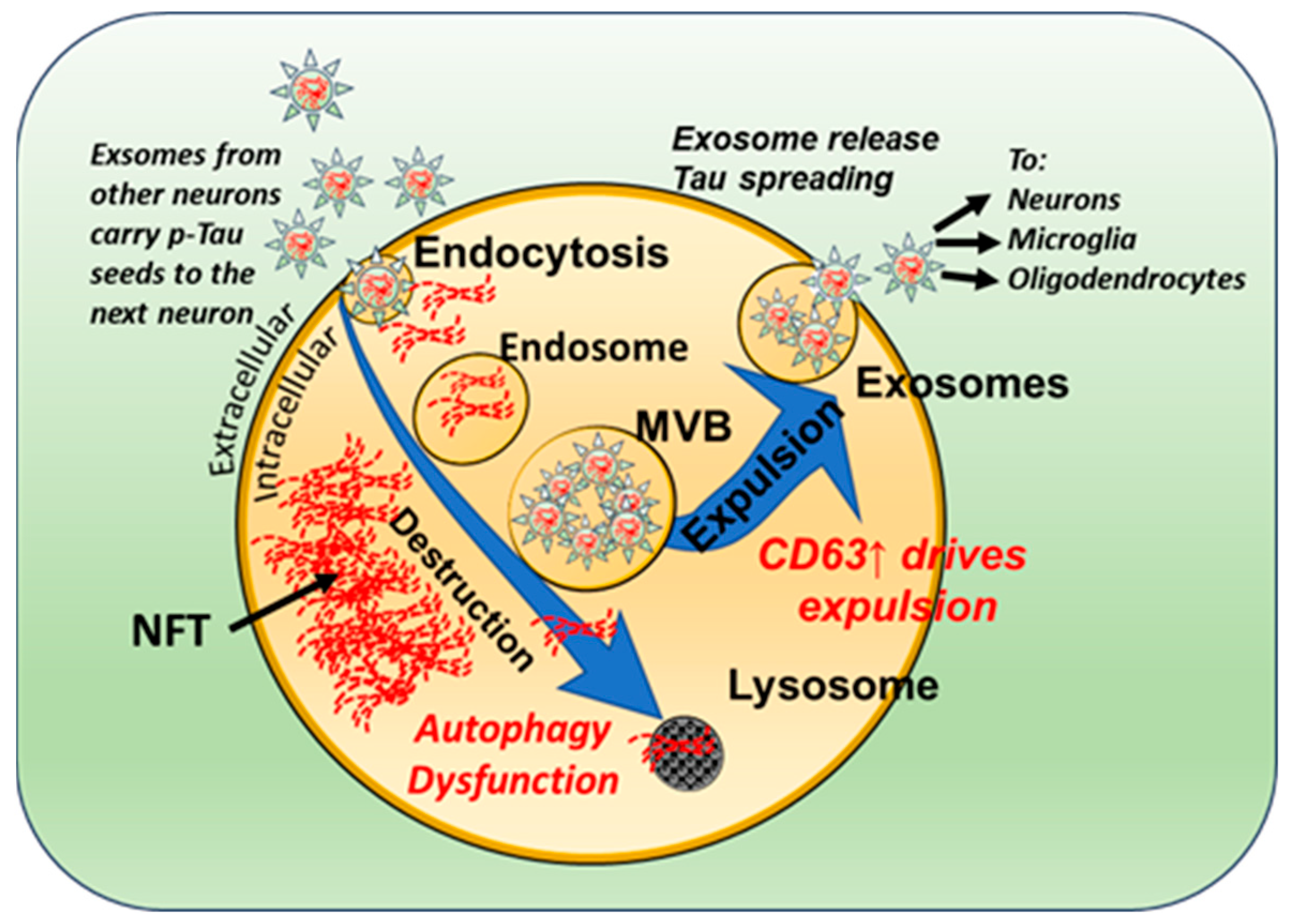
3.2. Neuronal Exosomes Harbor Pathogenic Tau Seeds
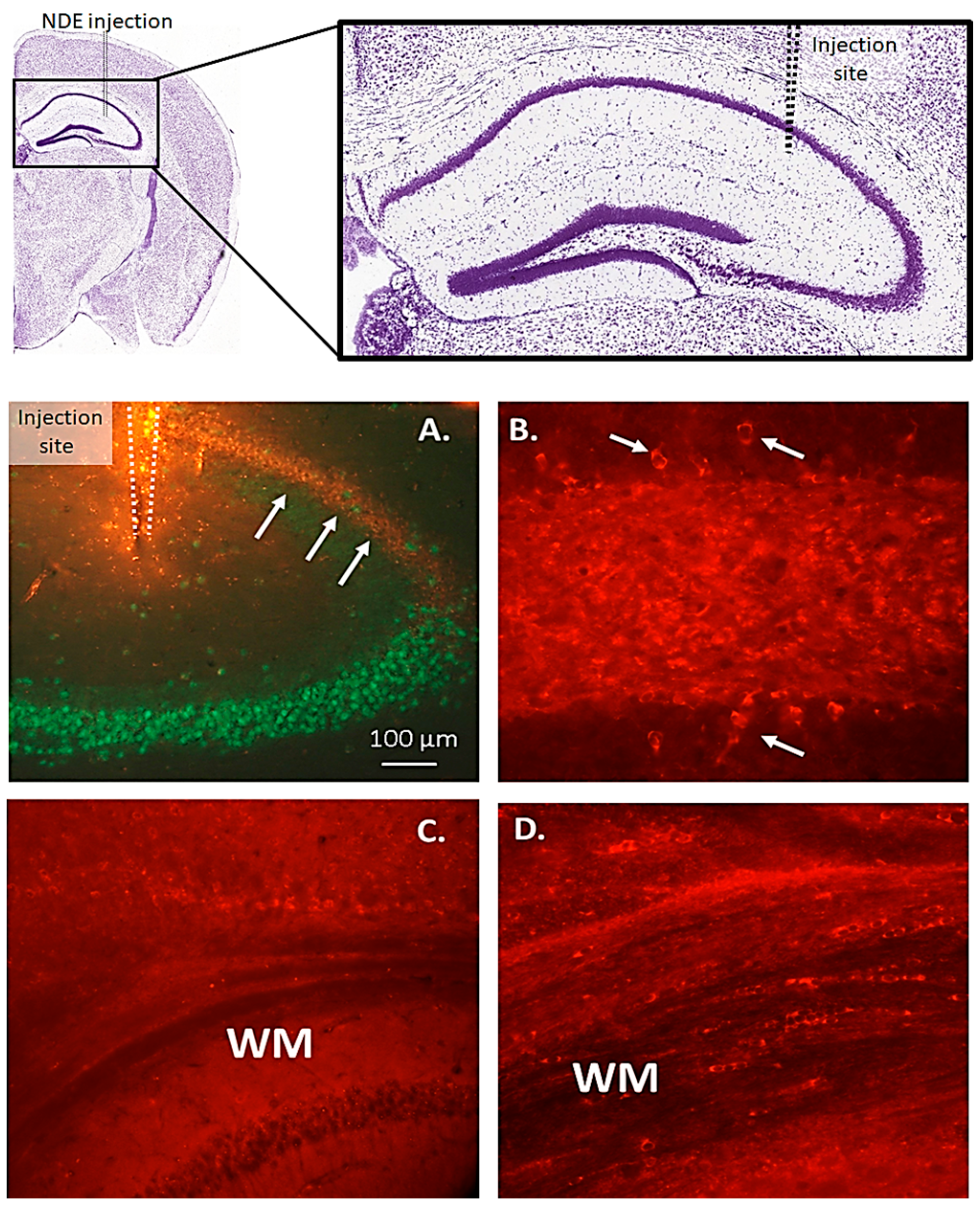
3.3. Tau Seeds Impart a Bystander-Spreading Effect in the Brain
4. Tau Biomarkers in Biofluids
4.1. Ultrasensitive Immunoassays
4.2. Tau Seeding and Aggregation Assays
4.3. Tau Binding Studies Using Positron Emission Tomography (PET) Ligands
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alldred, M.J.; Martini, A.C.; Patterson, D.; Hendrix, J.; Granholm, A.C. Aging with Down Syndrome—Where Are We Now and Where Are We Going? J. Clin. Med. 2021, 10, 4687. [Google Scholar] [CrossRef]
- Delabar, J.M.; Allinquant, B.; Bianchi, D.; Blumenthal, T.; Dekker, A.; Edgin, J.; O’Bryan, J.; Dierssen, M.; Potier, M.C.; Wiseman, F.; et al. Changing Paradigms in Down Syndrome: The First International Conference of the Trisomy 21 Research Society. Mol. Syndromol. 2016, 7, 251–261. [Google Scholar] [CrossRef]
- Head, E.; Lott, I.T.; Wilcock, D.M.; Lemere, C.A. Aging in Down Syndrome and the Development of Alzheimer’s Disease Neuropathology. Curr. Alzheimer Res. 2016, 13, 18–29. [Google Scholar] [CrossRef]
- Flores-Aguilar, L.; Iulita, M.F.; Kovecses, O.; Torres, M.D.; Levi, S.M.; Zhang, Y.; Askenazi, M.; Wisniewski, T.; Busciglio, J.; Cuello, A.C. Evolution of neuroinflammation across the lifespan of individuals with Down Syndrome. Brain 2020, 143, 3653–3671. [Google Scholar] [CrossRef]
- Fortea, J.; Zaman, S.H.; Hartley, S.; Rafii, M.S.; Head, E.; Carmona-Iragui, M. Alzheimer’s disease associated with Down Syndrome: A genetic form of dementia. Lancet Neurol. 2021, 20, 930–942. [Google Scholar] [CrossRef]
- Hartley, D.; Blumenthal, T.; Carrillo, M.; DiPaolo, G.; Esralew, L.; Gardiner, K.; Granholm, A.C.; Iqbal, K.; Krams, M.; Lemere, C.; et al. Down Syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimers Dement. 2015, 11, 700–709. [Google Scholar] [CrossRef]
- Bartley, M.G.; Marquardt, K.; Kirchhof, D.; Wilkins, H.M.; Patterson, D.; Linseman, D.A. Overexpression of amyloid-beta protein precursor induces mitochondrial oxidative stress and activates the intrinsic apoptotic cascade. J. Alzheimers Dis. 2012, 28, 855–868. [Google Scholar] [CrossRef]
- Helman, A.M.; Siever, M.; McCarty, K.L.; Lott, I.T.; Doran, E.; Abner, E.L.; Schmitt, F.A.; Head, E. Microbleeds and Cerebral Amyloid Angiopathy in the Brains of People with Down Syndrome with Alzheimer’s Disease. J. Alzheimers Dis. 2019, 67, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.L.; Isacson, O.; Nelson, M.; Bimonte-Nelson, H.; Seo, H.; Lin, L.; Ford, K.; Kindy, M.S.; Granholm, A.C. Regional alterations in amyloid precursor protein and nerve growth factor across age in a mouse model of Down’s syndrome. Neurosci. Res. 2003, 45, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Millan Sanchez, M.; Heyn, S.N.; Das, D.; Moghadam, S.; Martin, K.J.; Salehi, A. Neurobiological elements of cognitive dysfunction in Down Syndrome: Exploring the role of APP. Biol. Psychiatry 2012, 71, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Green, P.H.R.; Wang, T.C.; Kong, X.F. Interferon-Driven Immune Dysregulation in Down Syndrome: A Review of the Evidence. J. Inflamm. Res. 2021, 14, 5187–5200. [Google Scholar] [CrossRef]
- Powers, R.K.; Culp-Hill, R.; Ludwig, M.P.; Smith, K.P.; Waugh, K.A.; Minter, R.; Tuttle, K.D.; Lewis, H.C.; Rachubinski, A.L.; Granrath, R.E.; et al. Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat. Commun. 2019, 10, 4766. [Google Scholar] [CrossRef]
- Ram, G.; Chinen, J. Infections and immunodeficiency in Down Syndrome. Clin. Exp. Immunol. 2011, 164, 9–16. [Google Scholar] [CrossRef]
- Martini, A.C.; Helman, A.M.; McCarty, K.L.; Lott, I.T.; Doran, E.; Schmitt, F.A.; Head, E. Distribution of microglial phenotypes as a function of age and Alzheimer’s disease neuropathology in the brains of people with Down Syndrome. Alzheimers Dement. 2020, 12, e12113. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Arena, A.; Head, E.; Butterfield, D.A.; Perluigi, M. Disturbance of redox homeostasis in Down Syndrome: Role of iron dysmetabolism. Free Radic. Biol. Med. 2018, 114, 84–93. [Google Scholar] [CrossRef]
- Hendrix, J.A.; Amon, A.; Abbeduto, L.; Agiovlasitis, S.; Alsaied, T.; Anderson, H.A.; Bain, L.J.; Baumer, N.; Bhattacharyya, A.; Bogunovic, D.; et al. Opportunities, barriers, and recommendations in Down Syndrome research. Transl. Sci. Rare Dis. 2021, 5, 99–129. [Google Scholar] [CrossRef] [PubMed]
- Snyder, H.M.; Bain, L.J.; Brickman, A.M.; Carrillo, M.C.; Esbensen, A.J.; Espinosa, J.M.; Fernandez, F.; Fortea, J.; Hartley, S.L.; Head, E.; et al. Further understanding the connection between Alzheimer’s disease and Down Syndrome. Alzheimers Dement. 2020, 16, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Maxwell, A.M.; Castillo, E.; Aoyagi, A.; Graff, C.; Ingelsson, M.; Lannfelt, L.; Bird, T.D.; Keene, C.D.; Seeley, W.W.; et al. Abeta and tau prions feature in the neuropathogenesis of Down Syndrome. Proc. Natl. Acad. Sci. USA 2022, 119, e2212954119. [Google Scholar] [CrossRef]
- Hamlett, E.D.; Goetzl, E.J.; Ledreux, A.; Vasilevko, V.; Boger, H.A.; LaRosa, A.; Clark, D.; Carroll, S.L.; Carmona-Iragui, M.; Fortea, J.; et al. Neuronal exosomes reveal Alzheimer’s disease biomarkers in Down Syndrome. Alzheimers Dement. 2017, 13, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Hamlett, E.D.; LaRosa, A.; Mufson, E.J.; Fortea, J.; Ledreux, A.; Granholm, A.C. Exosome release and cargo in Down Syndrome. Dev. Neurobiol. 2019, 79, 639–655. [Google Scholar] [CrossRef]
- Ledreux, A.; Thomas, S.; Hamlett, E.D.; Trautman, C.; Gilmore, A.; Rickman Hager, E.; Paredes, D.A.; Margittai, M.; Fortea, J.; Granholm, A.C. Small Neuron-Derived Extracellular Vesicles from Individuals with Down Syndrome Propagate Tau Pathology in the Wildtype Mouse Brain. J. Clin. Med. 2021, 10, 3931. [Google Scholar] [CrossRef]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 2841. [Google Scholar] [CrossRef]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Meyer, V.; Dinkel, P.D.; Luo, Y.; Yu, X.; Wei, G.; Zheng, J.; Eaton, G.R.; Ma, B.; Nussinov, R.; Eaton, S.S.; et al. Single mutations in tau modulate the populations of fibril conformers through seed selection. Angew. Chem. Int. Ed. Engl. 2014, 53, 1590–1593. [Google Scholar] [CrossRef] [PubMed]
- Siddiqua, A.; Luo, Y.; Meyer, V.; Swanson, M.A.; Yu, X.; Wei, G.; Zheng, J.; Eaton, G.R.; Ma, B.; Nussinov, R.; et al. Conformational basis for asymmetric seeding barrier in filaments of three- and four-repeat tau. J. Am. Chem. Soc. 2012, 134, 10271–10278. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Maury, E.A.; Kirk, M.J.; Saqran, L.; Roe, A.; DeVos, S.L.; Nicholls, S.; Fan, Z.; Takeda, S.; Cagsal-Getkin, O.; et al. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 2015, 34, 3028–3041. [Google Scholar] [CrossRef]
- Weismiller, H.A.; Murphy, R.; Wei, G.; Ma, B.; Nussinov, R.; Margittai, M. Structural disorder in four-repeat Tau fibrils reveals a new mechanism for barriers to cross-seeding of Tau isoforms. J. Biol. Chem. 2018, 293, 17336–17348. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020, 26, 1256–1263. [Google Scholar] [CrossRef]
- Armstrong, R.A.; McKee, A.C.; Alvarez, V.E.; Cairns, N.J. Clustering of tau-immunoreactive pathology in chronic traumatic encephalopathy. J. Neural. Transm. 2017, 124, 185–192. [Google Scholar] [CrossRef]
- Capano, L.S.; Sato, C.; Ficulle, E.; Yu, A.; Horie, K.; Kwon, J.S.; Burbach, K.F.; Barthelemy, N.R.; Fox, S.G.; Karch, C.M.; et al. Recapitulation of endogenous 4R tau expression and formation of insoluble tau in directly reprogrammed human neurons. Cell Stem Cell 2022, 29, 918–932.e8. [Google Scholar] [CrossRef]
- Furman, J.L.; Vaquer-Alicea, J.; White, C.L., 3rd; Cairns, N.J.; Nelson, P.T.; Diamond, M.I. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. 2017, 133, 91–100. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Escudero, V.; Ruiz-Gabarre, D.; Gargini, R.; Perez, M.; Garcia, E.; Cuadros, R.; Hernandez, I.H.; Cabrera, J.R.; Garcia-Escudero, R.; Lucas, J.J.; et al. A new non-aggregative splicing isoform of human Tau is decreased in Alzheimer’s disease. Acta Neuropathol. 2021, 142, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Nothias, F.; Boyne, L.; Murray, M.; Tessler, A.; Fischer, I. The expression and distribution of tau proteins and messenger RNA in rat dorsal root ganglion neurons during development and regeneration. Neuroscience 1995, 66, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Nunez, J.; Fischer, I. Microtubule-associated proteins (MAPs) in the peripheral nervous system during development and regeneration. J. Mol. Neurosci. 1997, 8, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2020, 11, 595532. [Google Scholar] [CrossRef]
- Trushina, N.I.; Bakota, L.; Mulkidjanian, A.Y.; Brandt, R. The Evolution of Tau Phosphorylation and Interactions. Front. Aging Neurosci. 2019, 11, 256. [Google Scholar] [CrossRef]
- Ryoo, S.R.; Jeong, H.K.; Radnaabazar, C.; Yoo, J.J.; Cho, H.J.; Lee, H.W.; Kim, I.S.; Cheon, Y.H.; Ahn, Y.S.; Chung, S.H.; et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down Syndrome and Alzheimer disease. J. Biol. Chem. 2007, 282, 34850–34857. [Google Scholar] [CrossRef]
- Neumann, F.; Gourdain, S.; Albac, C.; Dekker, A.D.; Bui, L.C.; Dairou, J.; Schmitz-Afonso, I.; Hue, N.; Rodrigues-Lima, F.; Delabar, J.M.; et al. DYRK1A inhibition and cognitive rescue in a Down Syndrome mouse model are induced by new fluoro-DANDY derivatives. Sci. Rep. 2018, 8, 2859. [Google Scholar] [CrossRef]
- Laham, A.J.; Saber-Ayad, M.; El-Awady, R. DYRK1A: A Down Syndrome-related dual protein kinase with a versatile role in tumorigenesis. Cell Mol. Life Sci. 2021, 78, 603–619. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C.X. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down Syndrome. FASEB J. 2008, 22, 3224–3233. [Google Scholar] [CrossRef] [PubMed]
- Feki, A.; Hibaoui, Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, K.S.; Kim, A.K.; Choi, M.; Choi, K.; Kang, M.; Chi, S.W.; Lee, M.S.; Lee, J.S.; Lee, S.Y.; et al. A chemical with proven clinical safety rescues Down-syndrome-related phenotypes in through DYRK1A inhibition. Dis. Models Mech. 2016, 9, 839–848. [Google Scholar] [CrossRef]
- Melchior, B.; Mittapalli, G.K.; Lai, C.; Duong-Polk, K.; Stewart, J.; Guner, B.; Hofilena, B.; Tjitro, A.; Anderson, S.D.; Herman, D.S.; et al. Tau pathology reduction with SM07883, a novel, potent, and selective oral DYRK1A inhibitor: A potential therapeutic for Alzheimer’s disease. Aging Cell 2019, 18, e13000. [Google Scholar] [CrossRef]
- Zhu, B.; Parsons, T.; Foley, C.; Shaw, Y.; Dunckley, T.; Hulme, C.; Hodge, J.J.L. DYRK1A antagonists rescue degeneration and behavioural deficits of in vivo models based on amyloid-beta, Tau and DYRK1A neurotoxicity. Sci. Rep. 2022, 12, 15847. [Google Scholar] [CrossRef] [PubMed]
- Lester, E.; Ooi, F.K.; Bakkar, N.; Ayers, J.; Woerman, A.L.; Wheeler, J.; Bowser, R.; Carlson, G.A.; Prusiner, S.B.; Parker, R. Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron 2021, 109, 1675–1691.e1679. [Google Scholar] [CrossRef] [PubMed]
- Shmookler Reis, R.J.; Atluri, R.; Balasubramaniam, M.; Johnson, J.; Ganne, A.; Ayyadevara, S. “Protein aggregates” contain RNA and DNA, entrapped by misfolded proteins but largely rescued by slowing translational elongation. Aging Cell 2021, 20, e13326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, Y.; Eschmann, N.A.; Zhou, H.; Rauch, J.N.; Hernandez, I.; Guzman, E.; Kosik, K.S.; Han, S. RNA stores tau reversibly in complex coacervates. PLoS Biol. 2017, 15, e2002183. [Google Scholar] [CrossRef]
- Dinkel, P.D.; Holden, M.R.; Matin, N.; Margittai, M. RNA Binds to Tau Fibrils and Sustains Template-Assisted Growth. Biochemistry 2015, 54, 4731–4740. [Google Scholar] [CrossRef]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Guo, C.; Yalamanchili, H.K.; Abreha, M.; Al-Ouran, R.; Li, Y.; Dammer, E.B.; Lah, J.J.; Levey, A.I.; Bennett, D.A.; et al. Tau-Mediated Disruption of the Spliceosome Triggers Cryptic RNA Splicing and Neurodegeneration in Alzheimer’s Disease. Cell Rep. 2019, 29, 301–316.e310. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, C.G.; Mehrabian, M.; Wang, X.; Mueller, I.; Lubambo, I.B.; Jonkman, J.E.; Wang, H.; Schmitt-Ulms, G. The Human Tau Interactome: Binding to the Ribonucleoproteome, and Impaired Binding of the Proline-to-Leucine Mutant at Position 301 (P301L) to Chaperones and the Proteasome. Mol. Cell Proteom. 2015, 14, 3000–3014. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Crino, P.B.; Lee, V.M.; Eberwine, J.H.; Trojanowski, J.Q. Sequestration of RNA in Alzheimer’s disease neurofibrillary tangles and senile plaques. Ann. Neurol. 1997, 41, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Zwierzchowski-Zarate, A.N.; Mendoza-Oliva, A.; Kashmer, O.M.; Collazo-Lopez, J.E.; White, C.L., 3rd; Diamond, M.I. RNA induces unique tau strains and stabilizes Alzheimer’s disease seeds. J. Biol. Chem. 2022, 298, 102132. [Google Scholar] [CrossRef] [PubMed]
- Antcliff, A.; McCullough, L.D.; Tsvetkov, A.S. G-Quadruplexes and the DNA/RNA helicase DHX36 in health, disease, and aging. Aging 2021, 13, 25578–25587. [Google Scholar] [CrossRef] [PubMed]
- Vijay Kumar, M.J.; Morales, R.; Tsvetkov, A.S. G-quadruplexes and associated proteins in aging and Alzheimer’s disease. Front. Aging 2023, 4, 1164057. [Google Scholar] [CrossRef]
- Novikova, G.; Andrews, S.J.; Renton, A.E.; Marcora, E. Beyond association: Successes and challenges in linking non-coding genetic variation to functional consequences that modulate Alzheimer’s disease risk. Mol. Neurodegener. 2021, 16, 27. [Google Scholar] [CrossRef]
- Sivagurunathan, N.; Ambatt, A.T.S.; Calivarathan, L. Role of Long Non-coding RNAs in the Pathogenesis of Alzheimer’s and Parkinson’s Diseases. Curr. Aging Sci. 2022, 15, 84–96. [Google Scholar] [CrossRef]
- Canseco-Rodriguez, A.; Masola, V.; Aliperti, V.; Meseguer-Beltran, M.; Donizetti, A.; Sanchez-Perez, A.M. Long Non-Coding RNAs, Extracellular Vesicles and Inflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 3171. [Google Scholar] [CrossRef]
- Olufunmilayo, E.O.; Holsinger, R.M.D. Roles of Non-Coding RNA in Alzheimer’s Disease Pathophysiology. Int. J. Mol. Sci. 2023, 24, 2498. [Google Scholar] [CrossRef]
- Lan, Z.; Chen, Y.; Jin, J.; Xu, Y.; Zhu, X. Long Non-coding RNA: Insight Into Mechanisms of Alzheimer’s Disease. Front. Mol. Neurosci. 2021, 14, 821002. [Google Scholar] [CrossRef]
- Sabaie, H.; Amirinejad, N.; Asadi, M.R.; Jalaiei, A.; Daneshmandpour, Y.; Rezaei, O.; Taheri, M.; Rezazadeh, M. Molecular Insight Into the Therapeutic Potential of Long Non-coding RNA-Associated Competing Endogenous RNA Axes in Alzheimer’s Disease: A Systematic Scoping Review. Front. Aging Neurosci. 2021, 13, 742242. [Google Scholar] [CrossRef]
- Wang, E.; Thombre, R.; Shah, Y.; Latanich, R.; Wang, J. G-Quadruplexes as pathogenic drivers in neurodegenerative disorders. Nucleic Acids Res. 2021, 49, 4816–4830. [Google Scholar] [CrossRef]
- Begeman, A.; Son, A.; Litberg, T.J.; Wroblewski, T.H.; Gehring, T.; Huizar Cabral, V.; Bourne, J.; Xuan, Z.; Horowitz, S. G-Quadruplexes act as sequence-dependent protein chaperones. EMBO Rep. 2020, 21, e49735. [Google Scholar] [CrossRef] [PubMed]
- Kallweit, L.; Hamlett, E.D.; Saternos, H.; Gilmore, A.; Granholm, A.C.; Horowitz, S. A New Role for RNA G-quadruplexes in Aging and Alzheimer’s Disease. bioRxiv 2023. [Google Scholar] [CrossRef]
- Asamitsu, S.; Imai, Y.; Yabuki, Y.; Ikenoshita, S.; Takeuchi, M.; Kashiwagi, H.; Tanoue, Y.; Fukuda, T.; Shioda, N. Identification and immunohistochemical characterization of G-quadruplexes in mouse brain. Biochem. Biophys. Res. Commun. 2020, 531, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sierra, F.; Mondragon-Rodriguez, S.; Basurto-Islas, G. Truncation of tau protein and its pathological significance in Alzheimer’s disease. J. Alzheimers Dis. 2008, 14, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.X.; Iqbal, K.; Liu, F. Truncation of Tau selectively facilitates its pathological activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef]
- Hamano, T.; Enomoto, S.; Shirafuji, N.; Ikawa, M.; Yamamura, O.; Yen, S.H.; Nakamoto, Y. Autophagy and Tau Protein. Int. J. Mol. Sci. 2021, 22, 7475. [Google Scholar] [CrossRef] [PubMed]
- Mirbaha, H.; Chen, D.; Morazova, O.A.; Ruff, K.M.; Sharma, A.M.; Liu, X.; Goodarzi, M.; Pappu, R.V.; Colby, D.W.; Mirzaei, H.; et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 2018, 7, e36584. [Google Scholar] [CrossRef]
- Mirbaha, H.; Chen, D.; Mullapudi, V.; Terpack, S.J.; White, C.L., 3rd; Joachimiak, L.A.; Diamond, M.I. Seed-competent tau monomer initiates pathology in a tauopathy mouse model. J. Biol. Chem. 2022, 298, 102163. [Google Scholar] [CrossRef]
- Liou, Y.C.; Sun, A.; Ryo, A.; Zhou, X.Z.; Yu, Z.X.; Huang, H.K.; Uchida, T.; Bronson, R.; Bing, G.; Li, X.; et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 2003, 424, 556–561. [Google Scholar] [CrossRef]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef]
- Kondo, A.; Shahpasand, K.; Mannix, R.; Qiu, J.; Moncaster, J.; Chen, C.H.; Yao, Y.; Lin, Y.M.; Driver, J.A.; Sun, Y.; et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015, 523, 431–436. [Google Scholar] [CrossRef]
- Kenney, K.; Iacono, D.; Edlow, B.L.; Katz, D.I.; Diaz-Arrastia, R.; Dams-O’Connor, K.; Daneshvar, D.H.; Stevens, A.; Moreau, A.L.; Tirrell, L.S.; et al. Dementia After Moderate-Severe Traumatic Brain Injury: Coexistence of Multiple Proteinopathies. J. Neuropathol. Exp. Neurol. 2018, 77, 50–63. [Google Scholar] [CrossRef]
- Condello, C.; Merz, G.E.; Aoyagi, A.; DeGrado, W.F.; Prusiner, S.B. Abeta and Tau Prions Causing Alzheimer’s Disease. Methods Mol. Biol. 2023, 2561, 293–337. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, P.D.; Siddiqua, A.; Huynh, H.; Shah, M.; Margittai, M. Variations in filament conformation dictate seeding barrier beween three- and four-repeat tau. Biochemistry 2011, 50, 4330–4336. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.E.; Sengupta, U.; Kayed, R. Tau Oligomers as Pathogenic Seeds: Preparation and Propagation In Vitro and In Vivo. Methods Mol. Biol. 2017, 1523, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Gratuze, M.; Chen, Y.; Parhizkar, S.; Jain, N.; Strickland, M.R.; Serrano, J.R.; Colonna, M.; Ulrich, J.D.; Holtzman, D.M. Activated microglia mitigate Abeta-associated tau seeding and spreading. J. Exp. Med. 2021, 218, e20210542. [Google Scholar] [CrossRef]
- Mate De Gerando, A.; Welikovitch, L.A.; Khasnavis, A.; Commins, C.; Glynn, C.; Chun, J.E.; Perbet, R.; Hyman, B.T. Tau seeding and spreading in vivo is supported by both AD-derived fibrillar and oligomeric tau. Acta Neuropathol. 2023, 146, 191–210. [Google Scholar] [CrossRef]
- Das, D.; Phillips, C.; Hsieh, W.; Sumanth, K.; Dang, V.; Salehi, A. Neurotransmitter-based strategies for the treatment of cognitive dysfunction in Down Syndrome. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 54, 140–148. [Google Scholar] [CrossRef]
- Ponnusamy, R.; McNerney, M.W.; Moghadam, S.; Salehi, A. Assessing disease-modifying effects of norepinephrine in Down Syndrome and Alzheimer’s disease. Brain Res. 2019, 1702, 3–11. [Google Scholar] [CrossRef]
- Granholm, A.C.; Sanders, L.A.; Crnic, L.S. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down’s syndrome. Exp. Neurol. 2000, 161, 647–663. [Google Scholar] [CrossRef]
- Iulita, M.F.; Do Carmo, S.; Ower, A.K.; Fortress, A.M.; Flores Aguilar, L.; Hanna, M.; Wisniewski, T.; Granholm, A.C.; Buhusi, M.; Busciglio, J.; et al. Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain 2014, 137, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L.; Zammit, M.D.; West, N.R.; Christian, B.T.; Bhattacharyya, A. Basal Forebrain Cholinergic Neurons: Linking Down Syndrome and Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 703876. [Google Scholar] [CrossRef] [PubMed]
- Sendera, T.J.; Ma, S.Y.; Jaffar, S.; Kozlowski, P.B.; Kordower, J.H.; Mawal, Y.; Saragovi, H.U.; Mufson, E.J. Reduction in TrkA-immunoreactive neurons is not associated with an overexpression of galaninergic fibers within the nucleus basalis in Down’s syndrome. J. Neurochem. 2000, 74, 1185–1196. [Google Scholar] [CrossRef]
- Ball, M.J.; Nuttall, K. Neurofibrillary tangles, granulovacuolar degeneration, and neuron loss in Down Syndrome: Quantitative comparison with Alzheimer dementia. Ann. Neurol. 1980, 7, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.M.; van Scheppingen, J.; Milenkovic, I.; Anink, J.J.; Adle-Biassette, H.; Kovacs, G.G.; Aronica, E. mTOR Hyperactivation in Down Syndrome hippocampus appears early during development. J. Neuropathol. Exp. Neurol. 2014, 73, 671–683. [Google Scholar] [CrossRef]
- Milenkovic, I.; Stojanovic, T.; Aronica, E.; Fulop, L.; Bozso, Z.; Mate, Z.; Yanagawa, Y.; Adle-Biassette, H.; Lubec, G.; Szabo, G.; et al. GABAA receptor subunit deregulation in the hippocampus of human foetuses with Down Syndrome. Brain Struct. Funct. 2018, 223, 1501–1518. [Google Scholar] [CrossRef]
- Wegiel, J.; Kuchna, I.; Wisniewski, T.; de Leon, M.J.; Reisberg, B.; Pirttila, T.; Kivimaki, T.; Lehtimaki, T. Vascular fibrosis and calcification in the hippocampus in aging, Alzheimer disease, and Down Syndrome. Acta Neuropathol. 2002, 103, 333–343. [Google Scholar] [CrossRef]
- Yu, T.; Liu, C.; Belichenko, P.; Clapcote, S.J.; Li, S.; Pao, A.; Kleschevnikov, A.; Bechard, A.R.; Asrar, S.; Chen, R.; et al. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res. 2010, 1366, 162–171. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J. Prev. Alzheimers Dis. 2019, 6, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.E.; Miguel, J.C.; He, B.; Malek-Ahmadi, M.; Abrahamson, E.E.; Ikonomovic, M.D.; Lott, I.; Doran, E.; Alldred, M.J.; Ginsberg, S.D.; et al. Frontal cortex and striatal cellular and molecular pathobiology in individuals with Down Syndrome with and without dementia. Acta Neuropathol. 2019, 137, 413–436. [Google Scholar] [CrossRef] [PubMed]
- Joe, E.; Ringman, J.M. Cognitive symptoms of Alzheimer’s disease: Clinical management and prevention. BMJ 2019, 367, l6217. [Google Scholar] [CrossRef]
- Martorana, A.; Esposito, Z.; Koch, G. Beyond the cholinergic hypothesis: Do current drugs work in Alzheimer’s disease? CNS Neurosci. Ther. 2010, 16, 235–245. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Eady, N.; Sheehan, R.; Rantell, K.; Sinai, A.; Bernal, J.; Bohnen, I.; Bonell, S.; Courtenay, K.; Dodd, K.; Gazizova, D.; et al. Impact of cholinesterase inhibitors or memantine on survival in adults with Down Syndrome and dementia: Clinical cohort study. Br. J. Psychiatry 2018, 212, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Vana, L.; Kanaan, N.M.; Ugwu, I.C.; Wuu, J.; Mufson, E.J.; Binder, L.I. Progression of tau pathology in cholinergic Basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. Am. J. Pathol. 2011, 179, 2533–2550. [Google Scholar] [CrossRef]
- Phillips, C.; Fahimi, A.; Das, D.; Mojabi, F.S.; Ponnusamy, R.; Salehi, A. Noradrenergic System in Down Syndrome and Alzheimer’s Disease A Target for Therapy. Curr. Alzheimer Res. 2016, 13, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Chalermpalanupap, T.; Schroeder, J.P.; Rorabaugh, J.M.; Liles, L.C.; Lah, J.J.; Levey, A.I.; Weinshenker, D. Locus Coeruleus Ablation Exacerbates Cognitive Deficits, Neuropathology, and Lethality in P301S Tau Transgenic Mice. J. Neurosci. 2018, 38, 74–92. [Google Scholar] [CrossRef] [PubMed]
- Chalermpalanupap, T.; Weinshenker, D.; Rorabaugh, J.M. Down but Not Out: The Consequences of Pretangle Tau in the Locus Coeruleus. Neural Plast. 2017, 2017, 7829507. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, D. Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci. 2018, 41, 211–223. [Google Scholar] [CrossRef]
- Samuels, E.R.; Szabadi, E. Functional neuroanatomy of the noradrenergic locus coeruleus: Its roles in the regulation of arousal and autonomic function part I: Principles of functional organisation. Curr. Neuropharmacol. 2008, 6, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [Google Scholar] [CrossRef]
- Reilly, P.; Winston, C.N.; Baron, K.R.; Trejo, M.; Rockenstein, E.M.; Akers, J.C.; Kfoury, N.; Diamond, M.; Masliah, E.; Rissman, R.A.; et al. Novel human neuronal tau model exhibiting neurofibrillary tangles and transcellular propagation. Neurobiol. Dis. 2017, 106, 222–234. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e601. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016, 30, 3853–3859. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef]
- Meyer, V.; Dinkel, P.D.; Rickman Hager, E.; Margittai, M. Amplification of Tau fibrils from minute quantities of seeds. Biochemistry 2014, 53, 5804–5809. [Google Scholar] [CrossRef] [PubMed]
- Guix, F.X.; Corbett, G.T.; Cha, D.J.; Mustapic, M.; Liu, W.; Mengel, D.; Chen, Z.; Aikawa, E.; Young-Pearse, T.; Kapogiannis, D.; et al. Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2018, 19, 663. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Gotz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef] [PubMed]
- Ugbode, C.; Fort-Aznar, L.; Sweeney, S.T. Leaky endosomes push tau over the seed limit. J. Biol. Chem. 2019, 294, 18967–18968. [Google Scholar] [CrossRef]
- Yan, M.; Zheng, T. Role of the endolysosomal pathway and exosome release in tau propagation. Neurochem. Int. 2021, 145, 104988. [Google Scholar] [CrossRef]
- Gauthier, S.A.; Perez-Gonzalez, R.; Sharma, A.; Huang, F.K.; Alldred, M.J.; Pawlik, M.; Kaur, G.; Ginsberg, S.D.; Neubert, T.A.; Levy, E. Enhanced exosome secretion in Down Syndrome brain—A protective mechanism to alleviate neuronal endosomal abnormalities. Acta Neuropathol. Commun. 2017, 5, 65. [Google Scholar] [CrossRef]
- Koychev, I.; Jansen, K.; Dette, A.; Shi, L.; Holling, H. Blood-Based ATN Biomarkers of Alzheimer’s Disease: A Meta-Analysis. J. Alzheimers Dis. 2021, 79, 177–195. [Google Scholar] [CrossRef]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. 2016, 3, 63–72. [Google Scholar] [CrossRef]
- Winston, C.N.; Aulston, B.; Rockenstein, E.M.; Adame, A.; Prikhodko, O.; Dave, K.N.; Mishra, P.; Rissman, R.A.; Yuan, S.H. Neuronal Exosome-Derived Human Tau is Toxic to Recipient Mouse Neurons in vivo. J. Alzheimers Dis. 2018, 67, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Aguilo Garcia, M.; Carmona, M.; Andres-Benito, P.; Torrejon-Escribano, B.; Garcia-Esparcia, P.; Del Rio, J.A. Involvement of Oligodendrocytes in Tau Seeding and Spreading in Tauopathies. Front. Aging Neurosci. 2019, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Darricau, M.; Dou, C.; Kinet, R.; Zhu, T.; Zhou, L.; Li, X.; Bedel, A.; Claverol, S.; Tokarski, C.; Katsinelos, T.; et al. Tau seeds from Alzheimer’s disease brains trigger tau spread in macaques while oligomeric-Aβ mediates pathology maturation. Alzheimers Dement. 2024, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bassil, F.; Brown, H.J.; Pattabhiraman, S.; Iwasyk, J.E.; Maghames, C.M.; Meymand, E.S.; Cox, T.O.; Riddle, D.M.; Zhang, B.; Trojanowski, J.Q.; et al. Amyloid-Beta (Abeta) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Abeta Pathology. Neuron 2020, 105, 260–275.e266. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Zelaya, M.V.; Aguilo Garcia, M.; Carmona, M.; Lopez-Gonzalez, I.; Andres-Benito, P.; Lidon, L.; Gavin, R.; Garcia-Esparcia, P.; Del Rio, J.A. Relevance of host tau in tau seeding and spreading in tauopathies. Brain Pathol. 2020, 30, 298–318. [Google Scholar] [CrossRef]
- Salehi, A.; Ashford, J.W.; Mufson, E.J. The Link between Alzheimer’s Disease and Down Syndrome. A Historical Perspective. Curr. Alzheimer Res. 2016, 13, 2–6. [Google Scholar] [CrossRef]
- McCrea, M.; Broglio, S.P.; McAllister, T.W.; Gill, J.; Giza, C.C.; Huber, D.L.; Harezlak, J.; Cameron, K.L.; Houston, M.N.; McGinty, G.; et al. Association of Blood Biomarkers With Acute Sport-Related Concussion in Collegiate Athletes: Findings From the NCAA and Department of Defense CARE Consortium. JAMA Netw. Open 2020, 3, e1919771. [Google Scholar] [CrossRef]
- Petersen, M.E.; Rafii, M.S.; Zhang, F.; Hall, J.; Julovich, D.; Ances, B.M.; Schupf, N.; Krinsky-McHale, S.J.; Mapstone, M.; Silverman, W.; et al. Plasma Total-Tau and Neurofilament Light Chain as Diagnostic Biomarkers of Alzheimer’s Disease Dementia and Mild Cognitive Impairment in Adults with Down Syndrome. J. Alzheimers Dis. 2021, 79, 671–681. [Google Scholar] [CrossRef]
- Carmona-Iragui, M.; Alcolea, D.; Barroeta, I.; Videla, L.; Munoz, L.; Van Pelt, K.L.; Schmitt, F.A.; Lightner, D.D.; Koehl, L.M.; Jicha, G.; et al. Diagnostic and prognostic performance and longitudinal changes in plasma neurofilament light chain concentrations in adults with Down Syndrome: A cohort study. Lancet Neurol. 2021, 20, 605–614. [Google Scholar] [CrossRef]
- Janelidze, S.; Christian, B.T.; Price, J.; Laymon, C.; Schupf, N.; Klunk, W.E.; Lott, I.; Silverman, W.; Rosas, H.D.; Zaman, S.; et al. Detection of Brain Tau Pathology in Down Syndrome Using Plasma Biomarkers. JAMA Neurol. 2022, 79, 797–807. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Hamlett, E.D.; Ledreux, A.; Potter, H.; Chial, H.J.; Patterson, D.; Espinosa, J.M.; Bettcher, B.M.; Granholm, A.C. Exosomal biomarkers in Down Syndrome and Alzheimer’s disease. Free Radic. Biol. Med. 2018, 114, 110–121. [Google Scholar] [CrossRef]
- Kocisko, D.A.; Come, J.H.; Priola, S.A.; Chesebro, B.; Raymond, G.J.; Lansbury, P.T.; Caughey, B. Cell-free formation of protease-resistant prion protein. Nature 1994, 370, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Saijo, E.; Metrick, M.A., 2nd; Newell, K.; Sigurdson, C.J.; Zanusso, G.; Ghetti, B.; Caughey, B. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol. 2019, 137, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, R.; Jin, N.; Chu, D.; Gu, J.; Tung, Y.C.; Hu, Z.; Gong, C.X.; Iqbal, K. Two simple assays for assessing the seeding activity of proteopathic tau. Front. Aging Neurosci. 2023, 15, 1073774. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Gu, J.; Wu, R.; Chu, D.; Tung, Y.C.; Wegiel, J.; Wisniewski, T.; Gong, C.X.; Iqbal, K.; Liu, F. Tau seeding activity in various regions of Down Syndrome brain assessed by two novel assays. Acta Neuropathol. Commun. 2022, 10, 132. [Google Scholar] [CrossRef]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef]
- Rafii, M.S.; Lukic, A.S.; Andrews, R.D.; Brewer, J.; Rissman, R.A.; Strother, S.C.; Wernick, M.N.; Pennington, C.; Mobley, W.C.; Ness, S.; et al. PET Imaging of Tau Pathology and Relationship to Amyloid, Longitudinal MRI, and Cognitive Change in Down Syndrome: Results from the Down Syndrome Biomarker Initiative (DSBI). J. Alzheimers Dis. 2017, 60, 439–450. [Google Scholar] [CrossRef]
- Lao, P.J.; Handen, B.L.; Betthauser, T.J.; Mihaila, I.; Hartley, S.L.; Cohen, A.D.; Tudorascu, D.L.; Bulova, P.D.; Lopresti, B.J.; Tumuluru, R.V.; et al. Longitudinal changes in amyloid positron emission tomography and volumetric magnetic resonance imaging in the nondemented Down Syndrome population. Alzheimers Dement. 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Zammit, M.D.; Tudorascu, D.L.; Laymon, C.M.; Hartley, S.L.; Zaman, S.H.; Ances, B.M.; Johnson, S.C.; Stone, C.K.; Mathis, C.A.; Klunk, W.E.; et al. PET measurement of longitudinal amyloid load identifies the earliest stages of amyloid-beta accumulation during Alzheimer’s disease progression in Down Syndrome. Neuroimage 2021, 228, 117728. [Google Scholar] [CrossRef]
- Zammit, M.D.; Betthauser, T.J.; McVea, A.K.; Laymon, C.M.; Tudorascu, D.L.; Johnson, S.C.; Hartley, S.L.; Converse, A.K.; Minhas, D.S.; Zaman, S.H.; et al. Characterizing the emergence of amyloid and tau burden in Down Syndrome. Alzheimers Dement. 2024, 20, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278; discussion 278–284. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, I.; Jarc, J.; Dassler, E.; Aronica, E.; Iyer, A.; Adle-Biassette, H.; Scharrer, A.; Reischer, T.; Hainfellner, J.A.; Kovacs, G.G. The physiological phosphorylation of tau is critically changed in fetal brains of individuals with Down Syndrome. Neuropathol. Appl. Neurobiol. 2018, 44, 314–327. [Google Scholar] [CrossRef]
- Lemoine, L.; Ledreux, A.; Mufson, E.J.; Perez, S.E.; Simic, G.; Doran, E.; Lott, I.; Carroll, S.; Bharani, K.; Thomas, S.; et al. Regional binding of tau and amyloid PET tracers in Down Syndrome autopsy brain tissue. Mol. Neurodegener. 2020, 15, 68. [Google Scholar] [CrossRef] [PubMed]
- Murugan, N.A.; Chiotis, K.; Rodriguez-Vieitez, E.; Lemoine, L.; Agren, H.; Nordberg, A. Cross-interaction of tau PET tracers with monoamine oxidase B: Evidence from in silico modelling and in vivo imaging. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, K.M.; Yang, L.; Dong, Q.; Yu, J.T. Tauopathies: New perspectives and challenges. Mol. Neurodegener. 2022, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Albayram, O.; Kondo, A.; Mannix, R.; Smith, C.; Tsai, C.Y.; Li, C.; Herbert, M.K.; Qiu, J.; Monuteaux, M.; Driver, J.; et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat. Commun. 2017, 8, 1000. [Google Scholar] [CrossRef]
- Canete-Masse, C.; Carbo-Carrete, M.; Pero-Cebollero, M.; Cui, S.X.; Yan, C.G.; Guardia-Olmos, J. Abnormal degree centrality and functional connectivity in Down Syndrome: A resting-state fMRI study. Int. J. Clin. Health Psychol. 2023, 23, 100341. [Google Scholar] [CrossRef]
- Hamadelseed, O.; Chan, M.K.S.; Wong, M.B.F.; Skutella, T. Distinct neuroanatomical and neuropsychological features of Down Syndrome compared to related neurodevelopmental disorders: A systematic review. Front. Neurosci. 2023, 17, 1225228. [Google Scholar] [CrossRef]
- Utagawa, E.C.; Moreno, D.G.; Schafernak, K.T.; Arva, N.C.; Malek-Ahmadi, M.H.; Mufson, E.J.; Perez, S.E. Neurogenesis and neuronal differentiation in the postnatal frontal cortex in Down Syndrome. Acta Neuropathol. Commun. 2022, 10, 86. [Google Scholar] [CrossRef]
- Biswas, S.; Kalil, K. The Microtubule-Associated Protein Tau Mediates the Organization of Microtubules and Their Dynamic Exploration of Actin-Rich Lamellipodia and Filopodia of Cortical Growth Cones. J. Neurosci. 2018, 38, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, M.; Hassan, B.A. Amyloid precursor protein and neural development. Development 2014, 141, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Octave, J.N.; Pierrot, N.; Ferao Santos, S.; Nalivaeva, N.N.; Turner, A.J. From synaptic spines to nuclear signaling: Nuclear and synaptic actions of the amyloid precursor protein. J. Neurochem. 2013, 126, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Waheed, Z.; Choudhary, J.; Jatala, F.H.; Fatimah; Noor, A.; Zerr, I.; Zafar, S. The Role of Tau Proteoforms in Health and Disease. Mol. Neurobiol. 2023, 60, 5155–5166. [Google Scholar] [CrossRef]
- Song, C.; Shi, J.; Zhang, P.; Zhang, Y.; Xu, J.; Zhao, L.; Zhang, R.; Wang, H.; Chen, H. Immunotherapy for Alzheimer’s disease: Targeting beta-amyloid and beyond. Transl. Neurodegener. 2022, 11, 18. [Google Scholar] [CrossRef]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Rafii, M.S.; Zaman, S.; Handen, B.L. Integrating Biomarker Outcomes into Clinical Trials for Alzheimer’s Disease in Down Syndrome. J. Prev. Alzheimers Dis. 2021, 8, 48–51. [Google Scholar] [CrossRef]
- Aldecoa, I.; Barroeta, I.; Carroll, S.L.; Fortea, J.; Gilmore, A.; Ginsberg, S.D.; Guzman, S.J.; Hamlett, E.D.; Head, E.; Perez, S.E.; et al. Down Syndrome Biobank Consortium: A perspective. Alzheimers Dement. 2024, 1–11. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granholm, A.-C.; Hamlett, E.D. The Role of Tau Pathology in Alzheimer’s Disease and Down Syndrome. J. Clin. Med. 2024, 13, 1338. https://doi.org/10.3390/jcm13051338
Granholm A-C, Hamlett ED. The Role of Tau Pathology in Alzheimer’s Disease and Down Syndrome. Journal of Clinical Medicine. 2024; 13(5):1338. https://doi.org/10.3390/jcm13051338
Chicago/Turabian StyleGranholm, Ann-Charlotte, and Eric D. Hamlett. 2024. "The Role of Tau Pathology in Alzheimer’s Disease and Down Syndrome" Journal of Clinical Medicine 13, no. 5: 1338. https://doi.org/10.3390/jcm13051338
APA StyleGranholm, A.-C., & Hamlett, E. D. (2024). The Role of Tau Pathology in Alzheimer’s Disease and Down Syndrome. Journal of Clinical Medicine, 13(5), 1338. https://doi.org/10.3390/jcm13051338