Abstract
Objective: To assess the frequency and types of genetic mutations in patients with arrhythmias who underwent cardiac device implantation. Methods: Retrospective observational study, including 38 patients with different arrhythmias and cardiac arrest as a first cardiac event. Treatment modalities encompass pacemakers, transvenous defibrillators, loop recorders, subcutaneous defibrillators, and cardiac resynchronization therapy. All patients underwent genetic testing, using commercially available panels (106–174 genes). Outcome measures include mortality, arrhythmia recurrence, and device-related complications. Results: Clinical parameters revealed a family history of sudden cardiac death in 19 patients (50%), who were predominantly male (58%) and had a mean age of 44.5 years and a mean left ventricle ejection fraction of 40.3%. Genetic testing identified mutations in various genes, predominantly TMEM43 (11%). In two patients (3%) with arrhythmogenic cardiomyopathy, complete subcutaneous defibrillator extraction with de novo transvenous implantable cardioverter-defibrillator implantation was needed. The absence of multiple associations among severe gene mutations was crucial for cardiac resynchronization therapy response. Mortality in this group was around 3% in titin dilated cardiomyopathy patients. Conclusions: Integration of genetic testing into the decision-making process for patients with electronic devices represents a paradigm shift in personalized medicine. By identifying genetic markers associated with arrhythmia susceptibility, heart failure etiology, and cardiac resynchronization therapy response, clinicians can tailor device choices to optimize patient outcomes.
1. Introduction
Arrhythmias and cardiac arrest pose significant challenges in clinical management, requiring various cardiac devices and ablation procedures for treatment [1,2,3,4,5]. Understanding the genetics and clinical factors influencing outcomes is crucial for personalized treatment strategies [6]. Genetic testing has emerged as a valuable tool, providing insights into mutations associated with channelopathies and cardiomyopathies [6,7].
Sudden cardiac death (SCD) represents a devastating outcome, often occurring unexpectedly and leaving a profound impact on affected individuals and their families. While SCD can stem from various cardiac pathologies, channelopathies and cardiomyopathies emerge as significant contributors to this tragic event. Channelopathies encompass a group of genetic disorders characterized by abnormal ion channel function in cardiac cells, leading to arrhythmias and, in severe cases, to SCD. Similarly, cardiomyopathies entail structural abnormalities of the heart muscle, impairing its contractile function and elevating the risk of lethal arrhythmias.
It is estimated that there are about 7000 single-gene inherited disorders. Many genes responsible for hereditary cardiomyopathies, such as dilated cardiomyopathy (DCM, OMIM #604145), hypertrophic cardiomyopathy (HCM, OMIM #192600), and arrhythmogenic cardiomyopathy (ACM, OMIM #604400), as well as hereditary arrhythmias such as long QT syndromes (LQTS, OMIM #192500), Brugada syndrome (BrS, OMIM #601144), cardiac conduction defects (CCD, OMIM #115080), and catecholaminergic polymorphic ventricular tachycardia (CPVT OMIM #604772), have been identified.
Congenital LQTS is characterized by a prolonged QT interval on the baseline ECG, usually associated with T-wave abnormalities. Long QT syndrome genes can be classified into three main groups: pathogenic variants that reduce potassium outward currents, pathogenic variants that increase sodium inward currents, and pathogenic variants that increase calcium inward currents. Pathogenic variants related to potassium channels account for the vast majority of LQTS cases, with KCNQ1 and KCNH2 [8,9] being responsible for 80% of all genetically explained LQTS cases. Currently, the genes with definitive evidence include KCNQ1, KCNH2, SCN5A, CALM1, CALM2, and CALM3. The genes with moderate evidence are CACNA1C and KCNE1, and testing may be considered in patients with a high probability of diagnosis [10,11].
Brugada syndrome (BrS) is a hereditary disorder characterized by ST-segment elevation in the right precordial leads and malignant ventricular arrhythmias. This syndrome may account for approximately 18–28% of unexplained sudden cardiac arrests. Rare genetic variants in the SCN5A gene, leading to the loss of function of the cardiac sodium channel, are found in about 20% of the cases [12,13].
Cardiac conduction disease (CCD) is often age-dependent and is a heterogeneous progressive cardiac conduction disease (PCCD) disorder marked by impaired electrical impulse propagation in the sinoatrial node, atrioventricular (AV) node, and His–Purkinje system. On the surface ECG, sinus bradycardia, sinus pauses, prolonged P-wave duration, AV block, and different degrees of bundle branch block are typical features. The genes involved in CCD are SCN5A [14,15] and TRPM4 [16].
Hypertrophic cardiomyopathy (HCM) is a relatively common hereditary disorder marked by hypertrophy of the left ventricular wall that cannot be attributed to other conditions such as hypertension or valvular heart disease. Typically, the hypertrophy is asymmetric and mainly affects the intraventricular septum. Gene panels that are generally recommended include eight sarcomere genes, including MYH7, MYBPC3, TNNI3, TNNT2, TPM1, MYL2, MYL3, and ACTC1 [17]. This panel typically identifies a disease-causing variant in about 60% of familial cases [18].
Dilated cardiomyopathy (DCM) is characterized by the presence of left ventricular or biventricular dilatation and systolic dysfunction. It encompasses a wide range of genetic or acquired disorders. Approximately 100 genes have been identified as potentially related to DCM, with truncating variants in the titin gene (TTN) being the most common in DCM, accounting for up to 20% of cases [19]. Genetic testing panels should include the most prevalent genes such as TTN and TNNT2, as well as genes with prognostic or therapeutic implications, such as LMNA, FLNC, and DSP, or other genes such as NEXN, ACTC1, and ACTN2. The selection of genetic testing panels can be guided by the presence of specific extracardiac phenotypes, such as neuromuscular diseases (e.g., DMD and EMD), mitochondrial disease (e.g., NDUFB3), and congenital syndromes [20].
Arrhythmogenic cardiomyopathy (ACM) is mainly characterized by the replacement of myocardial tissue with fibrous or fibrofatty tissue, which can lead to progressive global or regional ventricular dysfunction with a high burden of ventricular arrhythmias. The recommended genetic test for ACM must include a minimal set of genes that have shown a clinical association with the disease. These genes, by frequency, include PKP2 (20–45%), DSP (2–15%), DSG2 (4–15%), DSC2 (2–7%), FLNC (3%), JUP (1%), TMEM43 (1%), PLN (1%), and DES (1–2%) [21]. Initial studies suggested that the RYR2 gene is part of the genetic basis of ACM. Apart from ACM, RYR2 mutations have been linked to CCD, DCM, and CPVT.
The identification of individuals at high risk of SCD due to channelopathies and cardiomyopathies poses a considerable clinical challenge. However, advances in genetic testing have revolutionized risk stratification, enabling healthcare providers to pinpoint underlying genetic mutations predisposing individuals to these.
Genetic testing in descendants of SCD victims plays a pivotal role in unraveling the basis of inherited cardiac conditions. The heritability of certain cardiac disorders, such as long QT syndrome, Brugada syndrome, and hypertrophic or arrhythmogenic cardiomyopathy, underscore the importance of identifying at-risk individuals within affected families [7,22].
The genetic make-up of individuals can influence their response to cardiac device therapy. Despite the potential benefits, incorporating genetic testing into routine clinical practice for patients with electronic cardiac devices has some challenges. Issues such as cost, accessibility, and the interpretation of genetic variants need to be addressed. Collaborative efforts between cardiologists, electrophysiologists, and genetic counsellors are essential to overcome this and integrate genetic testing into cardiac care.
2. Materials and Methods
- Study Design: Retrospective observational study.
- Inclusion criteria: (I) Patients diagnosed with arrhythmias as a first cardiac event (e.g., ventricular tachycardia, ventricular fibrillation, and 3rd-degree atrioventricular block) who are being treated with cardiac devices, such as pacemakers (PMKs), internal cardioverter-defibrillators (T-ICDs), subcutaneous internal cardioverter-defibrillators (S-ICDs), and undergoing genetic screening; (II) Patients with syncope of unknown cause, who are being treated with loop recorders and undergoing genetic screening; (III) Patients with cardiac resynchronization therapy (CRT) indications, heart failure (HF) belonging to New York Heart Association (NYHA) class II–IV, left ventricular ejection fraction (LVEF) ≤ 35%, QRS complex ≥ 130 ms, left bundle branch block (LBBB) pattern, and optimal pharmacological treatment 3 months prior to CRT, who are undergoing genetic testing.
- Exclusion criteria: patients with incomplete medical records or missing genetic data.
- Data collection:
- 4.1
- Patient demographics: age, gender, and family history of SCD.
- 4.2
- Clinical characteristics: symptoms, type of arrhythmias, and history of cardiac arrest.
- 4.3
- Cardiac imaging: (1) Echocardiographic measurements in all patients (valvular regurgitation and ejection fraction (EF)) and (2) Cardiac Magnetic Resonance Imaging (MRI) if available: ejection fraction, fibrosis, or scar.
- 4.4
- Interventions: type of cardiac device (PMK, ICD, S-ICD, CRT, or loop recorder) and type of ablation if it was performed.
- 4.5
- Genetic testing used next-generation sequencing panels. The testing focused on channelopathies and cardiomyopathies, used commercially available panels, ranged from 106–174 genes, and were chosen at the discretion of the attending physician.
Depending on availability and local collaboration protocols, certain patients had their genetic testing conducted at the Regional Center of Medical Genetics Dolj (CRGM Dolj). This was performed using the TruSightCardio panel Illumina (San Diego, CA, USA), which included 174 genes. Genomic DNA was isolated from the primary sample using commercial kits (Wizard® Genomic DNA Purification Kit, PureLink™ Genomic DNA, QIAsymphony DSP DNA). Preparation of sequencing libraries was performed according to the manufacturer’s recommendations. The protocol was based on enzymatic fragmentation and selective amplification of target areas. The library obtained was sequenced on the Illumina platform with the aim of obtaining coverage of at least 50× (depth) for germline variants, and at least 98% (coverage) of the targeted areas. Bioinformatics analysis was performed using a bioinformatics solution implemented locally within CRGM Dolj. Variants with convincing evidence of variant/gene–phenotype correlation according to established international databases (OMIM and Clin Var) were especially evaluated. All known modes of transmission for the presumed diseases were taken into account. Mendelian and variants with insufficient criteria for diagnosis were excluded from the report (e.g., heterozygous variants in genes known to be recessive). Bioinformatics tools such as GEMENI, (iGenomes GATK GRCh37 variants nf-core/sarek v2.7.1; Nextflow v21.04.1; BWA 0.7.17; GATK v4.1.7.0) as well as tapes/annovar, could be used for filtering and prioritization. WAS followed the AMCG variant classification, which reflected the probability that a variant is pathogenic in the following sequence: Benign, Likely Benign, Variant with Uncertain Significance (VUS—variant of unknown significance), Likely Pathogenic, and Pathogenic. Only those variants that correlate with the clinical phenotype were reported.
Some patients underwent genetic testing at the Genomic Center of the University of Medicine and Pharmacy Victor Babes Timisoara, utilizing a 174-gene sequencing panel, the TruSightCardio panel from Illumina (San Diego, CA, USA). Target enrichment was conducted with the TruSight Rapid Capture kit (Illumina). Sequence reads were aligned to the human reference genome, hg37, using the Burrows–Wheeler alignment (BWA) tool. Variants identified were annotated using ANNOVAR, as described in previous publications [23].
For some patients, NGS gene panels were utilized at Invitae laboratories (USA) to test for sequence and exon-level copy number variants, as previously described [24,25]. The prescribing physician selected one or more panels among the commercially available NGS panels at their discretion. The basic commercial panel included 106 genes, while larger panels, comprising up to 168 genes, covered a broader set of genes for the Invitae Arrhythmia and Cardiomyopathy Comprehensive Panel, Add-on Preliminary evidence Genes for Arrhythmia and Cardiomyopathy, and Add-on Sudden Unexpected Death in Epilepsy (SUDEP) Genes. Familial screening was offered to relatives of a proband with pathogenic and likely pathogenic variants. The patient and physician chose among the three laboratories based on different turnaround times and reimbursement considerations. The outcomes were device efficacy and device-related complications, arrhythmia recurrence, and mortality.
Statistical Analysis
Data are presented as mean ± standard deviation for continuous variables and as proportions for categorical variables. Continuous variables were compared between groups using an unpaired T-test (variables with normal distribution) or Chi-square test. A p-value < 0.05 was considered significant.
All the subjects included in the study gave their informed consent before inclusion. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of our institute (number 46/28.09.2018).
3. Results
Thirty-eight patients, 44.5 ± 13.1 y.o. (58% males), were included. All patients received an electronic cardiac device in a tertiary center between 2018–2023. The most frequent presentation that led to the diagnosis was ventricular tachycardia in 34% of the cases, followed by cardiac arrest in 26% of the cases. Demographic, clinical, and echocardiographic parameters are found in Table 1.

Table 1.
Demographic, clinical, and echocardiographic parameters. COPD—chronic obstructive pulmonary disease, SD—standard deviation, MR—mitral regurgitation, TR—tricuspid regurgitation.
Twenty patients (53%) underwent an MRI scan, and in 70% of cases, extensive fibrosis was identified during the scan. The baseline medication for all the patients is presented in Table 2.
Table 2.
Cardiological medication taken by the patients included in the study; NOAC—non-vitamin K antagonist oral anticoagulant, SGLT2—sodium-glucose co-transporter 2 inhibitors, ACEI—angiotensin-converting enzyme inhibitor, ARNI—angiotensin receptor neprylisin inhibitor.
In accordance with the cardiac pathology being the first cardiac manifestation, patients were implanted with an electronic device, as follows: six dual-chamber PMKs, 10 single-chamber ICDs, five dual-chamber ICDs, three subcutaneous ICDs, four loop recorders, 10 CRT,3 CRT-Pacemakers (CRT-P), and seven CRT-D defibrillators (CRT-D).
Nineteen patients (50%) had a family history of SCD. All the patients were genetically tested, focusing on channelopathies and cardiomyopathies and using commercially available panels, which ranged from 106–174 genes. The results are displayed in graph no. 1. Testing identified pathogenic (P) or likely pathogenic (LP) variants in 27 patients (71%), variants of unknown significance (VUS) in seven patients (18%), and negative results in four patients (11%) (Table 3).
Table 3.
Genetic testing in our group of patients.
According to the recommendations of the American College of Medical Genetics and Genomics (ACMG) [26], the patients included in our study were divided into two groups. The first group included 27 patients with positive genetic tests (pathogenic and likely pathogenic), and the second group included 11 patients either with negative genetic tests or with a variant of unknown significance. We considered TTN and LMNA as a separate group (14.81% of patients). The following genes were included in the sarcomeric motor genes group: MYBPC3, CTNNA3, MYH7, TNNI3K, and MYLK. This functional gene group recorded a positive genetic test in 22.22% of patients (Figure 1A). Regarding desmosomal genes, these included DSP, DSC2, and PKP2. This functional gene group recorded a positive test in 14.81% of the patients. Another functional gene group was represented by ion channel genes, which included the following genes: KCNQ1, RYR2, SCN5A, TRPM4, and CACNB2. This gene group registered positive genetic tests in 22.22% of the patients. In addition, genes from the cytoskeleton-Z-disk gene structural group included DMD, with a positive genetic test in only one patient (3.70%). Patients with mutations in the remaining genes that were screened were categorized into an “other genes” group. These genes include SGCD, TMEM43, FBN1, A2ML1, SOS1, AGL, NDUFB3, and EMD, and were registered in 22.22% of the patients with positive genetic tests. Regarding the patients with negative genetic tests or with a variant of unknown significance (only 11 patients out of a total of 38 patients), the following genes were identified: CACNB2 from the ion channel genes group, MYLK from the motor sarcomeric genes group, and A2ML1, SOS1, AGL, and NDUFB3 from the other genes group (Figure 1B).
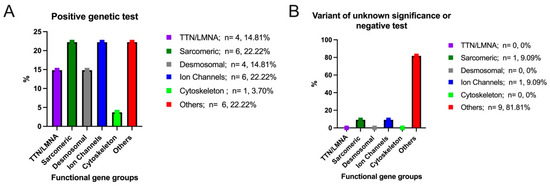
Figure 1.
Distribution of positive genetic test (A)/variant of unknown significance or negative test (B) in the overall study cohort.
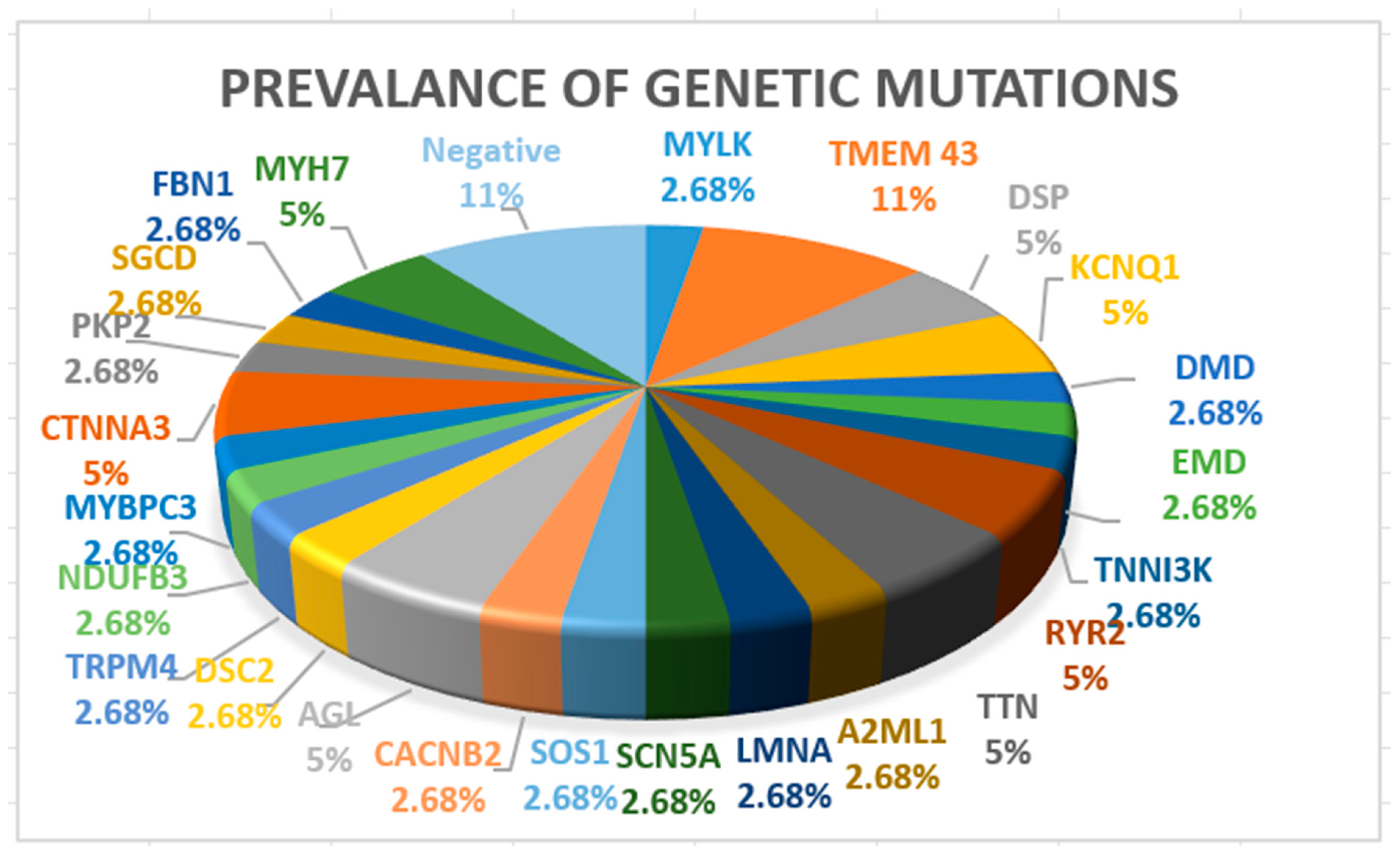
Seven patients (18%) had ACM, two patients (5%) had HCM, two had muscular dystrophies (one with Becker and one with Emery Dreifuss tip 1), three patients (8%) had channelopathies (two patients with LQTS type 1 and one patient with BrS syndrome), one patient (3%) had mitochondrial disease, four patients (11%) had CCD, and the rest had DCM. A more detailed image of genetic testing results is presented in Figure 2.

Figure 2.
Prevalence of genetic mutations.
TMEM43 mutations (11%) were the most prevalent due to being a disease-causing variant of arrhythmogenic cardiomyopathy. In four patients (11%), genetic testing was negative, two patients had idiopathic ventricular fibrillation, one patient had ACM, and one patient had DCM. In these patients, genetic retesting will be considered using an extensive cardiology panel.
The average follow-up was 4.7 ± 1.8 years, and the longest follow-up was 6 years. During follow-up, five patients (13%) needed ventricular tachycardia ablation, two patients (5%) underwent atrial fibrillation ablation, and one patient (3%) underwent ganglion denervation.
In ten patients (26%) with DCM, CRT was performed (seven CRT-D, three CRT-P). The assessment of responses to CRT was based on the following criteria [27,28]:
- -
- Clinical response to CRT, defined as improvement in NYHA functional class.
- -
- Echocardiographic response (defined as >5% increase in LVEF and decreased mitral regurgitation degree).
Patients were divided into two groups: super-responders (SRs) and non-SRs (responders and hyporesponders). SR patients were defined as those with a stable ejection fraction (LVEF) ≥ 45%. A detailed comparison regarding SRs and non-SRs characteristics is presented in Table 4.
Table 4.
A comparison regarding technical aspects, echocardiography parameters, and genetic mutation in SRs versus non-SRs; LBBB—left bundle branch block, SD—standard deviation, FO—follow-up, super-responders (SRs), non-SRs (responders and hyporesponders), typical pattern—Strauss Criteria, MR—mitral regurgitation.
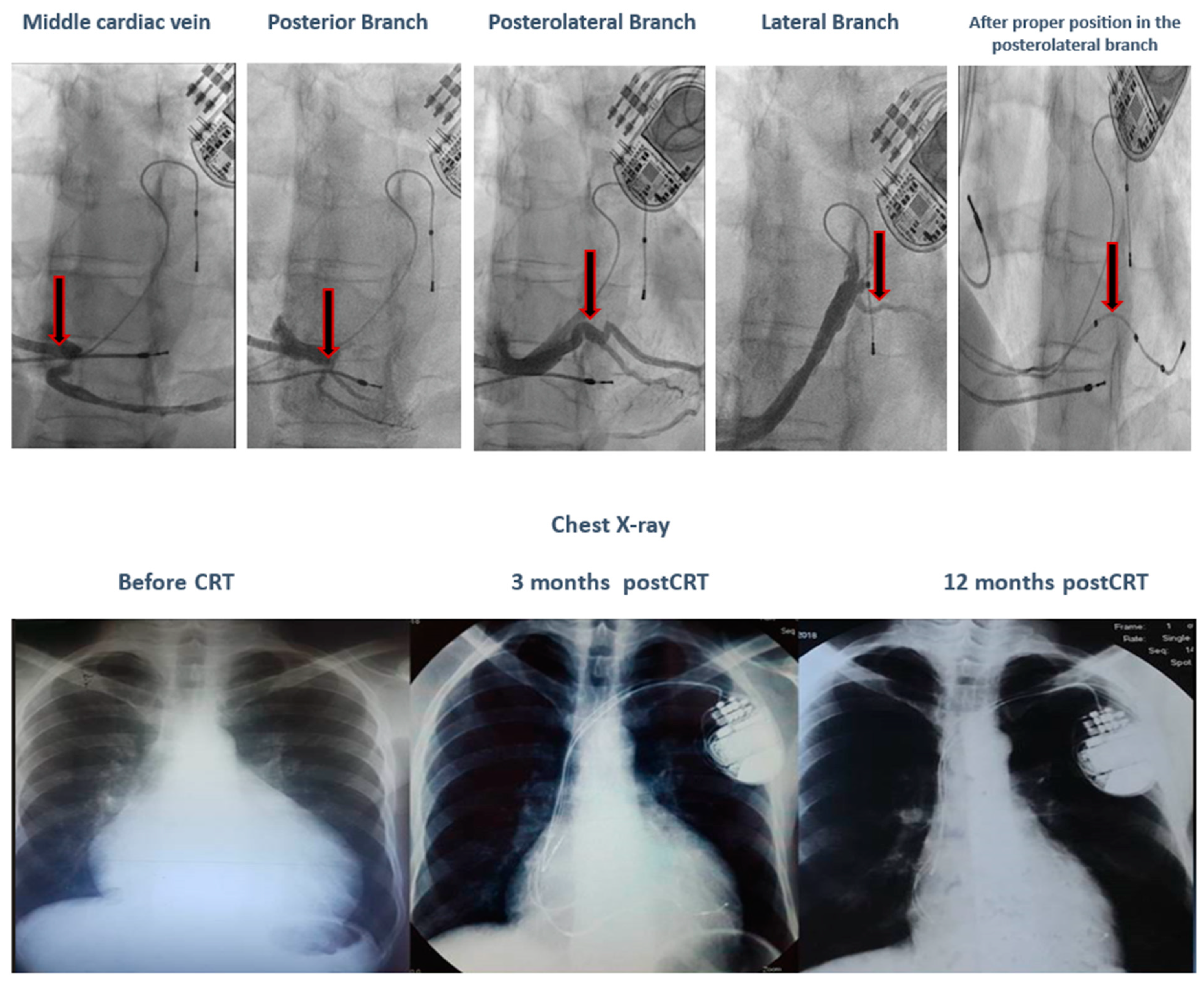
Comparing the SR vs. non-SR groups, we observe that the SR group has a younger average age, a 100% typical LBBB pattern, a wider QRS complex, and LV leads placed only in posterolateral or lateral positions. During a 12-month follow-up, none of the patients in the SR group experienced severe or moderate mitral regurgitation. In terms of genetic testing, there was a variation of mutations in the SR group, while the non-SR group had two patients with VUS-AGL and one patient with both TTN and TMEM43 pathogenic mutations. No deaths were recorded in the SR group. In the non-SR group, there was one cardiac death in the patient with both TTN and TMEM43 mutations due to refractory HF and electrical storm following a severe COVID infection, despite having the clinical and paraclinical criteria of a super-responder (nonischemic, younger age, typical LBBB pattern, wider QRS, and LV lead in the posterolateral position). Additionally, in the SR group, one patient with a TNNI3K gene mutation, who met the clinical and paraclinical criteria of a super-responder (nonischemic, younger age, typical LBBB pattern, and wider QRS) on maximal medical treatment, was initially a non-responder due to the inadequate positioning of the LV lead in the anterior wall. After proper positioning in the posterolateral wall, the patient became a super-responder, as illustrated in Figure 3.

Figure 3.
The evolution of a patient with a TNNI3K gene mutation after proper positioning of the LV lead in the posterolateral wall.
An upgrade to CRT-D was necessary in the non-SR group for one patient with a VUS-AGL mutation, a possible non-disease-causing variant. This patient had nonischemic DCM with an atypical LBBB pattern and a QRS duration of 140 ms, and was on maximal medical treatment when initially implanted with a CRT-P device. Two years after CRT implantation, due to repeated ventricular tachycardia, a CRT-D upgrade was needed, including replacement of the LV lead from the anterior position to the lateral wall. However, the patient, just like the other one with the same mutation, remained a non-responder.
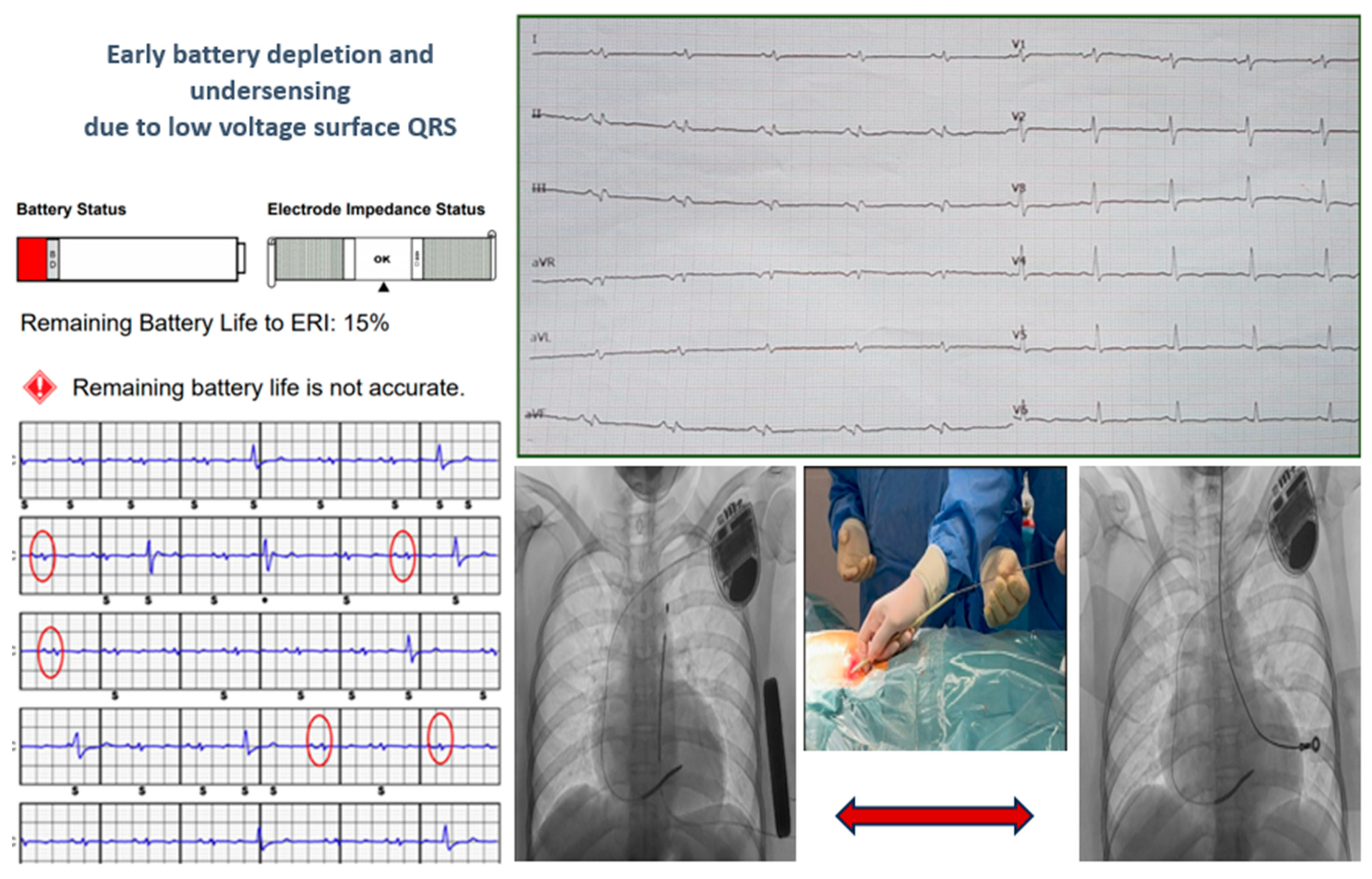
Two young patients, who experienced cardiac arrest due to ventricular fibrillation as their first cardiac event, received a subcutaneous implantable cardioverter-defibrillator (S-ICD). Genetic testing later diagnosed them with arrhythmogenic cardiomyopathy, revealing one DSP mutation and one TMEM43 mutation, both of which are pathogenic. During follow-up, they developed electrical storm due to multiple forms of ventricular tachycardia. Despite undergoing endoepicardial ablation procedures, they continued to experience ventricular tachycardia. Both patients experienced early battery depletion, and the patient with DSP mutation also showed under-sensing. Consequently, a transvenous ICD (T-ICD) was recommended with complete removal of the S-ICD system, as shown in Figure 4.
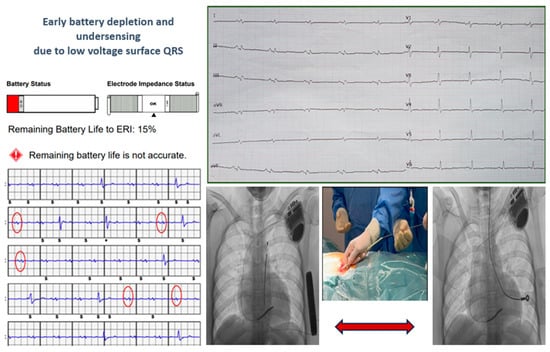
Figure 4.
The cases of two young patients with S-ICD who experienced early battery depletion and under-sensing due to low voltage surface QRS. A transvenous ICD (T-ICD) was recommended with complete removal of the S-ICD system. The red circles on the ECG are highlighting QRS underdetection by the S-ICD.
In the entire group of patients, there were two cardiac deaths, both in individuals with TTN mutations who suffered from refractory heart failure. Despite receiving maximum treatment, their condition progressed rapidly and was fulminant.
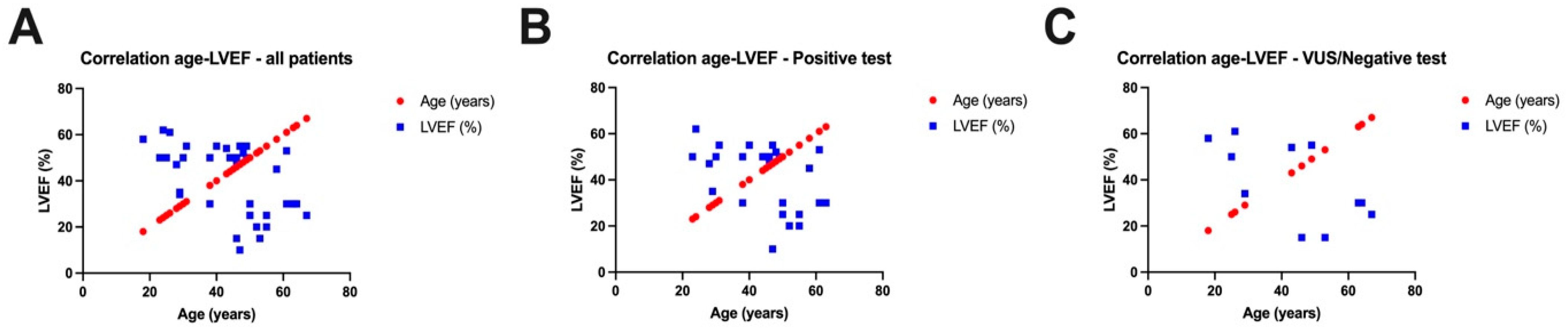
Assessing the correlation (Figure 5) between the age of the patients included in our study and the left ventricular ejection fraction (LVEF), we observed a negative correlation for all patients included in our study, meaning that the younger the patients, the higher the LVEF values, and the older the patients, the lower the LVEF values (r = −0.5210, p = 0.0008). Separating the two groups of patients, we noticed negative correlations both in patients with a positive genetic test (r = −0.4474, p = 0.0193) and in patients with a variant of unknown significance or a negative test (r = −0.6354, p = 0.0357).

Figure 5.
Correlations between left ventricular ejection fraction (LVEF) and age of all patients in the study (A), between LVEF and age of patients with positive genetic tests (B), and between LVEF and age of patients with a variant of unknown significance or a negative test (C).
4. Discussion
Genetic testing in patients with electronic cardiac devices holds significant promise for optimizing therapy and improving outcomes. As our understanding of the genetic basis of cardiac disorders continues to expand, integrating genetic information into the clinical decision-making process will become increasingly important. By doing so, clinicians can offer more personalized and effective treatments for patients with pacemakers, defibrillators, and resynchronization therapy devices.
Identifying a specific genetic trait can aid in patient management and guide clinical decisions. Patients with pathogenic LMNA variants consistently face a poor prognosis, particularly with a high risk of sudden cardiac death (SCD) due to conduction defects or ventricular arrhythmia. Preventive pacemaker (PM) or implantable cardioverter-defibrillator (ICD) therapy should be considered early for LMNA carriers, with ICD implantation algorithms that include pathogenic variant mechanisms (truncating vs. missense variant) due to the higher SCD risk [29,30]. Similarly, a higher risk of SCD is linked to pathogenic variants, especially truncated variants, in the FLNC, DES, RBM20, and PLN genes, warranting consideration for preventive ICD implantation in these patients [31,32]. Desmosomal pathogenic variants in individuals with dilated cardiomyopathy or biventricular arrhythmogenic cardiomyopathy are also associated with an increased risk of life-threatening ventricular arrhythmias and SCD [33].
Cardiomyopathies can be either inherited and/or acquired [34,35]. They may also be exacerbated by disease modifiers, which are conditions that can worsen or trigger cardiomyopathies (such as many cardiovascular comorbidities). Identifying an acquired cause of DCM does not rule out the presence of an underlying gene mutation; conversely, a genetic variant might require an additional acquired cause to clinically manifest [36,37].
Cardiac resynchronization therapy has emerged as a valuable treatment option for heart failure patients [38,39]. As technology advances, genetic testing plays an increasingly pivotal role in tailoring therapies to individual patients’ needs. One critical decision faced by clinicians is choosing between a traditional CRT-P and a CRT-D.
After the controversial Danish trial, the ICD benefit in primary prophylaxis in patients with non-ischemic cardiomyopathy is debatable in the presence of CRT, and it seems that CRT-P is not inferior to CRT-D [28,40].
The data indicate differences within the group we studied. All patients who underwent CRT and genetic testing had non-ischemic cardiomyopathy. Among them, 70% received a CRT-D device following a cardiac arrest caused by ventricular fibrillation or ventricular tachycardia as their first cardiac event, while the remaining 30% received a CRT-P device. One patient with a VUS-AGL mutation, a potentially non-disease-causing variant, required an upgrade to CRT-D due to repeated ventricular tachycardia two years post-implantation, which also involved the replacement of the LV lead from the anterior position to the lateral wall. Despite these interventions, this patient, like another with the same mutation, remained a non-responder. Another patient, despite having the clinical and paraclinical criteria of a super-responder (nonischemic, younger age, typical LBBB pattern, wider QRS, and LV lead in the posterolateral position), died due to refractory HF and electrical storm, having both TTN and TMEM43 mutations. It appears that patients with multiple mutations experience more severe disease and exhibit a weaker response to CRT. Furthermore, despite being a nonischemic DCM, a patient with a TMEM43 mutation, known for its extreme arrhythmogenicity, more often has an unfavorable outcome.
In human patients, mutations in the nuclear envelope protein TMEM43 are linked to severe diseases, including ACM type 5, a devastating cardiomyopathy that leads to malignant arrhythmias and heart failure. The TMEM43-p.S358L mutation has been identified as the genetic cause of an aggressive form of ACM, primarily affecting males. Despite extensive in vivo studies, the pathogenic mechanisms of TMEM43-associated ACM remain poorly understood. Various research groups have developed different models using mice and zebrafish, and induced pluripotent stem cells with TMEM43 mutations to study ACM [41,42,43]. However, both TMEM43-p.S358L knock-in and knock-out mice do not develop a cardiac phenotype under normal conditions [41], suggesting that the pathogenicity of this specific mutation requires enhancement through overexpression or additional genetic, epigenetic, or environmental factors in mice. In the study by Zink et al. [42], the first transgenic zebrafish model expressing two different potential pathogenic variants found in human patients under a heart-specific promoter was created, along with genetic mutants of TMEM43 in zebrafish. These zebrafish lines were characterized from early embryonic stages to adulthood. The mutant p.S358L TMEM43 was found to be unstable and partially redistributed into the cytoplasm in embryonic and adult hearts. Additionally, both TMEM43 variants exhibited cardiac morphological defects at juvenile stages and ultrastructural changes within the myocardium, along with dysregulated gene expression in adulthood.
Conversely, a patient with a TNNI3K gene mutation, who met the super-responder criteria (nonischemic, younger age, typical LBBB pattern, and wider QRS) on maximal medical treatment, was initially a non-responder due to the inadequate positioning of the LV lead in the anterior wall. After proper positioning in the posterolateral wall, the patient became a super-responder.
Considering our group of patients, in order to become a super-responder, meeting the clinical and paraclinical criteria of a super-responder (nonischemic DCM, younger age, typical LBBB pattern, wider QRS complex, and appropriate LV lead position) is necessary. Additionally, the absence of multiple associations with severe gene mutations is also crucial.
Two patients diagnosed with arrhythmogenic cardiomyopathy, who experienced cardiac arrest due to ventricular fibrillation as their first cardiac event, received subcutaneous implantable cardioverter-defibrillators (S-ICDs). During follow-up, they developed electrical storm due to multiple forms of ventricular tachycardia. Both patients experienced early battery depletion, and the patient with a DSP mutation also showed under-sensing. Consequently, a transvenous ICD (T-ICD) was recommended with complete removal of the S-ICD system. The oldest lead was 6 years old. Multiple simple manual tractions were necessary to release the generator and the proximal part of the lead. For the distal part, a mechanical dilator sheath (yellow sheath, inner ID/OD 8.5/10.7Fr) was used to free the lead. The systems were completely removed without complications.
Considering this, it might be preferable to implant a T-ICD when arrhythmogenic cardiomyopathy is suspected, even if the patient experienced cardiac arrest due to ventricular fibrillation as their first cardiac event.
Studies indicate that both T-ICD and S-ICD are effective in detecting and terminating life-threatening arrhythmias in arrhythmogenic cardiomyopathy (ACM); however, the absence of antitachycardia pacing (ATP) in S-ICDs can be a limitation for patients who frequently experience ventricular tachycardia that could otherwise be managed without a shock [44,45]. Guidelines generally support the use of either device in ACM patients, emphasizing personalized treatment based on patient risk profile, lifestyle, age, and arrhythmic burden [2]. As speculated in the literature in cases of severely impaired right ventricular function, a T-ICD is better than an S-ICD [46]. In the remaining cases, an S-ICD could be taken into account, especially in younger patients or those with a higher risk of lead-related complications.
Regarding mortality, two patients died during follow-up due to refractory heart failure, both of whom had titin mutations. A significant proportion of DCM cases are linked to genetic mutations, with titin mutations being the most common type [47]. Titin is a giant protein that plays a crucial role in the elasticity and stability of cardiac sarcomeres. Studies have shown that patients with this mutation have a worse prognosis, especially a higher incidence of heart failure progression and death, but the extent of the impact can vary based on individual patient factors [48,49]. Early genetic screening and tailored therapeutic strategies are essential in managing patients with TTN-related DCM to improve outcomes and reduce mortality, including more aggressive heart failure management and the use of ICD to prevent sudden cardiac death.
The severity and progression of DCM exhibit significant variability among individuals, not only in sporadic cases but also within members of the same family. This variability may be explained by the fact that the clinical phenotype is influenced not only by a single causative gene variant but also by the interaction with common variants in the genome, epigenetic factors, and environmental influences. Patients with DCM who carry pathogenic variants in the LMNA, RBM20, and DSP genes are at higher risk for heart failure progression and may require heart transplantation [33,50].
5. Conclusions
Genetic testing has the potential to revolutionize the decision-making process in patients with cardiac electronic devices. By identifying genetic markers associated with arrhythmia susceptibility, heart failure etiology, and CRT response, clinicians can tailor therapy to individual patient needs.
Author Contributions
Concept/design: E.-V.G., D.C. and C.V.; Data collection: E.-V.G., A.-A.F.-G., A.U. and G.T.; Data analysis/interpretation: C.V., R.V., L.P., D.C. and S.M.; Drafting article: E.-V.G., A.U., G.T., and D.C.; Critical revision of article: E.-V.G., D.C., A.M. and D.C.; visualization, S.M., E.-V.G., D.C. and C.V.; supervision, E.-V.G., D.C. and R.V.; project administration, E.-V.G., D.C. and C.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
All the subjects included in the study gave their informed consent before inclusion. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of our institute (number 46/28 September 2018).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data available on request due to restrictions (privacy and ethical reasons).
Acknowledgments
We would like to acknowledge Victor Babes University of Medicine and Pharmacy Timisoara for their support in covering the costs of publication for this research paper.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| A2ML1 | Alpha 2 Macroglobulin Like 1 |
| ACM | Arrhythmogenic Cardiomyopathy |
| ACMG | American College of Medical Genetics and Genomics |
| ACTC1 | Actin Alpha Cardiac Muscle 1 |
| ACTN2 | Actinin Alpha 2 |
| ACEI | Angiotensin-Coverting Enzyme Inhibitor |
| AGL | Amylo-Alpha-1, 6-Glucosidase, 4-Alpha-Glucanotransferase |
| ARNI | Angiotensin Receptor Neprylisin Inhibitor |
| ATP | Anti-Tachycardia Pacing |
| AV | Atrioventricular |
| BrS | Brugada |
| CACNB2 | Calcium Voltage-Gated Channel Auxiliary Subunit Beta 2 |
| CALM 1 | Calmodulin 1 |
| CALM 2 | Calmodulin 2 |
| CALM 3 | Calmodulin 3 |
| CCD | Cardiac Conduction Defects |
| COPD | Chronic Obstructive Pulmonary Disease |
| CPVT | Catecholaminergic Polymorphic Ventricular Tachycardia |
| CRT | Cardiac Resynchronization Therapy |
| CRT-D | Cardiac Resynchronization Therapy Defibrillator |
| CRT-P | Cardiac Resynchronization Therapy Pacemaker |
| CTNNA3 | Catenin Alpha 3 |
| DCM | Dilated Cardiomyopathy |
| DES | Desmin |
| DSG2 | Desmoglein 2 |
| DMD | Dystrophin |
| DSP | Desmoplakin |
| DSC2 | Desmocollin 2 |
| EF | Ejection Fraction |
| EMD | Emerin |
| FBN1 | Fibrillin 1 |
| FLNC | Filamin C |
| FO | Follow-Up |
| HCM | Hypertrophic Cardiomyopathy |
| HF | Heart Failure |
| ICD | Implantable Cardioverter Defibrillator |
| JUP | Junction Plakoglobin |
| KCNQ1 | Potassium Voltage Gated Channel Subfamily Q Member 1 |
| KCNH2 | Potassium Voltage Gated Channel Subfamily H Member 2 |
| LBBB | Left Bundle Branch Block |
| LP | Likely Pathogenic |
| LMNA | Lamin A/C |
| LQTs | Long QT Syndrome |
| LV | Left Ventricle |
| LVEF | Left Ventricular Ejection Fraction |
| MRI | Magnetic Resonance Imaging |
| MR | Mitral Regurgitation |
| MYBPC3 | Myosin Binding Protein C, Cardiac |
| MYH7 | Myosin Heavy Chain 7 |
| MYLK | Myosin Light Chain Kinase |
| MYL2 | Myosin Light Chain 2 |
| MYL3 | Myosin Light Chain 3 |
| NDUFB3 | NADH: Ubiquinone Oxidoreductase Subunit B3 |
| NEXN | Nexilin F-actin binding protein |
| NOAC | Non-Vitamin K Antagonist Oral Anticoagulant |
| NYHA | New York Heart Association |
| P | Pathogenic |
| PLN | Phospholamban |
| PKP2 | Plakophilin 2 |
| PMK | Pacemaker |
| RBM20 | RNA Binding Motif Protein 20 |
| RYR2 | Ryanodine Receptor 2 |
| SCD | Sudden Cardiac Death |
| SCN5A | Sodium Voltage Gated Channel Alpha Subunit 5 |
| SGCD | Sarcoglycan Delta |
| SGLT2 | Sodium-Glucose Co-transporter 2 Inhibitors |
| S-ICD | Subcutaneous Implantable Cardioverter Defibrillator |
| SOS1 | SOS Ras/Rac Guanine Nucleotide Exchange Factor 1 |
| SR | Superresponder |
| SD | Standard Deviation |
| T-ICD | Transvenous Implantable Cardioverter Defibrillator |
| TMEM43 | Transmembrane Protein 43 |
| TNNI3K | TNNI3 Interacting Kinase |
| TNNI3 | Troponin I3, Cardiac Type |
| TNNT2 | Troponin T2, Cardiac Type |
| TPM1 | Tropomyosin 1 |
| TR | Tricuspid Regurgitation |
| TRMP4 | Transient Receptor Potential Cation Channel Subfamily M Member 4 |
| TTN | Titin |
| TTN-DCM | Titin Related Dilated Cardiomyopathy |
| VUS | Variant of Uncertain Significance |
References
- Glikson, M.; Nielsen, J.C.; Kronborg, M.B.; Michowitz, Y.; Auricchio, A.; Barbash, I.M.; Barrabe, J.A.; Boriani, G.; Braunschweig, F.; Brignole, M.; et al. 2021 ESC Guidelines on cardiac pacing and cardiac resynchronization therapy: Developed by the Task Force on cardiac pacing and cardiac resynchronization therapy of the European Society of Cardiology (ESC) With the special contribution of the European Heart Rhythm Association (EHRA). Eur. Heart J. 2021, 42, 3427–3520. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; A Blom, N.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Developed by the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC) Endorsed by the Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef] [PubMed]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, J.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef] [PubMed]
- Lenarczyk, R.; Zeppenfeld, K.; Tfelt-Hansen, J.; Heinzel, F.R.; Deneke, T.; Ene, E.; Meyer, C.; Wilde, A.; Arbelo, E.; Jędrzejczyk-Patej, E.; et al. Management of patients with an electrical storm or clustered ventricular arrhythmias: A clinical consensus statement of the European Heart Rhythm Association of the ESC—Endorsed by the Asia-Pacific Heart Rhythm Society, Heart Rhythm Society, and Latin-American Heart Rhythm Society. EP Eur. 2024, 26, euae049. [Google Scholar] [CrossRef]
- Tzeis, S.; Gerstenfeld, E.P.; Kalman, J.; Saad, E.B.; Shamloo, A.S.; Andrade, J.G.; Barbhaiya, C.R.; Baykaner, T.; Boveda, S.; Calkins, H.; et al. 2024 European Heart Rhythm Association/Heart Rhythm Society/Asia Pacific Heart Rhythm Society/Latin American Heart Rhythm Society expert consensus statement on catheter and surgical ablation of atrial fibrillation. EP Eur. 2024, 26, euae043. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Semsarian, C.; Marquez, M.F.; Shamloo, A.S.; Ackerman, M.J.; Ashley, E.A.; Sternick, E.B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. EP Eur. 2022, 24, 1367. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; A Blom, N.; A de Boer, R.; et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H.; et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 2020, 141, 418–428. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shimizu, W.; Horie, M. Phenotypic manifestations of mutations in genes encoding subunits of cardiac potassium channels. Circ. Res. 2011, 109, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Odening, K.E.; Sanguinetti, M.C. Heritable arrhythmias associated with abnormal function of cardiac potassium channels. Cardiovasc. Res. 2020, 116, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Ackerman, M.J.; Antzelevitch, C.; Bezzina, C.R.; Borggrefe, M.; Cuneo, B.F.; Wilde, A.A.M. Inherited cardiac arrhythmias. Nat. Rev. Dis. Primers 2020, 6, 58. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Probst, V.; Wilde, A.A.; Barc, J.; Sacher, F.; Babuty, D.; Mabo, P.; Mansourati, J.; Le Scouarnec, S.; Kyndt, F.; Le Caignec, C.; et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ. Cardiovasc. Genet. 2009, 2, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.-B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049, Erratum in Nat. Genet. 2013, 45, 1409. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Neu, A.; Eiselt, M.; Paul, M.; Sauter, K.; Stallmeyer, B.; Isbrandt, D.; Schulze-Bahr, E. A homozygous SCN5A mutation in a severe, recessive type of cardiac conduction disease. Hum. Mutat. 2010, 31, E1609-21. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.B.; Gando, I.; Bu, L.; Cecchin, F.; Coetzee, W. A homozygous SCN5A mutation associated with atrial standstill and sudden death. Pacing Clin. Electrophysiol. 2018, 41, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Daumy, X.; Amarouch, M.-Y.; Lindenbaum, P.; Bonnaud, S.; Charpentier, E.; Bianchi, B.; Nafzger, S.; Baron, E.; Fouchard, S.; Thollet, A.; et al. Targeted resequencing identifies TRPM4 as a major gene predisposing to progressive familial heart block type I. Int. J. Cardiol. 2016, 207, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.; et al. For the SHaRe Investigators. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mazzarotto, F.; Tayal, U.; Buchan, R.J.; Midwinter, W.; Wilk, A.; Whiffin, N.; Govind, R.; Mazaika, E.; de Marvao, A.; Dawes, T.J.; et al. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation 2020, 141, 387–398. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Brodehl, A. Insights into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011, 8, 1308–1339. [Google Scholar] [CrossRef]
- Chirita-Emandi, A.; Andreescu, N.; Zimbru, C.G.; Tutac, P.; Arghirescu, S.; Serban, M.; Puiu, M. Challenges in reporting pathogenic/potentially pathogenic variants in 94 cancer predisposing genes—In pediatric patients screened with NGS panels. Sci. Rep. 2020, 10, 223. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lincoln, S.E.; Kobayashi, Y.; Anderson, M.J.; Yang, S.; Desmond, A.J.; Mills, M.A.; Nilsen, G.B.; Jacobs, K.B.; Monzon, F.A.; Kurian, A.W.; et al. A Systematic Comparison of Traditional and Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Genes in More Than 1000 Patients. J. Mol. Diagn. 2015, 17, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, S.E.; Truty, R.; Lin, C.-F.; Zook, J.M.; Paul, J.; Ramey, V.H.; Salit, M.; Rehm, H.L.; Nussbaum, R.L.; Lebo, M.S. A Rigorous Interlaboratory Examination of the Need to Confirm Next-Generation Sequencing–Detected Variants with an Orthogonal Method in Clinical Genetic Testing. J. Mol. Diagn. 2019, 21, 318–329. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Burri, H.; Prinzen, F.W.; Gasparini, M.; Leclercq, C. Left univentricular pacing for cardiac resynchronization therapy. Europace 2017, 19, 912–919. [Google Scholar] [CrossRef]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbæk, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Thuillot, M.; Maupain, C.; Gandjbakhch, E.; Waintraub, X.; Hidden-Lucet, F.; Isnard, R.; Ader, F.; Rouanet, S.; Richard, P.; Charron, P. External validation of risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers. Eur. J. Heart Fail 2019, 21, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic genotypes in familial dilated cardiomyopathy: Implications for genetic testing and clinical management. Heart Lung Circ. 2019, 28, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Ferro, M.D.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Béhin, A.; Charron, P.; Dunand, M.; Richard, P.; Meune, C.; Vicart, P.; Laforêt, P.; Stojkovic, T.; Bécane, H.M.; et al. High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: A 10-year longitudinal study. Neuromuscul. Disord. 2012, 22, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Ferro, M.D.; Altinier, A.; et al. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Arbustini, E.; Narula, N.; Dec, G.W.; Reddy, K.S.; Greenberg, B.; Kushwaha, S.; Marwick, T.; Pinney, S.; Bellazzi, R.; Favalli, V.; et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: Endorsed by the World Heart Federation. J. Am. Coll. Cardiol. 2013, 62, 2046–2072. [Google Scholar] [CrossRef]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; De Groote, P.; Imazio, M.; et al. MProposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef]
- Bondue, A.; Arbustini, E.; Bianco, A.; Ciccarelli, M.; Dawson, D.; De Rosa, M.; Hamdani, N.; Hilfiker-Kleiner, D.; Meder, B.; Leite-Moreira, A.F.; et al. Complex roads from genotype to phenotype in dilated cardiomyopathy: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1287–1303. [Google Scholar] [CrossRef]
- Hazebroek, M.; Kemna, M.; Schalla, S.; Wijk, S.S.-V.; Gerretsen, S.; Dennert, R.; Merken, J.; Kuznetsova, T.; Staessen, J.; Rocca, H.B.-L.; et al. Prevalence and prognostic relevance of cardiac involvement in ANCA-associated vasculitis: Eosinophilic granulomatosis with polyangiitis and granulomatosis with polyangiitis. Int. J. Cardiol. 2015, 199, 170–179. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 4901. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2024, 45, 53. [Google Scholar] [CrossRef]
- Beggs, S.A.S.; Jhund, P.S.; Jackson, C.E.; McMurray, J.J.V.; Gardner, R.S. Non-ischaemic cardiomyopathy, sudden death and implantable defibrillators: A review and meta-analysis. Heart 2018, 104, 144–150. [Google Scholar] [CrossRef]
- Stroud, M.J.; Fang, X.; Zhang, J.; Guimarães-Camboa, N.; Veevers, J.; Dalton, N.D.; Gu, Y.; Bradford, W.H.; Peterson, K.L.; Evans, S.M.; et al. Luma is not essential for murine cardiac development and function. Cardiovasc. Res. 2018, 114, 378–388. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zink, M.; Seewald, A.; Rohrbach, M.; Brodehl, A.; Liedtke, D.; Williams, T.; Childs, S.J.; Gerull, B. Decreased survival and cardiac performance of mutant TMEM43 in transgenic zebrafish. Circulation 2018, 138 (Suppl. 1), A15878. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ratnavadivel, S.; de Toledo, M.S.; Rasmussen, T.B.; Šarić, T.; Gummert, J.; Zenke, M.; Milting, H. Human pluripotent stem cell line (HDZi001-A) derived from a patient carrying the ARVC-5 associated mutation TMEM43-p S358L. Stem Cell Res. 2020, 48, 101957. [Google Scholar] [CrossRef] [PubMed]
- Healey, J.S.; Krahn, A.D.; Bashir, J.; Amit, G.; McIntyre, W.F.; Tsang, B.; Joza, J.; Exner, D.V.; Birnie, D.H.; Sadek, M.; et al. Perioperative Safety andarly Patient and Device Outcomes Among Subcutaneous Versus Transvenous Implantable Cardioverter Defibrillator Implantations: A Randomized, Multicenter Trial. Ann. Intern. Med. 2022, 175, 1658–1665. [Google Scholar] [CrossRef]
- Wang, W.; Gasperetti, A.; Sears, S.F.; Tichnell, C.; Murray, B.; Tandri, H.; James, C.A.; Calkins, H. Subcutaneous and Transvenous Defibrillators in Arrhythmogenic Right Ventricular Cardiomyopathy: A Comparison of Clinical and Quality-of-Life Outcomes. JACC Clin. Electrophysiol. 2023, 9, 394–402. [Google Scholar] [CrossRef]
- Honarbakhsh, S.; Protonotarios, A.; Monkhouse, C.; Hunter, R.J.; Elliott, P.M.; Lambiase, P.D. Right ventricular function is a predictor for sustained ventricular tachycardia requiring anti-tachycardic pacing in arrhythmogenic ventricular cardiomyopathy: Insight into transvenous vs. subcutaneous implantable cardioverter defibrillator insertion. Europace 2023, 25, euad073. [Google Scholar] [CrossRef]
- McNally, E.M.; Mestroni, L. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef]
- Chauveau, C.; Rowell, J.; Ferreiro, A. A rising titan: TTN review and mutation update. Hum. Mutat. 2014, 35, 1046–1059. [Google Scholar] [CrossRef]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-phenotype associations in dilated cardiomyopathy: Meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).