The Complex Immunological Alterations in Patients with Type 2 Diabetes Mellitus on Hemodialysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
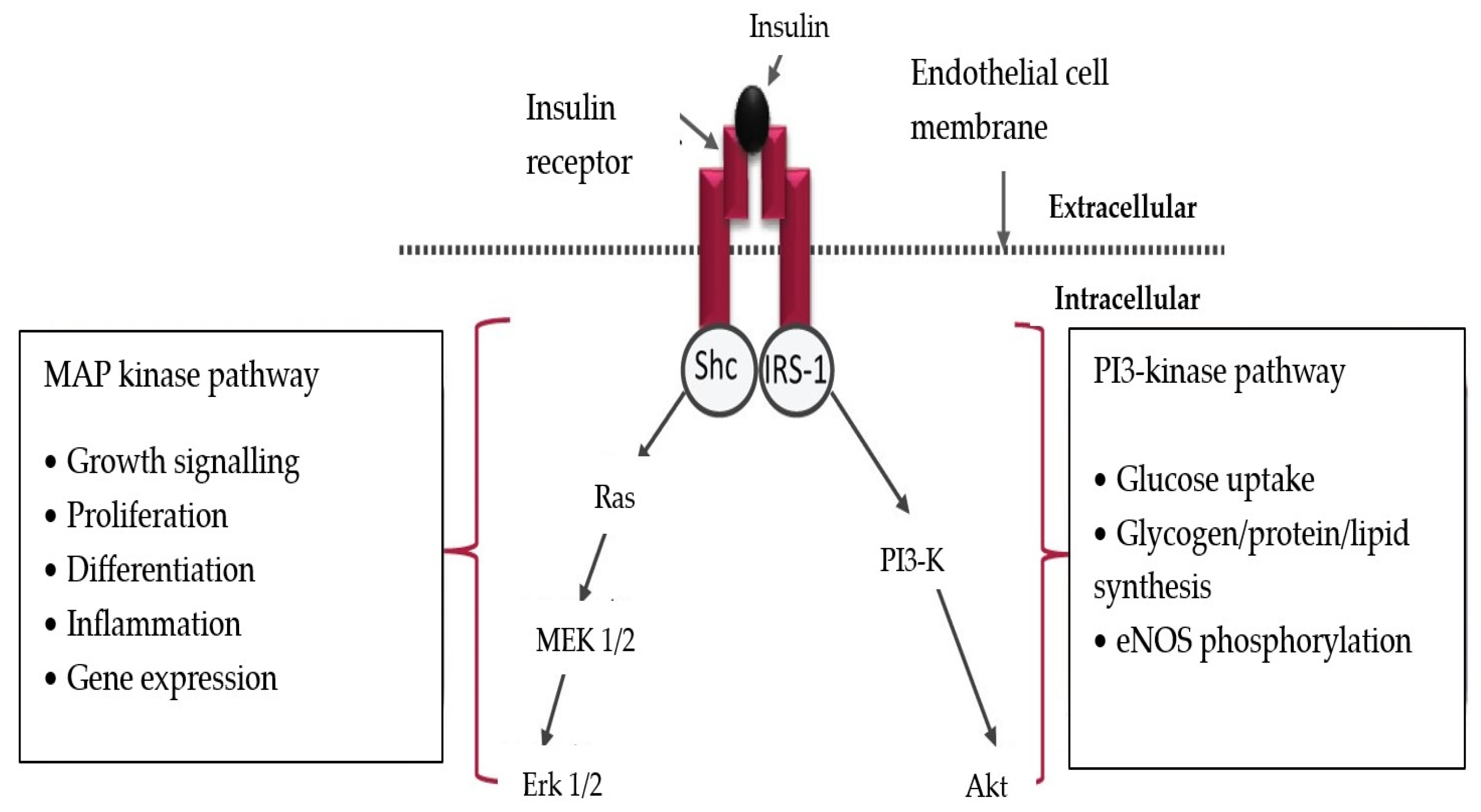
2. General Mechanisms of Endothelial Alterations in Patients with Type 2 Diabetes Mellitus
3. Participation of Endothelium in Diabetic Inflammatory Status
4. Proinflammatory Signaling in T2DM
5. The Role of Vascular Inflammation-Associated Oxidative Stress in T2DM
6. The Molecular Mechanisms of Increasing Insulin Resistance Induced by Inflammation
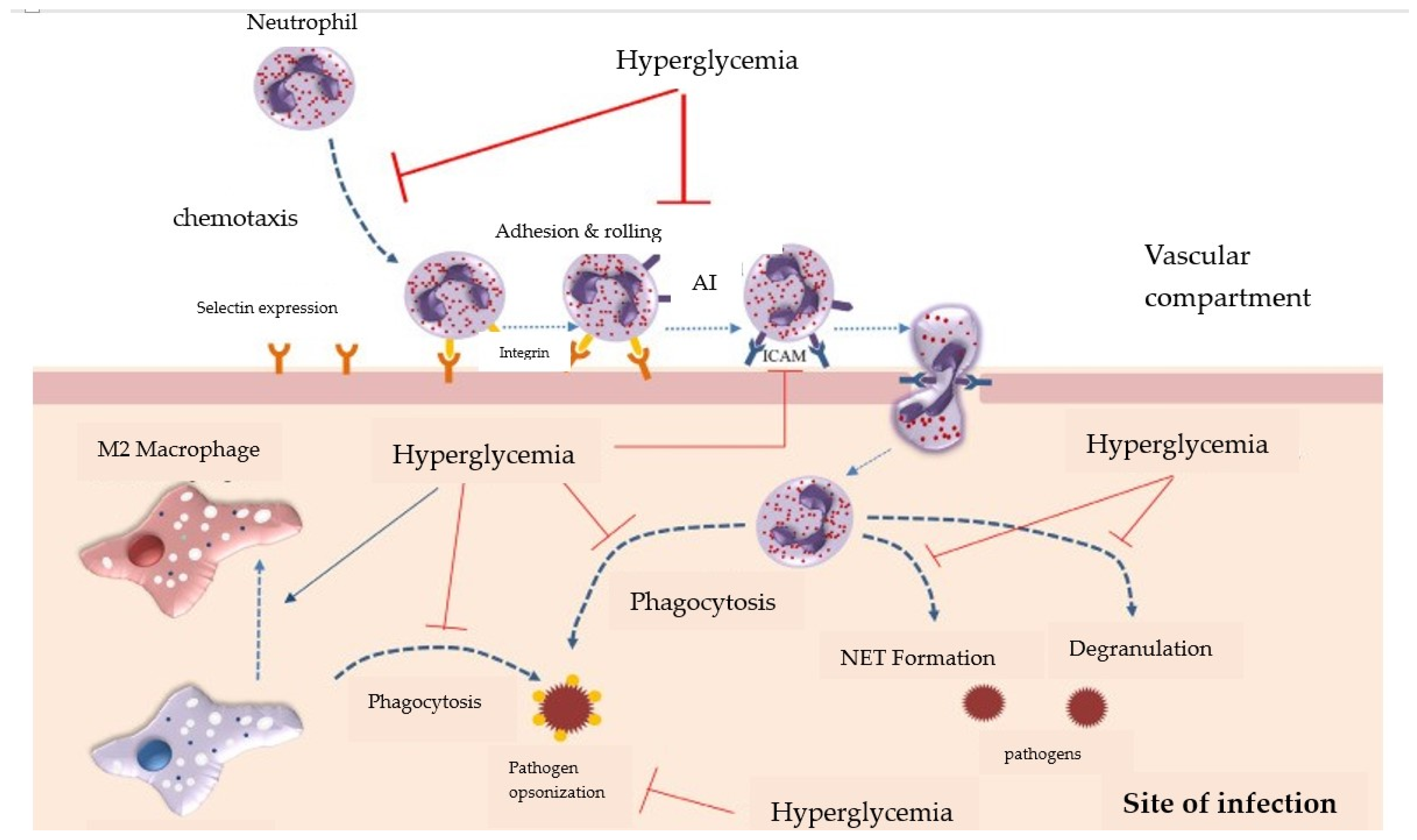
7. The Inhibition of Leukocyte Chemotaxis in T2DM
8. Alterations of Antigen Recognition in T2DM
9. Neutrophil Dysfunctions
10. Macrophage Disfunction
11. Natural Killer Lymphocyte Disfunction
12. The Impact of Hemodialysis on the Immunity of Patients with Diabetes
12.1. Innate Immunity Alteration in Patients on Hemodialysis
12.1.1. Complement System Alterations
- -
- The administration of citrate, which inhibits platelet activation also blocks complement activity, through calcium chelation in the hemodialysis circuit [82];
- -
- Administrating a C1 inhibitor has significantly reduced complement activation and IL-6 and TNFα proinflammatory cytokine release, as well as von Willebrand factor secretion [77];
- -
- Blocking the complement system at C3 level through compstatin has led to increased dialysis membrane biocompatibility [83].
12.1.2. Neutrophils and Monocyte–Macrophages Alterations
12.1.3. The Amplification of Chronic Inflammation in Patients with Diabetes Undergoing Hemodialysis
12.2. Acquired Immunity Alterations in Patients Undergoing Hemodialysis
12.2.1. T Lymphocyte Alterations
12.2.2. B Lymphocyte Alterations
13. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aguirre, F.; Brown, A.; Cho, N.H.; Dahlquist, G.; Dodd, S.; Dunning, T.; Hirst, S.M.; Hwang, C.; Magliano, D.; Patterson, C.; et al. IDF Diabetes Atlas, 6th ed.; International Diabetes Federation: Brussels, Belgium, 2013; 160 p. [Google Scholar]
- Ronacher, K.; Joosten, S.A.; van Crevel, R.; Dockrell, H.M.; Walzl, G.; Ottenhoff, T.H. Acquired immunodeficiencies and tuberculosis: Focus on HIV/AIDS and diabetes mellitus. Immunol. Rev. 2015, 264, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.I. Diabetes and Tuberculosis. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Vrieling, F.; Ronacher, K.; Kleynhans, L.; van den Akker, E.; Walzl, G.; Ottenhoff, T.H.M.; Joosten, S.A. Patients with concurrent tuberculosis and diabetes have a pro-atherogenic plasma lipid profile. EBioMedicine 2018, 32, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Prada-Medina, C.A.; Fukutani, K.F.; Pavan Kumar, N.; Gil-Santana, L.; Babu, S.; Lichtenstein, F.; West, K.; Sivakumar, S.; Menon, P.A.; Viswanathan, V.; et al. Systems immunology of diabetes-tuberculosis comorbidity reveals signatures of disease complications. Sci. Rep. 2017, 7, 1999. [Google Scholar] [CrossRef] [PubMed]
- Kornum, J.B.; Thomsen, R.W.; Riis, A.; Lervang, H.-H.; Schønheyder, H.C.; Sørensen, H.T. Type 2 diabetes and pneumonia outcomes: A population-based cohort study. Diabetes Care 2007, 30, 2251–2257. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.L.; Neuzil, K.M.; Thompson, W.W.; Shay, D.K.; Yu, O.; Hanson, C.A.; Jackson, L.A. The burden of community-acquired pneumonia in seniors: Results of a population-based study. Clin. Infect. Dis. 2004, 39, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Benfield, T.; Jensen, J.S.; Nordestgaard, B.G. Influence of diabetes and hyperglycaemia on infectious disease hospitalisation and outcome. Diabetologia 2007, 50, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.; Boavida, J.M.; Raposo, J.F.; Froes, F.; Nunes, B.; Ribeiro, R.T.; Macedo, M.P.; Penha-Gonçalves, C. Diabetes hinders community-acquired pneumonia outcomes in hospitalized patients. BMJ Open Diabetes Res. Care 2016, 4, e000181. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, O.; Elias, M.; Chazan, B.; Saliba, W. Urinary tract infections in patients with type 2 diabetes mellitus: Review of prevalence, diagnosis, and management. Diabetes Metab. Syndr. Obes. 2015, 8, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Boyko, E.J.; Fihn, S.D.; Scholes, D.; Chen, C.-L.; Normand, E.H.; Yarbro, P. Diabetes and the risk of acute urinary tract infection among postmenopausal women. Diabetes Care 2002, 25, 1778–1783. [Google Scholar] [CrossRef]
- Jenkins, T.C.; Knepper, B.C.; Jason Moore, S.; Saveli, C.C.; Pawlowski, S.W.; Perlman, D.M.; McCollister, B.D.; Burman, W.J. Comparison of the microbiology and antibiotic treatment among diabetic and nondiabetic patients hospitalized for cellulitis or cutaneous abscess. J. Hosp. Med. 2014, 9, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Dryden, M.; Baguneid, M.; Eckmann, C.; Corman, S.; Stephens, J.; Solem, C.; Li, J.; Charbonneau, C.; Baillon-Plot, N.; Haider, S. Pathophysiology and burden of infection in patients with diabetes mellitus and peripheral vascular disease: Focus on skin and soft-tissue infections. Clin. Microbiol. Infect. 2015, 21 (Suppl. 2), S27–S32. [Google Scholar] [CrossRef] [PubMed]
- Suaya, J.A.; Eisenberg, D.F.; Fang, C.; Miller, L.G. Skin and soft tissue infections and associated complications among commercially insured patients aged 0-64 years with and without diabetes in the U.S. PLoS ONE 2013, 8, e60057. [Google Scholar] [CrossRef] [PubMed]
- Levick, J.R. An Introduction to Cardiovascular Physiology, 4th ed.; Distributed in the United States of America by Oxford University Press: Arnold, CA, USA, 2003. [Google Scholar]
- International Diabetes Federation. IDF Clinical Practice Recommendations for Managing Type 2 Diabetes in Primary Care. 2017. Available online: https://idf.org/ (accessed on 13 May 2024).
- Muoio, D.M.; Newgard, C.B. Mechanisms of disease: Molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Banday, M.Z.; Sameer, A.S.; Nissar, S. Pathophysiology of diabetes: An overview. Avicenna J. Med. 2020, 10, 174–188. [Google Scholar] [CrossRef]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C., Jr.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Lodish, H.F. Insulin resistance in adipose tissue: Direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev. 2003, 14, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammatory pathways and insulin action. Int. J. Obes. Relat. Metab. Disord. 2003, 27 (Suppl. 3), S53–S55. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.B. The complex role of adipokines in obesity, inflammation, and autoimmunity. Clin. Sci. 2021, 135, 731–752. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Kim, B.Y.; Mok, J.O.; Kang, S.K.; Kim, C.H. Association between serum adipocytokine levels and microangiopathies in patients with type 2 diabetes mellitus. J. Diabetes Investig. 2014, 5, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, S.K.; Boguszewska, K.; Szewczuk, M.; Kaźmierczak-Barańska, J.; Karwowski, B.T. 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine (8-oxodG) and 8-Hydroxy-2′-Deoxyguanosine (8-OHdG) as a Potential Biomarker for Gestational Diabetes Mellitus (GDM) Development. Molecules 2020, 25, 202. [Google Scholar] [CrossRef] [PubMed]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef]
- McDonald, M.J.; Shapiro, R.; Bleichman, M.; Solway, J.; Bunn, H.F. Glycosylated minor components of human adult hemoglobin. Purification, identification, and partial structural analysis. J. Biol. Chem. 1978, 253, 2327–2332. [Google Scholar] [CrossRef] [PubMed]
- Štambuk, T.; Gornik, O. Protein Glycosylation in Diabetes. Adv. Exp. Med. Biol. 2021, 1325, 285–305. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Guillausseau, P.J. Diabetes, advanced glycation endproducts and vascular disease. Vasc. Med. 1998, 3, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Bitla, A.R.; Devi, N.H.; Kiranmayi, V. Molecular mechanisms underlying diabetic microvascular complications. J. Clin. Sci. Res. 2016, 5, 112–123. [Google Scholar] [CrossRef]
- Lee, A.Y.; Chung, S.S. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999, 13, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Dills, W.L., Jr. Protein fructosylation: Fructose and the Maillard reaction. Am. J. Clin. Nutr. 1993, 58 (Suppl. 6), 779S–787S. [Google Scholar] [CrossRef] [PubMed]
- Villaverde, A.; Estévez, M. Carbonylation of myofibrillar proteins through the maillard pathway: Effect of reducing sugars and reaction temperature. J. Agric. Food Chem. 2013, 61, 3140–3147. [Google Scholar] [CrossRef] [PubMed]
- Allarakha, S.; Ahmad, P.; Ishtikhar, M.; Zaheer, M.S.; Siddiqi, S.S.; Moinuddin; Ali, A. Fructosylation generates neo-epitopes on human serum albumin. IUBMB Life 2015, 67, 338–347. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Liu, S.; Gao, M.; Wang, W.; Chen, K.; Huang, L.; Liu, Y. Diabetic vascular diseases: Molecular mechanisms and therapeutic strategies. Signal Transduct. Target. Ther. 2023, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Tronc, F.; Wassef, M.; Esposito, B.; Henrion, D.; Glagov, S.; Tedgui, A. Role of NO in flow-induced remodeling of the rabbit common carotid artery. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1256–1262. [Google Scholar] [CrossRef]
- Sumpio, B.E.; Riley, J.T.; Dardik, A. Cells in focus: Endothelial cell. Int. J. Biochem. Cell Biol. 2002, 34, 1508–1512. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.Y.; Lin, Y.W.; Clemont, A.; Feener, E.P.; Hein, K.D.; Igarashi, M.; Yamauchi, T.; White, M.F.; King, G.L. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J. Clin. Investig. 1999, 104, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.C.; Porter, K.E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diab. Vasc. Dis. Res. 2013, 10, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, C.; King, G. Mechanisms of Disease: Endothelial dysfunction in insulin resistance and diabetes. Nat. Clin. Pr. Endocrinol. Metab. 2007, 3, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Wu, Y.; Xu, J.; Xie, Z.; Dong, Y.; Zhang, M.; Zou, M.H. Reactive nitrogen species induced by hyperglycemia suppresses Akt signaling and triggers apoptosis by upregulating phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome 10) in an LKB1-dependent manner. Circulation 2007, 116, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A. Chapter 6—Direct and indirect actions of insulin: Role of insulin receptor, glucose transporters (GLUTs), and sodium-glucose linked transporters (SGLTs). In Understanding Insulin and Insulin Resistance; Gupta, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; ISBN 9780128202340. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Chawla, A. Alternative macrophage activation and metabolism. Annu. Rev. Pathol. 2011, 6, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I.; Ruslami, R. Type 2 Diabetes and its Impact on the Immune System. Curr. Diabetes Rev. 2020, 16, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.; Ketheesan, N.; Martens, G.W.; West, K.; Lien, E.; Kornfeld, H. Defects in early cell recruitment contribute to the increased susceptibility to respiratory Klebsiella pneumoniae infection in diabetic mice. Microbes Infect. 2016, 18, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Maratha, A.; Siednienko, J.; Natarajan, A.; Gajanayake, T.; Hoashi, S.; Miggin, S. Analysis of inflammatory cytokine and TLR expression levels in Type 2 Diabetes with complications. Sci. Rep. 2017, 7, 7633. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.S.; Weakley, S.M.; Yao, Q.; Chen, C. Resistin: Functional roles and therapeutic considerations for cardiovascular disease. Br. J. Pharmacol. 2012, 165, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Maemura, K.; Takeda, N.; Harada, T.; Nojiri, T.; Imai, Y.; Manabe, I.; Utsunomiya, K.; Nagai, R. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: A new insight into adipocytokine-endothelial cell interactions. Biochem. Biophys. Res. Commun. 2004, 314, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, G.; Ohta, M.; Ichida, Y.; Obayashi, H.; Shigeta, M.; Yamasaki, M.; Fukui, M.; Yoshikawa, T.; Nakamura, N. Increased serum resistin levels in patients with type 2 diabetes are not linked with markers of insulin resistance and adiposity. Acta Diabetol. 2005, 42, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Ding, W.; Li, K.; Pang, Y.; Tang, C. Plasma resistin associated with myocardium injury in patients with acute coronary syndrome. Circ. J. 2008, 72, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Perner, A.; Nielsen, S.E.; Rask-Madsen, J. High glucose impairs superoxide production from isolated blood neutrophils. Intensive Care Med. 2003, 29, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Stegenga, M.E.; van der Crabben, S.N.; Blümer, R.M.E.; Levi, M.; Meijers, J.C.; Serlie, M.J.; Tanck, M.W.; Sauerwein, H.P.; van der Poll, T. Hyperglycemia enhances coagulation and reduces neutrophil degranulation, whereas hyperinsulinemia inhibits fibrinolysis during human endotoxemia. Blood 2008, 112, 82–89. [Google Scholar] [CrossRef]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- de Souza Ferreira, C.; Araújo, T.H.; Ângelo, M.L.; Pennacchi, P.C.; Okada, S.S.; de Araújo Paula, F.B.; Migliorini, S.; Rodrigues, M.R. Neutrophil dysfunction induced by hyperglycemia: Modulation of myeloperoxidase activity. Cell Biochem. Funct. 2012, 30, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.I.; Twahirwa, M.; Rahbar, M.H.; Schlesinger, L.S. Phagocytosis via complement or Fc-gamma receptors is compromised in monocytes from type 2 diabetes patients with chronic hyperglycemia. PLoS ONE 2014, 9, e92977. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, S.; Lindsay, J.; Ingram, R.; Xu, H.; Chen, M. Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol. 2018, 19, 24. [Google Scholar] [CrossRef] [PubMed]
- Berrou, J.; Fougeray, S.; Venot, M.; Chardiny, V.; Gautier, J.F.; Dulphy, N.; Toubert, A.; Peraldi, M.N. Natural killer cell function, an important target for infection and tumor protection, is impaired in type 2 diabetes. PLoS ONE 2013, 8, e62418. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, T.; Ninomiya, T.; Jha, V.; Neal, B.; Patrice, H.M.; Okpechi, I.; Zhao, M.H.; Lv, J.; Garg, A.X.; Knight, J.; et al. Worldwide access to treatment for end-stage kidney disease: A systematic review. Lancet 2015, 385, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, L. Inflammation and Cardiovascular Disease Associated With Hemodialysis for End-Stage Renal Disease. Front. Pharmacol. 2022, 13, 800950. [Google Scholar] [CrossRef]
- Locatelli, F.; Zoccali, C.; SIR SIN Study Investigators. Clinical policies on the management of chronic kidney disease patients in Italy. Nephrol. Dial. Transplant. 2008, 23, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Jofré, R.; Rodriguez-Benitez, P.; López-Gómez, J.M.; Pérez-Garcia, R. Inflammatory syndrome in patients on hemodialysis. J. Am. Soc. Nephrol. 2006, 17, S274–S280. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An. evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Burton, D.G.A.; Stolzing, A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res. Rev. 2018, 43, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Plowden, J.; Renshaw-Hoelscher, M.; Engleman, C.; Katz, J.; Sambhara, S. Innate immunity in aging: Impact on macrophage function. Aging Cell 2004, 3, 161–167. [Google Scholar] [CrossRef]
- Ducloux, D.; Legendre, M.; Bamoulid, J.; Saas, P.; Courivaud, C.; Crepin, T. End-Stage Renal Disease-Related Accelerated Immune Senescence: Is Rejuvenation of the Immune System a Therapeutic Goal? Front. Med. (Lausanne) 2021, 8, 720402. [Google Scholar] [CrossRef] [PubMed]
- Canaud, B.; Kooman, J.P.; Selby, N.M.; Taal, M.W.; Francis, S.; Maierhofer, A.; Kopperschmidt, P.; Collins, A.; Kotanko, P. Dialysis-Induced Cardiovascular and Multiorgan Morbidity. Kidney Int. Rep. 2020, 5, 1856–1869. [Google Scholar] [CrossRef] [PubMed]
- Vahdat, S. The complex effects of adipokines in the patients with kidney disease. J. Res. Med. Sci. 2018, 23, 60. [Google Scholar] [CrossRef] [PubMed]
- Melchior, P.; Erlenkötter, A.; Zawada, A.M.; Delinski, D.; Schall, C.; Stauss-Grabo, M.; Kennedy, J.P. Complement activation by dialysis membranes and its association with secondary membrane formation and surface charge. Artif. Organs 2021, 45, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Stepniewska, J.; Dolegowska, B.; Golembiewska, E.; Marchelek-Mysliwiec, M.; Domanski, M.; Ciechanowski, K.; Zair, L. The activation of complement system in different types of renal replacement therapy. J. Physiol. Pharmacol. 2020, 71, 275–281. [Google Scholar] [CrossRef]
- Poppelaars, F.; Faria, B.; Gaya da Costa, M.; Franssen, C.F.M.; van Son, W.J.; Berger, S.P.; Daha, M.R.; Seelen, M.A. The Complement. System in Dialysis: A Forgotten Story? Front. Immunol. 2018, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.; Chang, T.I.; Cushman, W.C.; Furth, S.L.; Hou, F.F.; Ix, J.H.; Knoll, G.A.; Muntner, P.; Pecoits-Filho, R.; Sarnak, M.J.; et al. Executive summary of the KDIGO 2021 Clinical Practice Guideline for the Management of Blood Pressure in Chronic Kidney Disease. Kidney Int. 2021, 99, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.L.; Pilely, K.; Lund, K.P.; Warming, P.E.; Plesner, L.L.; Iversen, K.K.; Garred, P. Hemodialysis leads to plasma depletion of lectin complement pathway initiator molecule ficolin-2. Hemodial. Int. 2021, 25, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Mares, J.; Richtrova, P.; Hricinova, A.; Tuma, Z.; Moravec, J.; Lysak, D.; Matejovic, M. Proteomic profiling of blood-dialyzer interactome reveals involvement of lectin complement pathway in hemodialysis-induced inflammatory response. Proteom. Clin. Appl. 2010, 4, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Seong, E.Y.; Lee, W.; Kim, S.; Ahn, H.S.; Yeom, J.; Kim, K.; Kwon, C.H.; Song, S.H. Comparative analysis of therapeutic effects between medium cut-off and high flux dialyzers using metabolomics and proteomics: Exploratory, prospective study in hemodialysis. Sci. Rep. 2021, 11, 17335. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Sandholm, K.; Jonsson, N.; Nilsson, A.; Wieslander, A.; Grundström, G.; Hancock, V.; Ekdahl, K.N. Low concentrations of citrate reduce complement and granulocyte activation in vitro in human blood. Clin. Kidney J. 2015, 8, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kourtzelis, I.; Markiewski, M.M.; Doumas, M.; Rafail, S.; Kambas, K.; Mitroulis, I.; Panagoutsos, S.; Passadakis, P.; Vargemezis, V.; Magotti, P.; et al. Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood 2010, 116, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Fukushi, T.; Yamamoto, T.; Yoshida, M.; Fujikura, E.; Miyazaki, M.; Nakayama, M. Enhanced neutrophil apoptosis accompanying myeloperoxidase release during hemodialysis. Sci. Rep. 2020, 10, 21747. [Google Scholar] [CrossRef] [PubMed]
- Bieber, S.; Muczynski, K.A.; Lood, C. Neutrophil Activation and Neutrophil Extracellular Trap Formation in Dialysis Patients. Kidney Med. 2020, 2, 692–698.e1. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Fujieda, H.; Meguro, H.; Ueno, Y.; Aoki, T.; Miwa, K.; Kainoh, M. Biocompatibility of Polysulfone Hemodialysis Membranes and Its Mechanisms: Involvement of Fibrinogen and Its Integrin Receptors in Activation of Platelets and Neutrophils. Artif. Organs 2018, 42, E246–E258. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Carmona, A.; Agüera, M.L.; Luna-Ruiz, C.; Buendía, P.; Calleros, L.; García-Jerez, A.; Rodríguez-Puyol, M.; Arias, M.; Arias-Guillen, M.; de Arriba, G.; et al. Markers of endothelial damage in patients with chronic kidney disease on hemodialysis. Am. J. Physiol. Renal Physiol. 2017, 312, F673–F681. [Google Scholar] [CrossRef] [PubMed]
- Borges Bonan, N.; Schepers, E.; Pecoits-Filho, R.; Dhondt, A.; Pletinck, A.; De Somer, F.; Vanholder, R.; Van Biesen, W.; Moreno-Amaral, A.; Glorieux, G. Contribution of the uremic milieu to an increased pro-inflammatory monocytic phenotype in chronic kidney disease. Sci. Rep. 2019, 9, 10236. [Google Scholar] [CrossRef] [PubMed]
- Liakopoulos, V.; Jeron, A.; Shah, A.; Bruder, D.; Mertens, P.R.; Gorny, X. Hemodialysis-related changes in phenotypical features of monocytes. Sci. Rep. 2018, 8, 13964. [Google Scholar] [CrossRef]
- Nockher, W.A.; Wiemer, J.; Scherberich, J.E. Haemodialysis monocytopenia: Differential sequestration kinetics of CD14+CD16+ and CD14++ blood monocyte subsets. Clin. Exp. Immunol. 2001, 123, 49–55. [Google Scholar] [CrossRef]
- Sester, U.; Sester, M.; Heine, G.; Kaul, H.; Girndt, M.; Köhler, H. Strong depletion of CD14(+)CD16(+) monocytes during haemodialysis treatment. Nephrol. Dial. Transplant. 2001, 16, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Kakuta, T.; Komaba, H.; Takagi, N.; Takahashi, Y.; Suzuki, H.; Hyodo, T.; Nagaoka, M.; Tanaka, R.; Iwao, S.; Ishida, M.; et al. A Prospective Multicenter Randomized Controlled Study on Interleukin-6 Removal and Induction by a new Hemodialyzer With Improved Biocompatibility in Hemodialysis Patients: A Pilot Study. Ther. Apher. Dial. 2016, 20, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Kamińska, J.; Stopiński, M.; Mucha, K.; Jędrzejczak, A.; Gołębiowski, M.; Niewczas, M.A.; Pączek, L.; Foroncewicz, B. IL 6 but not TNF is linked to coronary artery calcification in patients with chronic kidney disease. Cytokine 2019, 120, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Sanchis, P.; Ho, C.Y.; Liu, Y.; Beltran, L.E.; Ahmad, S.; Jacob, A.P.; Furmanik, M.; Laycock, J.; Long, D.A.; Shroff, R.; et al. Arterial “inflammaging” drives vascular calcification in children on dialysis. Kidney Int. 2019, 95, 958–972. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Schmidt, I.M.; Sabbisetti, V.; Tio, M.C.; Opotowsky, A.R.; Waikar, S.S. Plasma Endothelin-1 and Risk of Death and Hospitalization in Patients Undergoing Maintenance Hemodialysis. Clin. J. Am. Soc. Nephrol. 2020, 15, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.; Akazaki, S.; Nagano, Y.; Ueda, A.; Chang-Il Lee, M.; Aoyagi, K.; Oowada, S.; Sato, K. Hemodialysis raises oxidative stress through carbon-centered radicals despite improved biocompatibility. J. Clin. Biochem. Nutr. 2021, 69, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef] [PubMed]
- Wann, J.G.; Hsu, Y.H.; Yang, C.C.; Lin, C.S.; Tai, D.W.; Chen, J.S.; Hsiao, C.W.; Chen, C.F. Neutrophils in acidotic haemodialysed patients have lower intracellular pH and inflamed state. Nephrol. Dial. Transplant. 2007, 22, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Morena, M.; Cristol, J.P.; Senécal, L.; Leray-Moragues, H.; Krieter, D.; Canaud, B. Oxidative stress in hemodialysis patients: Is. NADPH oxidase complex the culprit? Kidney Int. Suppl. 2002, 61, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Montero, R.; Gómez-López, V.E.; Guerrero-Pavón, F.; Carmona-Muñoz, A.; Romero-Saldaña, M.; Ranchal-Sanchez, A.; Aljama-García, P. Influence of Tunneled Hemodialysis-Catheters on Inflammation and Mortality in Dialyzed Patients. Int. J. Environ. Res. Public Health 2021, 18, 7605. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, L.; Nopp, A.; Jacobson, S.H.; Hylander, B.; Lundahl, J. Hemodialysis Patients Display a Declined Proportion of Th2 and Regulatory T Cells in Parallel with a High Interferon-γ Profile. Nephron 2017, 136, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Meijers, R.W.; Litjens, N.H.; de Wit, E.A.; Langerak, A.W.; van der Spek, A.; Baan, C.C.; Weimar, W.; Betjes, M.G. Uremia causes premature ageing of the T cell compartment in end-stage renal disease patients. Immun. Ageing 2012, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Litjens, N.H.; van Druningen, C.J.; Betjes, M.G. Progressive loss of renal function is associated with activation and depletion of naive T lymphocytes. Clin. Immunol. 2006, 118, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Crépin, T.; Legendre, M.; Carron, C.; Vachey, C.; Courivaud, C.; Rebibou, J.M.; Ferrand, C.; Laheurte, C.; Vauchy, C.; Gaiffe, E.; et al. Uraemia-induced immune senescence and clinical outcomes in chronic kidney disease patients. Nephrol. Dial. Transplant. 2020, 35, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Borges, A.; Borges, M.; Fernandes, J.; Nascimento, H.; Sameiro-Faria, M.; Miranda, V.; Reis, F.; Belo, L.; Costa, E.; Santos-Silva, A. Apoptosis of peripheral CD4(+) T-lymphocytes in end-stage renal disease patients under hemodialysis and rhEPO therapies. Ren. Fail. 2011, 33, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Lisowska, K.A.; Pindel, M.; Pietruczuk, K.; Kuźmiuk-Glembin, I.; Storoniak, H.; Dębska-Ślizień, A.; Witkowski, J.M. The influence of a single hemodialysis procedure on human T lymphocytes. Sci. Rep. 2019, 9, 5041. [Google Scholar] [CrossRef] [PubMed]
- Lisowska, K.A.; Dębska-Ślizień, A.; Jasiulewicz, A.; Heleniak, Z.; Bryl, E.; Witkowski, J.M. Hemodialysis affects phenotype and proliferation of CD4-positive T lymphocytes. J. Clin. Immunol. 2012, 32, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Xiang, F.; Cao, X.; Chen, X.; Zhang, Z.; Ding, X.; Zou, J.; Shen, B. Decreased Peripheral Naïve T Cell Number and Its Role in Predicting Cardiovascular and Infection Events in Hemodialysis Patients. Front. Immunol. 2021, 12, 644627. [Google Scholar] [CrossRef] [PubMed]
- Betjes, M.G.; Meijers, R.W.; de Wit, L.E.; Litjens, N.H. A killer on the road: Circulating CD4(+)CD28null T cells as cardiovascular risk factor in ESRD patients. J. Nephrol. 2012, 25, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Chung, B.H.; Jeon, E.J.; Kim, B.M.; Choi, B.S.; Park, C.W.; Kim, Y.S.; Cho, S.G.; Cho, M.L.; Yang, C.W. B cell-associated immune profiles in patients with end-stage renal disease (ESRD). Exp. Mol. Med. 2012, 44, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Esposito, P.; Rampino, T.; Gregorini, M.; Gabanti, E.; Bianzina, S.; Canton, A.D. Mechanisms underlying sCD40 production in hemodialysis patients. Cell. Immunol. 2012, 278, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, G.H.; Rosenstiel, P.; Arlt, A.; Till, A.; Bräutigam, K.; Schäfer, H.; Rose-John, S.; Seegert, D.; Schreiber, S. Soluble tumor necrosis factor (TNF) receptor-1 induces apoptosis via reverse TNF signaling and autocrine transforming growth factor-beta1. FASEB J. 2005, 19, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Garbers, C.; Rabe, B.; Rose-John, S.; Scheller, J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011, 90, 484–494. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trandafir, M.-F.; Savu, O.I.; Gheorghiu, M. The Complex Immunological Alterations in Patients with Type 2 Diabetes Mellitus on Hemodialysis. J. Clin. Med. 2024, 13, 3687. https://doi.org/10.3390/jcm13133687
Trandafir M-F, Savu OI, Gheorghiu M. The Complex Immunological Alterations in Patients with Type 2 Diabetes Mellitus on Hemodialysis. Journal of Clinical Medicine. 2024; 13(13):3687. https://doi.org/10.3390/jcm13133687
Chicago/Turabian StyleTrandafir, Maria-Florina, Octavian Ionel Savu, and Mihaela Gheorghiu. 2024. "The Complex Immunological Alterations in Patients with Type 2 Diabetes Mellitus on Hemodialysis" Journal of Clinical Medicine 13, no. 13: 3687. https://doi.org/10.3390/jcm13133687
APA StyleTrandafir, M.-F., Savu, O. I., & Gheorghiu, M. (2024). The Complex Immunological Alterations in Patients with Type 2 Diabetes Mellitus on Hemodialysis. Journal of Clinical Medicine, 13(13), 3687. https://doi.org/10.3390/jcm13133687