
Targeted Therapy for SLE—What Works, What Doesn’t, What’s Next


Abstract
1. Introduction
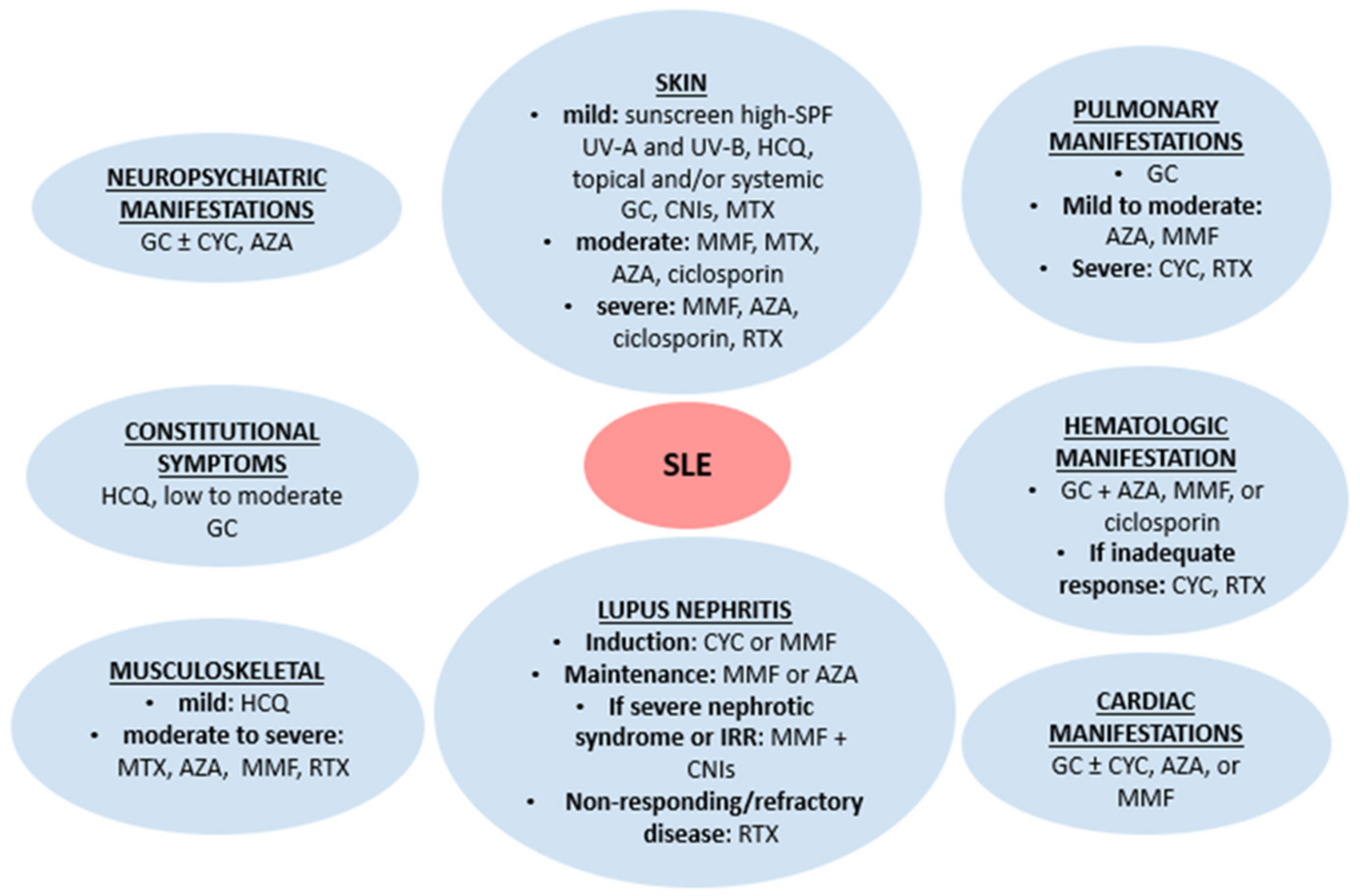
2. Conventional Lupus Treatment
3. Brief Overview of the Immunopathology of SLE and How This Guides the Development of Relevant Biologic Drugs
4. Development and Current Usage of Biological Drugs Approved for the Treatment of SLE
4.1. Rituximab
4.2. Belimumab
4.3. Anifrolumab
5. Reviewing Those Drugs That Have Achieved Phase II Drug Trial Endpoints and Are in or about to Start Phase III Trials
5.1. Obinutuzumab
5.2. Dapirolizumab
5.3. Deucravacitinib
5.4. Litifilimab
5.5. Atacicept
5.6. Telitacicept
5.7. Other Drugs
6. Combination Therapies
7. Pediatric SLE
8. Resume of Drugs in Phase I Clinical Trials
9. Discussion
10. Conclusions
11. Future directions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bruce, I.N.; O’Keeffe, A.G.; Farewell, V.; Hanly, J.G.; Manzi, S.; Su, L.; Gladman, D.D.; Bae, S.-C.; Sanchez-Guerrero, J.; Romero-Diaz, J.; et al. Factors associated with damage accrual in patients with systemic lupus erythematosus: Results from the Systemic Lupus International Collaborating Clinics (SLICC) Inception Cohort. Ann. Rheum. Dis. 2015, 74, 1706–1713. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, B.; Marozoff, S.; Li, L.; Sayre, E.C.; Zubieta, J.A.A. All-cause and cause-specific mortality in systemic lupus erythematosus: A population-based study. Rheumatology 2021, 61, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Merrell, M.; Shulman, L.E. Determination of prognosis in chronic disease, illustrated by systemic lupus erythematosus. J. Chronic Dis. 1955, 1, 12–32. [Google Scholar] [CrossRef] [PubMed]
- Bernatsky, S.; Boivin, J.-F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef] [PubMed]
- Borchers, A.T.; Keen, C.L.; Shoenfeld, Y.; Gershwin, M. Surviving the butterfly and the wolf: Mortality trends in systemic lupus erythematosus. Autoimmun. Rev. 2004, 3, 423–453. [Google Scholar] [CrossRef]
- Gladman, D.D.; Urowitz, M.B.; Rahman, P.; Ibañez, M.; Tam, L.-S. Accrual of Organ Damage Over Time in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2003, 30, 1955–1959. [Google Scholar]
- Frodlund, M.; Reid, S.; Wetterö, J.; Dahlström; Sjöwall, C.; Leonard, D. The majority of Swedish systemic lupus erythematosus patients are still affected by irreversible organ impairment: Factors related to damage accrual in two regional cohorts. Lupus 2019, 28, 1261–1272. [Google Scholar] [CrossRef]
- Segura, B.T.; Bernstein, B.S.; McDonnell, T.; Wincup, C.; Ripoll, V.M.; Giles, I.; Isenberg, D.; Rahman, A. Damage accrual and mortality over long-term follow-up in 300 patients with systemic lupus erythematosus in a multi-ethnic British cohort. Rheumatology 2019, 59, 524–533. [Google Scholar] [CrossRef]
- Durcan, L.; O’Dwyer, T.; Petri, M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet 2019, 393, 2332–2343. [Google Scholar] [CrossRef]
- Burmester, G.R.; Bijlsma, J.W.J.; Cutolo, M.; McInnes, I.B. Managing rheumatic and musculoskeletal diseases — past, present and future. Nat. Rev. Rheumatol. 2017, 13, 443–448. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Adamichou, C.; Koutsoviti, S.; Panopoulos, S.; Staveri, C.; Klagou, A.; Tsalapaki, C.; Pantazi, L.; Konsta, S.; Mavragani, C.P.; et al. Low disease activity—irrespective of serologic status at baseline—associated with reduction of corticosteroid dose and number of flares in patients with systemic lupus erythematosus treated with belimumab: A real-life observational study. Semin. Arthritis Rheum. 2018, 48, 467–474. [Google Scholar] [CrossRef]
- Agnihotri, P.; Fazel-Rezai, R.; Kaabouch, N. Comparative analysis of various brain imaging techniques. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2010, 2010, 3029–3032. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, R.; Araújo, C.; Martins-Coelho, G.; Isenberg, D. Use of Rituximab in Systemic Lupus Erythematosus: A Single Center Experience Over 14 Years. Arthritis Care Res. 2017, 69, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Yusof, Y.M.; Shaw, D.; El-Sherbiny, Y.M.; Dunn, E.; Rawstron, A.C.; Emery, P.; Vital, E.M. Predicting and managing primary and secondary non-response to rituximab using B-cell biomarkers in systemic lupus erythematosus. Ann. Rheum. Dis. 2017, 76, 1829–1836. [Google Scholar] [CrossRef]
- Collins, C.E.; Cortes-Hernández, J.; Garcia, M.A.; Von Kempis, J.; Schwarting, A.; Touma, Z.; Kurtinecz, M.; Gairy, K. Real-World Effectiveness of Belimumab in the Treatment of Systemic Lupus Erythematosus: Pooled Analysis of Multi-Country Data from the OBSErve Studies. Rheumatol. Ther. 2020, 7, 949–965. [Google Scholar] [CrossRef] [PubMed]
- Freitas, S.; Ruiz, M.M.; Carneiro, A.C.; Isenberg, D.A. Why do some patients with systemic lupus erythematosus fail to respond to B-cell depletion using rituximab? Clin. Exp. Rheumatol. 2020, 38, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, A.; Lukawska, J.; Cambridge, G.; Isenberg, D.; Leandro, M. Adverse infusion reactions to rituximab in systemic lupus erythematosus: A retrospective analysis. BMC Rheumatol. 2019, 3, 32. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Kostopoulou, M.; Alunno, A.; Aringer, M.; Bajema, I.; Boletis, J.N.; Cervera, R.; Doria, A.; Gordon, C.; Govoni, M.; et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 736–745. [Google Scholar] [CrossRef]
- Gordon, C.; Amissah-Arthur, M.-B.; Gayed, M.; Brown, S.; Bruce, I.N.; D’cruz, D.; Empson, B.; Griffiths, B.; Jayne, D.; Khamashta, M.; et al. The British Society for Rheumatology guideline for the management of systemic lupus erythematosus in adults. Rheumatology 2018, 57, e1–e45. [Google Scholar] [CrossRef]
- Ruiz-Irastorza, G.; Ramos-Casals, M.; Zeron, P.B.; Khamashta, M.A. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: A systematic review. Ann. Rheum. Dis. 2010, 69, 20–28. [Google Scholar] [CrossRef]
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kaplan, M.J. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat. Rev. Nephrol. 2016, 12, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.Y.H.; Chan, T.M. B Cell Abnormalities in Systemic Lupus Erythematosus and Lupus Nephritis—Role in Pathogenesis and Effect of Immunosuppressive Treatments. Int. J. Mol. Sci. 2019, 20, 6231. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R. Distinct B Cell Receptor Functions Are Determined by Phosphorylation. PLoS Biol. 2006, 4, e231. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef]
- Polyak, M.J.; Li, H.; Shariat, N.; Deans, J.P. CD20 Homo-oligomers Physically Associate with the B Cell Antigen Receptor. J. Biol. Chem. 2008, 283, 18545–18552. [Google Scholar] [CrossRef]
- E Mei, H.; Schmidt, S.; Dörner, T. Rationale of anti-CD19 immunotherapy: An option to target autoreactive plasma cells in autoimmunity. Arthritis Res. Ther. 2012, 14, S1. [Google Scholar] [CrossRef]
- O’Keefe, T.L.; Williams, G.T.; Batista, F.D.; Neuberger, M.S. Deficiency in CD22, a B Cell–specific Inhibitory Receptor, Is Sufficient to Predispose to Development of High Affinity Autoantibodies. J. Exp. Med. 1999, 189, 1307–1313. [Google Scholar] [CrossRef]
- Smith, K.G.C.; Clatworthy, M.R. FcγRIIB in autoimmunity and infection: Evolutionary and therapeutic implications. Nat. Rev. Immunol. 2010, 10, 328–343. [Google Scholar] [CrossRef]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef]
- Leandro, M.J.; Edwards, J.C.; Cambridge, G.; Ehrenstein, M.; Isenberg, D. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 2673–2677. [Google Scholar] [CrossRef] [PubMed]
- Cambridge, G.; Leandro, M.J.; Teodorescu, M.; Manson, J.; Rahman, A.; Isenberg, D.; Edwards, J.C. B cell depletion therapy in systemic lupus erythematosus: Effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum. 2006, 54, 3612–3622. [Google Scholar] [CrossRef] [PubMed]
- Khattri, S.; Zandman-Goddard, G.; Peeva, E. B-cell directed therapies in antiphospholipid antibody syndrome — New directions based on murine and human data. Autoimmun. Rev. 2012, 11, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-X.; Fan, Y.-T.; Peng, B.-W. Distinct mechanisms underlying therapeutic potentials of CD20 in neurological and neuromuscular disease. Pharmacol. Ther. 2022, 238, 108180. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.H.; Pourfathollah, A.A. CD40 and Tolerance Induction. Iran. J. Allergy Asthma Immunol. 2012, 11, 1–13. [Google Scholar]
- Qamar, N.; Fuleihan, R.L. The Hyper IgM Syndromes. Clin. Rev. Allergy Immunol. 2014, 46, 120–130. [Google Scholar] [CrossRef]
- Menard, L.C.; Habte, S.; Gonsiorek, W.; Lee, D.; Banas, D.; Holloway, D.A.; Manjarrez-Orduno, N.; Cunningham, M.; Stetsko, D.; Casano, F.; et al. B cells from African American lupus patients exhibit an activated phenotype. JCI Insight 2016, 1, e87310. [Google Scholar] [CrossRef]
- Duarte, J.H. ICOS sustains pathogenic T-cell survival in SLE mouse model. Nat. Rev. Rheumatol. 2015, 11, 260. [Google Scholar] [CrossRef]
- Thien, M.; Phan, T.; Gardam, S.; Amesbury, M.; Basten, A.; Mackay, F.; Brink, R. Excess BAFF Rescues Self-Reactive B Cells from Peripheral Deletion and Allows Them to Enter Forbidden Follicular and Marginal Zone Niches. Immunity 2004, 20, 785–798. [Google Scholar] [CrossRef]
- Carter, L.M.; Isenberg, D.; Ehrenstein, M.R. Elevated serum B-cell activating factor (BAFF / BLyS) is associated with rising anti-dsDNA antibody levels and flare following B-cell depletion therapy in systemic lupus erythematosus: BAFF and SLE Relapse Following Rituximab. Arthritis Rheum. 2013, 65, 2672–2679. [Google Scholar] [CrossRef]
- Michaelson, J.S.; Wisniacki, N.; Burkly, L.C.; Putterman, C. Role of TWEAK in Lupus Nephritis: A bench-to-bedside review. J. Autoimmun. 2012, 39, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Min, H.-K.; Kim, S.-M.; Park, J.-S.; Byun, J.-K.; Lee, J.; Kwok, S.-K.; Park, Y.-W.; Cho, M.-L.; Park, S.-H. Fn14-Fc suppresses germinal center formation and pathogenic B cells in a lupus mouse model via inhibition of the TWEAK/Fn14 Pathway. J. Transl. Med. 2016, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Parodis, I.; Zickert, A.; Sundelin, B.; Axelsson, M.; Gerhardsson, J.; Svenungsson, E.; Malmström, V.; Gunnarsson, I. Evaluation of B lymphocyte stimulator and a proliferation inducing ligand as candidate biomarkers in lupus nephritis based on clinical and histopathological outcome following induction therapy. Lupus Sci. Med. 2015, 2, e000061. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; Avery, L.; Menk, A.V.; Gordon, R.A.; Smita, S.; Kane, L.P.; Chikina, M.; Delgoffe, G.M.; Shlomchik, M.J. Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J. Clin. Investig. 2018, 128, 4884–4897. [Google Scholar] [CrossRef]
- Zhou, M.; Guo, C.; Li, X.; Huang, Y.; Li, M.; Zhang, T.; Zhao, S.; Wang, S.; Zhang, H.; Yang, N. JAK/STAT signaling controls the fate of CD8+CD103+ tissue-resident memory T cell in lupus nephritis. J. Autoimmun. 2020, 109, 102424. [Google Scholar] [CrossRef]
- Ripoll; de Ramon, L.; Draibe, J.; Merino, A.; Bolaños, N.; Goma, M.; Cruzado, J.M.; Grinyó, J.M.; Torras, J. JAK3-STAT pathway blocking benefits in experimental lupus nephritis. Arthritis Res. Ther. 2016, 18, 134. [Google Scholar] [CrossRef]
- Ittah, M.; Miceli-Richard, C.; Gottenberg, J.E.; Lavie, F.; Lazure, T.; Ba, N.; Sellam, J.; Lepajolec, C.; Mariette, X. B cell-activating factor of the tumor necrosis factor family (BAFF) is expressed under stimulation by interferon in salivary gland epithelial cells in primary Sjögren’s syndrome. Arthritis Res. Ther. 2006, 8, R51. [Google Scholar] [CrossRef]
- Crow, M.K. Type I Interferon in the Pathogenesis of Lupus. J. Immunol. 2014, 192, 5459–5468. [Google Scholar] [CrossRef]
- Schiller, M.; Parcina, M.; Heyder, P.; Foermer, S.; Ostrop, J.; Leo, A.; Heeg, K.; Herrmann, M.; Lorenz, H.-M.; Bekeredjian-Ding, I. Induction of Type I IFN Is a Physiological Immune Reaction to Apoptotic Cell-Derived Membrane Microparticles. J. Immunol. 2012, 189, 1747–1756. [Google Scholar] [CrossRef]
- Higgs, B.W.; Liu, Z.; White, B.; Zhu, W.; I White, W.; Morehouse, C.; Brohawn, P.; A Kiener, P.; Richman, L.; Fiorentino, D.; et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann. Rheum. Dis. 2011, 70, 2029–2036. [Google Scholar] [CrossRef]
- Oke, V.; Gunnarsson, I.; Dorschner, J.; Eketjäll, S.; Zickert, A.; Niewold, T.B.; Svenungsson, E. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res. Ther. 2019, 21, 107. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.C.W.; Cambridge, G. Sustained improvement in rheumatoid arthritis following a protocol designed to deplete B lymphocytes. Rheumatology 2001, 40, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Krenn, V.; Souto-Carneiro, M.M.; Kim, H.J.; Berek, C.; Starostik, P.; König, A.; Harms, H.; Müller-Hermelink, H.K. Histopathology and molecular pathology of synovial B-lymphocytes in rheumatoid arthritis. Histol. Histopathol. 2000, 15, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yu, Y.; Gao, Y.; Zhao, F.; Liang, Z.; Gao, J. Comparative Effectiveness of Rituximab and Common Induction Therapies for Lupus Nephritis: A Systematic Review and Network Meta-Analysis. Front. Immunol. 2022, 13, 859380. [Google Scholar] [CrossRef] [PubMed]
- Cobo-Ibáñez, T.; Loza-Santamaría, E.; Pego-Reigosa, J.M.; Marqués, A.O.; Rúa-Figueroa; Fernandez-Nebro, A.; Cáliz, R.C.; Longo, F.J.L.; Muñoz-Fernández, S. Efficacy and safety of rituximab in the treatment of non-renal systemic lupus erythematosus: A systematic review. Semin. Arthritis Rheum. 2014, 44, 175–185. [Google Scholar] [CrossRef]
- Pongtarakulpanit, N.; Pisitkun, P.; Ngamjanyaporn, P. Efficacy and safety of rituximab biosimilar in refractory lupus. Lupus Sci. Med. 2020, 7, e000442. [Google Scholar] [CrossRef]
- da Costa, R.Q.; Aguirre-Alastuey, M.E.; Isenberg, D.A.; Saracino, A.M. Assessment of Response to B-Cell Depletion Using Rituximab in Cutaneous Lupus Erythematosus. JAMA Dermatol. 2018, 154, 1432–1440. [Google Scholar] [CrossRef]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.-J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase ii/iii systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The lupus nephritis assessment with rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef]
- Ramos, L.; Isenberg, D. Rituximab: The Lupus Journey. Curr. Treat. Options Rheumatol. 2015, 1, 30–41. [Google Scholar] [CrossRef]
- Ehrenstein, M.; Wing, M.R.E.C. The BAFFling effects of rituximab in lupus: Danger ahead? Nat. Rev. Rheumatol. 2016, 12, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Fanouriakis, A.; Kostopoulou, M.; Cheema, K.; Anders, H.-J.; Aringer, M.; Bajema, I.; Boletis, J.; Frangou, E.; A Houssiau, F.; Hollis, J.; et al. 2019 Update of the Joint European League Against Rheumatism and European Renal Association–European Dialysis and Transplant Association (EULAR/ERA–EDTA) recommendations for the management of lupus nephritis. Ann. Rheum. Dis. 2020, 79, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, X.; Xue, H.; Lv, H.; Yu, L.; Wu, X.; Wang, Q.; Wu, H.; Han, F.; Xue, J. Rituximab as maintenance therapy following remission induction in relapsing or refractory systemic lupus erythematosus. Rheumatology 2023, 62, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Kerlavage, A.R.; Fleischmann, R.D.; A Fuldner, R.; Bult, C.J.; Lee, N.H.; Kirkness, E.F.; Weinstock, K.G.; Gocayne, J.D.; White, O. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature 1995, 377, 3–174. [Google Scholar] [PubMed]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J. Mice Transgenic for Baff Develop Lymphocytic Disorders along with Autoimmune Manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef] [PubMed]
- Cheema, G.S.; Roschke, V.; Hilbert, D.M.; Stohl, W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001, 44, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Stohl, W.; Hilbert, D.M. The discovery and development of belimumab: The anti-BLyS–lupus connection. Nat. Biotechnol. 2012, 30, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Stohl, W.; Furie, R.A.; Lisse, J.R.; Mckay, J.; Merrill, J.T.; Petri, M.A.; Ginzler, E.M.; Chatham, W.W.; McCune, W.J.; et al. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum. 2009, 61, 1168–1178. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzmán, R.M.; E Gallacher, A.; Hall, S.; A Levy, R.; E Jimenez, R.; Li, E.K.-M.; Thomas, M.; Kim, H.-Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Zhang, F.; Bae, S.-C.; Bass, D.; Chu, M.; Egginton, S.; Gordon, D.; A Roth, D.; Zheng, J.; Tanaka, Y. A pivotal phase III, randomised, placebo-controlled study of belimumab in patients with systemic lupus erythematosus located in China, Japan and South Korea. Ann. Rheum. Dis. 2018, 77, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Ginzler, E.; Barbosa, L.S.G.; D’Cruz, D.; Furie, R.; Maksimowicz-McKinnon, K.; Oates, J.; Santiago, M.B.; Saxena, A.; Sheikh, S.; Bass, D.L.; et al. Phase III/IV, Randomized, Fifty-Two –Week Study of the Efficacy and Safety of Belimumab in Patients of Black African Ancestry With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2022, 74, 112–123. [Google Scholar] [CrossRef] [PubMed]
- I Brunner, H.; Abud-Mendoza, C.; O Viola, D.; Penades, I.C.; Levy, D.; Anton, J.; E Calderon, J.; Chasnyk, V.G.; A Ferrandiz, M.; Keltsev, V.; et al. Safety and efficacy of intravenous belimumab in children with systemic lupus erythematosus: Results from a randomised, placebo-controlled trial. Ann. Rheum. Dis. 2020, 79, 1340–1348. [Google Scholar] [CrossRef]
- Stohl, W.; Schwarting, A.; Okada, M.; Scheinberg, M.; Doria, A.; Hammer, A.E.; Kleoudis, C.; Groark, J.; Bass, D.; Fox, N.L.; et al. Efficacy and Safety of Subcutaneous Belimumab in Systemic Lupus Erythematosus: A Fifty-Two–Week Randomized, Double-Blind, Placebo-Controlled Study. Arthritis Rheumatol. 2017, 69, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Dooley, M.; Houssiau, F.; Aranow, C.; D’cruz, D.; Askanase, A.; Roth, D.; Zhong, Z.; Cooper, S.; Freimuth, W.; Ginzler, E. Effect of belimumab treatment on renal outcomes: Results from the phase 3 belimumab clinical trials in patients with SLE. Lupus 2013, 22, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Rovin, B.H.; Houssiau, F.; Malvar, A.; Teng, Y.O.; Contreras, G.; Amoura, Z.; Yu, X.; Mok, C.-C.; Santiago, M.B.; et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N. Engl. J. Med. 2020, 383, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Parodis, I.; Vital, E.M.; Hassan, S.-U.; Jönsen, A.; A Bengtsson, A.; Eriksson, P.; Leonard, D.; Gunnarsson, I.; Rönnblom, L.; Sjöwall, C. De novo lupus nephritis during treatment with belimumab. Rheumatology 2020, 60, 4348–4354. [Google Scholar] [CrossRef]
- Sjöwall, C.; Cöster, L. Belimumab may not prevent lupus nephritis in serologically active patients with ongoing non-renal disease activity. Scand. J. Rheumatol. 2014, 43, 428–430. [Google Scholar] [CrossRef]
- Staveri, C.; Karokis, D.; Liossis, S.-N.C. New onset of lupus nephritis in two patients with SLE shortly after initiation of treatment with belimumab. Semin. Arthritis Rheum. 2017, 46, 788–790. [Google Scholar] [CrossRef]
- Sheikh, S.Z.; A Scheinberg, M.; Wei, J.C.-C.; Tegzova, D.; Stohl, W.; de Toledo, R.A.; Mucenic, T.; Banfi, M.R.A.; Maksimowicz-McKinnon, K.; Abud-Mendoza, C.; et al. Mortality and adverse events of special interest with intravenous belimumab for adults with active, autoantibody-positive systemic lupus erythematosus (BASE): A multicentre, double-blind, randomised, placebo-controlled, phase 4 trial. Lancet Rheumatol. 2021, 3, e122–e130. [Google Scholar] [CrossRef]
- Xie, W.; Huang, H.; Zhan, S.; Zhang, Z. Risk of psychiatric disorders and all-cause mortality with belimumab therapy in patients with systemic lupus erythematosus: A meta-analysis of randomised controlled trials. Lupus Sci. Med. 2021, 8, e000534. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, M.; Tummala, R.; Streicher, K.; da Costa, A.N.; Brohawn, P.Z. The Pathogenesis, Molecular Mechanisms, and Therapeutic Potential of the Interferon Pathway in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 11286. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef] [PubMed]
- A Furie, R.; Morand, E.F.; Bruce, I.N.; Manzi, S.; Kalunian, K.C.; Vital, E.M.; Ford, T.L.; Gupta, R.; Hiepe, F.; Santiago, M.; et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): A randomised, controlled, phase 3 trial. Lancet Rheumatol. 2019, 1, e208–e219. [Google Scholar] [CrossRef]
- Bruce, I.N.; A Furie, R.; Morand, E.F.; Manzi, S.; Tanaka, Y.; Kalunian, K.C.; Merrill, J.T.; Puzio, P.; Maho, E.; Kleoudis, C.; et al. Concordance and discordance in SLE clinical trial outcome measures: Analysis of three anifrolumab phase 2/3 trials. Ann. Rheum. Dis. 2022, 81, 962–969. [Google Scholar] [CrossRef]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.-C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Mössner, E.; Brünker, P.; Moser, S.; Püntener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; van Puijenbroek, E.; et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell–mediated B-cell cytotoxicity. Blood 2010, 115, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- A Furie, R.; Aroca, G.; Cascino, M.D.; Garg, J.P.; Rovin, B.H.; Alvarez, A.; Fragoso-Loyo, H.; Zuta-Santillan, E.; Schindler, T.; Brunetta, P.; et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: A randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2022, 81, 100–107. [Google Scholar] [CrossRef]
- Robles-Carrillo, L.; Meyer, T.; Hatfield, M.; Desai, H.; Dávila, M.; Langer, F.; Amaya, M.; Garber, E.; Francis, J.L.; Hsu, Y.-M.; et al. Anti-CD40L Immune Complexes Potently Activate Platelets In Vitro and Cause Thrombosis in FCGR2A Transgenic Mice. J. Immunol. 2010, 185, 1577–1583. [Google Scholar] [CrossRef]
- Tocoian, A.; Buchan, P.; Kirby, H.; Soranson, J.; Zamacona, M.; Walley, R.; Mitchell, N.; Esfandiari, E.; Wagner, F.; Oliver, R. First-in-human trial of the safety, pharmacokinetics and immunogenicity of a PEGylated anti-CD40L antibody fragment (CDP7657) in healthy individuals and patients with systemic lupus erythematosus. Lupus 2015, 24, 1045–1056. [Google Scholar] [CrossRef]
- Chamberlain, C.; Colman, P.J.; Ranger, A.M.; Burkly, L.C.; I Johnston, G.; Otoul, C.; Stach, C.; Zamacona, M.; Dörner, T.; Urowitz, M.; et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann. Rheum. Dis. 2017, 76, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- A Furie, R.; Bruce, I.N.; Dörner, T.; Leon, M.G.; Leszczyński, P.; Urowitz, M.; Haier, B.; Jimenez, T.; Brittain, C.; Liu, J.; et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology 2021, 60, 5397–5407. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Deucravacitinib: First Approval. Drugs 2022, 82, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.; Pike, M.; Merrill, J.T.; van Vollenhoven, R.; Werth, V.P.; Hobar, C.; Delev, N.; Shah, V.; Sharkey, B.; Wegman, T.; et al. Deucravacitinib, a Tyrosine Kinase 2 Inhibitor, in Systemic Lupus Erythematosus: A Phase II, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2023, 75, 242–252. [Google Scholar] [CrossRef]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef]
- Dzionek, A.; Fuchs, A.; Schmidt, P.; Cremer, S.; Zysk, M.; Miltenyi, S.; Buck, D.W.; Schmitz, J. BDCA-2, BDCA-3, and BDCA-4: Three Markers for Distinct Subsets of Dendritic Cells in Human Peripheral Blood. J. Immunol. 2000, 165, 6037–6046. [Google Scholar] [CrossRef]
- Blomberg, S.; Eloranta, M.-L.; Magnusson, M.; Alm, G.V.; Rönnblom, L. Expression of the markers BDCA-2 and BDCA-4 and production of interferon-α by plasmacytoid dendritic cells in systemic lupus erythematosus: BDCA-2/4 Expression and IFNα Production in SLE. Arthritis Rheum. 2003, 48, 2524–2532. [Google Scholar] [CrossRef]
- Furie, R.A.; van Vollenhoven, R.F.; Kalunian, K.; Navarra, S.; Romero-Diaz, J.; Werth, V.P.; Huang, X.; Clark, G.; Carroll, H.; Meyers, A.; et al. Trial of Anti-BDCA2 Antibody Litifilimab for Systemic Lupus Erythematosus. N. Engl. J. Med. 2022, 387, 894–904. [Google Scholar] [CrossRef]
- Kaegi, C.; Steiner, U.C.; Wuest, B.; Crowley, C.; Boyman, O. Systematic Review of Safety and Efficacy of Atacicept in Treating Immune-Mediated Disorders. Front. Immunol. 2020, 11, 433. [Google Scholar] [CrossRef]
- Carter, R.H.; Zhao, H.; Liu, X.; Pelletier, M.; Chatham, W.; Kimberly, R.; Zhou, T. Expression and occupancy of BAFF-R on B cells in systemic lupus erythematosus. Arthritis Rheum. 2005, 52, 3943–3954. [Google Scholar] [CrossRef]
- Isenberg, D.; Gordon, C.; Licu, D.; Copt, S.; Rossi, C.P.; Wofsy, D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann. Rheum. Dis. 2014, 74, 2006–2015. [Google Scholar] [CrossRef]
- Merrill, J.T.; Wallace, D.J.; Wax, S.; Kao, A.; Fraser, P.A.; Chang, P.; Isenberg, D.; on behalf of the ADDRESS II Investigators. Efficacy and Safety of Atacicept in Patients with Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 70, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Ginzler, E.M.; Wax, S.; Rajeswaran, A.; Copt, S.; Hillson, J.; Ramos, E.; Singer, N.G. Atacicept in combination with MMF and corticosteroids in lupus nephritis: Results of a prematurely terminated trial. Arthritis Res. Ther. 2012, 14, R33. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Teng, Y.K.O.; Ginzler, E.M.; Arriens, C.; Caster, D.J.; Romero-Diaz, J.; Gibson, K.; Kaplan, J.; Lisk, L.; Navarra, S.; et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): A double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2021, 397, 2070–2080. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; A Isenberg, D.; Morand, E.F.; Vazquez–Mateo, C.; Kao, A.H.; Aydemir, A.; Pudota, K.; Ona, V.; Aranow, C.; Merrill, J.T. Safety and clinical activity of atacicept in the long-term extension of the phase 2b ADDRESS II study in systemic lupus erythematosus. Rheumatology 2021, 60, 5379–5389. [Google Scholar] [CrossRef]
- Shi, F.; Xue, R.; Zhou, X.; Shen, P.; Wang, S.; Yang, Y. Telitacicept as a BLyS/APRIL dual inhibitor for autoimmune disease. Immunopharmacol. Immunotoxicol. 2021, 43, 666–673. [Google Scholar] [CrossRef]
- Dhillon, S. Telitacicept: First Approval. Drugs 2021, 81, 1671–1675. [Google Scholar] [CrossRef]
- A Human Recombinant Fusion Protein Targeting B Lymphocyte Stimulator (BlyS) and a Proliferation-Inducing Ligand (APRIL), Telitacicept (RC18), in Systemic Lupus Erythematosus (SLE): Results of a Phase 2b Study. ACR Meeting Abstracts n.d. Available online: https://acrabstracts.org/abstract/a-human-recombinant-fusion-protein-targeting-b-lymphocyte-stimulator-blys-and-a-proliferation-inducing-ligand-april-telitacicept-rc18-in-systemic-lupus-erythematosus-sle-results-of-a-phase/ (accessed on 25 March 2023).
- Telitacicept, a Human Recombinant Fusion Protein Targeting B Lymphocyte Stimulator (BlyS) and a Proliferation-Inducing Ligand (APRIL), in Systemic Lupus Erythematosus (SLE): Results of a Phase 3 Study. ACR Meeting Abstracts n.d. Available online: https://acrabstracts.org/abstract/telitacicept-a-human-recombinant-fusion-protein-targeting-b-lymphocyte-stimulator-blys-and-a-proliferation-inducing-ligand-april-in-systemic-lupus-erythematosus-sle-results-of-a-phase-3-study/ (accessed on 25 March 2023).
- Chen, R.; Fu, R.; Lin, Z.; Huang, C.; Huang, W. The efficacy and safety of telitacicept for the treatment of systemic lupus erythematosus: A real life observational study. Lupus 2023, 32, 94–100. [Google Scholar] [CrossRef]
- Merrill, J.T.; Van Vollenhoven, R.F.; Buyon, J.; A Furie, R.; Stohl, W.; Morgan-Cox, M.; Dickson, C.; Anderson, P.W.; Lee, C.; Berclaz, P.-Y.; et al. Efficacy and safety of subcutaneous tabalumab, a monoclonal antibody to B-cell activating factor, in patients with systemic lupus erythematosus: Results from ILLUMINATE-2, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 332–340. [Google Scholar] [CrossRef]
- A Isenberg, D.; Petri, M.; Kalunian, K.C.; Tanaka, Y.; Urowitz, M.B.; Hoffman, R.W.; Morgan-Cox, M.; Iikuni, N.; Silk, M.; Wallace, D.J. Efficacy and safety of subcutaneous tabalumab in patients with systemic lupus erythematosus: Results from ILLUMINATE-1, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 323–331. [Google Scholar] [CrossRef]
- A Furie, R.; Leon, G.; Thomas, M.; A Petri, M.; Chu, A.D.; Hislop, C.; Martin, R.S.; A Scheinberg, M. A phase 2, randomised, placebo-controlled clinical trial of blisibimod, an inhibitor of B cell activating factor, in patients with moderate-to-severe systemic lupus erythematosus, the PEARL-SC study. Ann. Rheum. Dis. 2015, 74, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Kalunian, K.; A Petri, M.; Strand, V.; A Houssiau, F.; Pike, M.; Kilgallen, B.; Bongardt, S.; Barry, A.; Kelley, L.; et al. Efficacy and safety of epratuzumab in patients with moderate/severe active systemic lupus erythematosus: Results from EMBLEM, a phase IIb, randomised, double-blind, placebo-controlled, multicentre study. Ann. Rheum. Dis. 2014, 73, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Shanahan, W.R.; Scheinberg, M.; Kalunian, K.C.; Wofsy, D.; Martin, R.S. Phase III trial results with blisibimod, a selective inhibitor of B-cell activating factor, in subjects with systemic lupus erythematosus (SLE): Results from a randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2018, 77, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Clowse, M.E.B.; Wallace, D.J.; Furie, R.A.; Petri, M.A.; Pike, M.C.; Leszczyński, P.; Neuwelt, C.M.; Hobbs, K.; Keiserman, M.; Duca, L.; et al. Efficacy and Safety of Epratuzumab in Moderately to Severely Active Systemic Lupus Erythematosus: Results from Two Phase III Randomized, Double-Blind, Placebo-Controlled Trials. Arthritis Rheumatol. 2017, 69, 362–375. [Google Scholar] [CrossRef]
- Kraaij, T.; Kamerling, S.W.; de Rooij, E.N.; van Daele, P.L.; Bredewold, O.W.; Bakker, J.A.; Bajema, I.M.; Scherer, H.U.; Toes, R.E.; Huizinga, T.J.; et al. The NET-effect of combining rituximab with belimumab in severe systemic lupus erythematosus. J. Autoimmun. 2018, 91, 45–54. [Google Scholar] [CrossRef]
- Atisha-Fregoso, Y.; Malkiel, S.; Harris, K.M.; Byron, M.; Ding, L.; Kanaparthi, S.; Barry, W.T.; Gao, W.; Ryker, K.; Tosta, P.; et al. Phase II Randomized Trial of Rituximab Plus Cyclophosphamide Followed by Belimumab for the Treatment of Lupus Nephritis. Arthritis Rheumatol. 2021, 73, 121–131. [Google Scholar] [CrossRef]
- Shipa, M.M.; Embleton-Thirsk, A.; Parvaz, M.M.; Santos, L.R.; Muller, M.P.; Chowdhury, M.K.; Isenberg, D.A.; Doré, B.C.J.; Gordon, M.C.; Ehrenstein, M.M.R.; et al. Effectiveness of Belimumab After Rituximab in Systemic Lupus Erythematosus: A Randomized Controlled Trial. Ann. Intern. Med. 2021, 174, 1647–1657. [Google Scholar] [CrossRef]
- Aranow, C. Efficacy and Safety of Subcutaneous Belimumab (BEL) and Rituximab (RTX) Sequential Therapy in Patients with Systemic Lupus Erythematosus: The Phase 3, Randomized, Placebo-Controlled BLISS-BELIEVE Study. ACR Meeting Abstracts n.d. Available online: https://acrabstracts.org/abstract/efficacy-and-safety-of-subcutaneous-belimumab-bel-and-rituximab-rtx-sequential-therapy-in-patients-with-systemic-lupus-erythematosus-the-phase-3-randomized-placebo-controlled-bliss-believe-stud/ (accessed on 20 March 2023).
- Lim, S.M.; Pyo, K.-H.; A Soo, R.; Cho, B.C. The promise of bispecific antibodies: Clinical applications and challenges. Cancer Treat. Rev. 2021, 99, 102240. [Google Scholar] [CrossRef]
- Kang, C. Mosunetuzumab: First Approval. Drugs 2022, 82, 1229–1234. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Torres-Moya, R.; Reyes, G.; Molinero, C.; Prada, D.; Lopez, A.M.; Hernandez, I.M.; Hernandez, M.V.; Martinez, J.P.; Hernandez, X.; et al. A clinical exploratory study with itolizumab, an anti-CD6 monoclonal antibody, in patients with rheumatoid arthritis. Results Immunol. 2012, 2, 204–211. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Costa, F.; Giuliani, N.; Pistoia, V.; Malavasi, F. CD38: A Target for Immunotherapeutic Approaches in Multiple Myeloma. Front. Immunol. 2018, 9, 2722. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Peng, Y.; Li, H.; Zhang, X. From monogenic lupus to TLR7/MyD88-targeted therapy. Innovation 2022, 3, 100299. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.; Bassi, R.; Chang, P.; Kao, A.; Jayne, D.; Wofsy, D.; Fleuranceau-Morel, P. Integrated safety profile of atacicept: An analysis of pooled data from the atacicept clinical trial programme. Rheumatol. Adv. Pr. 2019, 3, rkz021. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Obinutuzumab | |
|---|---|
| Clinical trial name | NOBILITY (NCT02550652) |
| Primary endpoint What was it? Was it achieved? | CRR at 52 W CRR, n (%) OBI vs. PBO: 22 (35) vs. 14 (23), p = 0.115 (statistically significant, alpha level 0.2) |
| Eligibility criteria | -SLE (ACR classification criteria—1997) -Class III or IV LN -UPCR >1 -eGFR ≥30 mL/min/1.73 m2 -18–75 years |
| Treatment scheme | IV OBI 1000 mg or IV PBO on day 1, W2, W24, W26 + MMF + GC |
| Number of patients who received the treatment | 63 (OBI) vs. 62 (PBO) |
| Study duration | 104 weeks |
| Key secondary endpoints | Clinical -ORR at 52 W, n (%) OBI vs. PBO: 35 (56) vs. 22 (36), p = 0.025 -ORR at 104 W, n (%) OBI vs. PBO: 34 (54) vs. 18 (29), p = 0.005 -CRR at 104 W in patients with class IV LN, n (%) OBI vs. PBO: 23 (47) vs. 7 (16), p = 0.001 -CRR at 104 W in patients with BL UPCR ≥3, n (%) OBI vs. PBO: 8 (31) vs. 2 (10), p = 0.098 Laboratory -Change in C3 from BL at 52 W, mean (SE) OBI vs. PBO: 30 (3.4) vs. 12 (3.5), p < 0.001 -Change in C3 from BL at 104 W, mean (SE) OBI vs. PBO: 29 (3.4) vs. 11 (3.4), p < 0.001 -Change in C4 from BL at 52 W, mean (SE) OBI vs. PBO: 9.7 (1.3) vs. 0.8 (1.3), p < 0.001 -Change in C4 from BL at 104 W, mean (SE) OBI vs. PBO: 9.6 (1.3) vs. 0.4 (1.3), p < 0.001 -Change in log anti-dsDNA titre from BL at 52 W, mean (SE) OBI vs. PBO: −0.91 (0.12) vs. −0.10 (0.12), p < 0.001 -Change in log anti-dsDNA titre from BL at 104 W, mean (SE) OBI vs. PBO: −1.1 (0.13) vs. −0.05 (0.13), p < 0.001 |
| Safety | Any AE, n (%) OBI vs. PBO: 58 (91) vs. 54 (89) Serious infectious AE, n (%) OBI vs. PBO: 5 (8) vs. 11 (18) Infusion-related reactions, n (%) OBI vs. PBO: 10 (16) vs. 6 (10) |
| Dapirolizumab | |
|---|---|
| Clinical trial name and/or identification | RISE (NCT02804763) |
| Primary endpoint What was it? Was it achieved? | Identifying a dose-response relationship. Responses defined by BICLA at W24. Not met (p = 0.07) |
| Eligibility criteria | -SLE (SLICC classification criteria) -Positive anti-dsDNA antibodies and/or low complement and/or an ANA titre 1:80, in combination with historical positivity for anti-dsDNA and/or positivity for anti-ENA [anti-Sm, anti-SSA, anti-SSB or anti-RNP] -Moderate to severe disease activity: ≥1 BILAG A or ≥2 BILAG B domains; SLEDAI-2K ≥ 6, and a SLEDAI-2K ≥4 excluding points from laboratory values 4 -≥18 years |
| Key exclusion criteria | -class III or IV LN -severe neuropsychiatric SLE -history of thromboembolic events within 12 months of screening |
| Treatment scheme | DZP IV or PBO IV every 4 weeks up to W20 |
| Number of patients who received the treatment | 45 PBO, 45 DZP 6 mg/kg, 45 DZP 24 mg/kg, 47 DZP 45 mg/kg |
| Study duration | 48 W (24 W treatment period + 24 W observational period) |
| Key secondary endpoints | Clinical * -BILAG 2004 W24 (mean change from BL PBO, DZP 6 mg/kg, DZP 24 mg/kg, DZP 45 mg/kg): −10.5, −10.9, −14.0, −12.0 (p <0.05) -BILAG 2004 W48 (mean change from BL PBO, DZP 6 mg/kg, DZP 24 mg/kg, DZP 45 mg/kg): −10.6, −11.5, −12.9, −12.7 (p <0.05) |
| Safety | Any TEAE, n (%) (PBO, DZP 6 mg/kg, DZP 24 mg/kg, DZP 45 mg/kg): 28 (62.2), 29 (64.4), 35 (77.8), 34 (72.3) Serious TEAEs, n (%) (PBO, DZP 6 mg/kg, DZP 24 mg/kg, DZP 45 mg/kg): 5 (11.1), 2 (4.4), 4 (8.9), 5 (10.6) Thromboembolic events, n (%) (PBO, DZP 6 mg/kg, DZP 24 mg/kg, DZP 45 mg/kg): 3 (6.7), 0, 1 (2.2), 0 |
| Deucravacitinib | |
|---|---|
| Clinical trial name and/or identification | PAISLEY (NCT03252587) |
| Primary endpoint What was it? Was it achieved? | SRI-4 response at W32 SRI-4 responses, n (%), PBO vs. DEU 3 mg BID: 31 (34.4) vs. 53 (58.2), p <0.001 SRI-4 responses, n (%), PBO vs. DEU 6 mg BID: 31 (34.4) vs. 46 (49.5), p = 0.02 SRI-4 responses, n (%), PBO vs. DEU 12 mg QD: 31 (34.4) vs. 40 (44.9), p = 0.08 |
| Eligibility criteria | -SLE (SLICC classification criteria) -≥1 positive test for ANA, anti-dsDNA Ab, anti-Sm -≥1 BILAG A or ≥2 BILAG B from the musculoskeletal or mucocutaneous domain -SLEDAI-2K ≥6 -18–75 years -taking ≥1 antimalarial or immunosuppressant |
| Key exclusion criteria | -active, severe LN -active neuropsychiatric SLE -history of herpes zoster, herpes simplex, or influenza infection within 12 weeks before randomization -history of disseminated or complicated herpes zoster infection |
| Treatment scheme | 4 arms -PBO + SOC -DEU 3 mg BID for 48 W + SOC -DEU 6 mg BID for 48 W + SOC -DEU 12 mg QD for 48 W + SOC |
| Number of patients who received the treatment | 90 PBO, 91 DEU 3 mg BID, 93 DEU 6 mg BID, 89 DEU 12 mg QD |
| Study duration | 48 W |
| Key secondary endpoints | Clinical -SRI-4 responders 48 W, n (%) PBO vs. DEU 3 mg BID: 31 (34.4) vs. 52 (57.1), p <0.001 -BICLA responders 48 W, n (%) PBO vs. DEU 3 mg BID: 23 (25.6) vs. 43 (47.3), p = 0.001 -LLDAS responders 48 W, n (%) PBO vs. DEU 3 mg BID: 12 (13.3) vs. 33 (36.3), p <0.001 -CLASI-50 responders 48 W, n (%) PBO vs. DEU 3 mg BID: 4 (16.7) vs. 16 (69.6), p <0.001 -Mean change from BL in the joint count, n (%) PBO vs. DEU 3 mg BID: −7.6 vs. −8.9, p = 0.001 |
| Safety | Any AE, n (%) PBO, DEU 3 mg BID, DEU 6 mg BID, DEU 12 mg QD: 79 (87.8), 85 (93.4), 81 (87.1), 75 (84.3) SAEs, n (%) PBO, DEU 3 mg BID, DEU 6 mg BID, DEU 12 mg QD: 11 (12.2), 7 (7.7), 8 (8.6), 7 (7.9) Herpes zoster, n (%) PBO, DEU 3 mg BID, DEU 6 mg BID, DEU 12 mg QD: 4 (4.4), 3 (3.3), 3 (3.2), 2 (2.2) Cutaneous AEs, % PBO, DEU 3 mg BID, DEU 6 mg BID, DEU 12 mg QD: 13.3, 16.5, 34.4, 33.7 |
| Litifilimab | |
|---|---|
| Clinical trial name and/or identification | LILIAC (NCT02847598) |
| Primary endpoint What was it? Was it achieved? | Control of joint involvement at W24 LSM (±SE) absolute changes from BL in the total number of active joints litifilimab vs. PBO: −15.0 ± 1.2 vs. −11.6 ± 1.3. LSM difference litifilimab vs. PBO: −3.4 (p = 0.04) |
| Eligibility criteria | -SLE (ACR classification criteria 1997) -ANA ≥ 1:80 and/or anti-dsDNA Ab ≥ 30 UI/mL -CLASI-A ≥ 8 (protocol version 1) -≥1 active skin lesion (protocol version 2, no requirements for CLASI-A) -≥4 tender and swollen joints (protocol version 2) -SLEDAI-2K ≥ 4 -18–75 years |
| Key exclusion criteria | -active LN -active neuropsychiatric SLE |
| Treatment scheme | PBO sc or litifilimab 450 mg sc at W 0, 2, 4, 8, 12, 16, 20 + SOC |
| Number of patients who received the treatment | 56 PBO, 64 litifilimab 450 mg |
| Study duration | 24 W treatment period + 12W observation period |
| Key secondary endpoints | Clinical -Decrease of ≥7 points in CLASI-A at W24, LSM difference (95% CI) litifilimab vs. PBO: 21.6 (0.1 to 43.1) -SRI-4 responders at W24, LSM difference (95% CI) litifilimab vs. PBO: 26.4 (9.5 to 43.2) -Change in SLEDAI-2K at W24, LSM difference (95% CI) litifilimab vs. PBO: –1.7 (–3.0 to –0.5) |
| Safety | Any AE, n (%) pooled litifilimab vs. PBO: 45 (59) vs. 38 (68) Serious AEs, n (%) pooled litifilimab vs. PBO: 4 (5) vs. 6 (11) |
| Atacicept | ||
|---|---|---|
| Clinical trial name and/or identification | APRIL-SLE (NCT00624338) | ADDRESS II (NCT01972568) |
| Primary endpoint What was it? Was it achieved? | The proportion of BILAG A or B flares Not met. Flare rate atacicept 75 mg vs. PBO: 58% vs. 54% (p = 0.543) PBO vs. atacicept 150 mg (post-hoc analysis): Flare rate: 37% vs. 54% (p = 0.002) | SRI-4 responders at 24 W Not met. Atacicept 75 mg vs. PBO: 57.8% vs. 44.0% (p = 0.045) Atacicept 150 mg vs. PBO: 53.8% vs. 44.0% (p =0.121) Using day 1 as BL (sensitivity analysis): p < 0.05 for SRI-4 of both atacicept doses vs. PBO Serologically active patients: -SRI-4 responses atacicept 75 mg vs. PBO: 62.1% vs. 24.1% (p = 0.003) -SRI-4 responses atacicept 150 mg vs. PBO: 61.5% VS. 24.1% (p = 0.002) HDA at BL: SRI-4 responses atacicept 150 mg vs. PBO: 62.7% vs. 42.3% (p = 0.029) |
| Eligibility criteria | -≥4/11 ACR classification criteria (1997) -ANA ≥1:80 and/or anti-dsDNA Ab ≥30 IU/mL -active SLE (BILAG A or B that led to a change in steroid dose) -SLEDAI-2K ≥ 6 -≥16 years | -≥4/11 ACR classification criteria (1997) -ANA ≥1:80 and/or anti-dsDNA Ab ≥30 IU/mL -≥1 BILAG A or ≥2 BILAG B from the -SLEDAI-2K ≥ 6 -disease duration ≥ 6 months -≥18 years |
| Key exclusion criteria | -therapy with CYC, MMF, CNIs, LEF, 6-MP, or thalidomide within 3 months of the screening -severe CNS lupus -active moderate-to-severe GN | -CYC within 3 months of the screening -severe CNS lupus -active severe GN |
| Treatment scheme | 3 arms: -PBO + SOC -Atacicept 75 mg sc BIW for 4 weeks, then QW + SOC -Atacicept 150 mg sc BIW for 4 weeks, then QW + SOC (the enrollment in this group was terminated prematurely) | 3 arms: -PBO + SOC -Atacicept 75 mg sc QW + SOC -Atacicept 150 mg sc QW + SOC |
| Number of patients who received the treatment | 157 PBO, 159 atacicept 75 mg, 145 atacicept 150 mg | 100 PBO, 102 atacicept 75 mg, 104 atacicept 150 mg |
| Study duration | 52 W | 24 W treatment period + 24 W safety follow-up period |
| Key secondary endpoints | Clinical -Time to first flare atacicept 75 mg vs. PBO (ITT analysis set), HR (95% CI): 0.98 (0.69 to 1.40), p = 0.929 -Time to first flare atacicept 75 mg vs. PBO (PC analysis set), HR (95% CI): 0.83 (0.53 to 1.29), p = 0.404 PBO vs. atacicept 150 mg (post-hoc analysis): -Time to first flare atacicept 150 mg vs. PBO (ITT analysis set), HR (95% CI): 0.56 (0.36 to 0.87), p = 0.009 -Time to first flare atacicept 150 mg vs. PBO (PC analysis set), HR (95% CI): 0.41 (0.24 to 0.70), p = 0.001 Immunological -Anti-dsDNA Ab change from BL, atacicept 150 mg, atacicept 75 mg, PBO: −38%, −31%, +14% -C3 change, LSM change atacicept 75 mg vs. PBO: 0.076 (p < 0.001) -C3 change, LSM change atacicept 150 mg vs. PBO: 0.138 (p < 0.001) -C4 change, LSM change atacicept 75 mg vs. PBO: 0.046 (p < 0.001) -C4 change, LSM change atacicept 150 mg vs. PBO: 0.066 (p < 0.001) | SRI-6 responders atacicept 150 mg vs. PBO: 54.9% vs. 28.8% (p = 0.005) -BILAG-A flares atacicept 75 mg vs. PBO (all pts): HR 0.24 (95% CI 0.07 to 0.87, p = 0.019) -BILAG-A flares atacicept 150 mg vs. PBO (all pts): HR 0.54 (95% CI 0.21 to 1.39, p = 0.198) -BILAG-A flares atacicept 75 mg vs. PBO (HDA): HR 0.08 (95% CI 0.01 to 0.59, p = 0.002) -BILAG-A flares atacicept 150 mg vs. PBO (HDA): HR 0.32 (95% CI 0.10 to 0.99, p = 0.038) Immunological: -Anti-dsDNA Ab median % change from BL atacicept 75 mg, 150 mg, PBO: −23.6%, −28.2%, +16.0% -C3 median % change from BL atacicept 75 mg, 150 mg, PBO: +5.3%, +22.1%, +1.5% -C4 median % change from BL atacicept 75 mg, 150 mg, PBO: +64.5%, +128.6%, 0 |
| Safety | Any AE (PBO, atacicept 75 mg, atacicept 150 mg): 79.9%, 83.4%, 83.3% Serious AEs (PBO, atacicept 75 mg, atacicept 150 mg): 17.5%, 19.1%, 16.0% | TEAEs (PBO, atacicept 75 mg, atacicept 150 mg): 72.0%, 81.4%, 80.8% Serious TEAEs (PBO, atacicept 75 mg, atacicept 150 mg): 12.0%, 5.8%, 8.8% |
| Telitacicept | |
|---|---|
| Clinical trial name and/or identification | NCT02885610 |
| Primary endpoint What was it? Was it achieved? | SRI-4 responders at 48 W -SRI-4 responders (%) telitacicept 80 mg vs. PBO: 71.0 vs. 33.9 (p < 0.0001) -SRI-4 responders (%) telitacicept 160 mg vs. PBO: 68.3 vs. 33.9 (p = 0.0001) -SRI-4 responders (%) telitacicept 240 mg vs. PBO: 75.8 vs. 33.9 (p <0.0001) |
| Eligibility criteria | -≥4/11 ACR classification criteria (1997) -SELENA-SLEDAI ≥ 8 -positive ANA and/or anti-dsDNA Ab -18–65 years |
| Key exclusion criteria | -severe LN -CNS involvement |
| Treatment scheme | 4 arms: -PBO + SOC -telitacicept 80 mg sc QW for 48 weeks + SOC -telitacicept 160 mg sc QW for 48 weeks + SOC -telitacicept 240 mg sc QW for 48 weeks + SOC |
| Number of patients who received the treatment | 62 telitacicept 80 mg, 63 telitacicept 160 mg, 62 telitacicept 240 mg, 62 PBO |
| Study duration | 48 W |
| Key secondary endpoints | Proportion of patients with ≥ 4 points reduction in SELENA-SLEDAI W48: -telitacicept 80 mg vs. PBO (%): 75.8 vs. 50.0 (p = 0.003) -telitacicept 160 mg vs. PBO (%): 77.8 vs. 50.0 (p = 0.001) -telitacicept 240 mg vs. PBO (%): 79.0 vs. 50.0 (p <0.001) Proportion of patients without worsening PGA W48: -telitacicept 80 mg vs. PBO (%): 96.8 vs. 75.8 (p <0.001) -telitacicept 160 mg vs. PBO (%): 92.1 vs. 75.8 (p = 0.013) -telitacicept 160 mg vs. PBO (%): 96.8 vs. 75.8 (p <0.001) |
| Safety | Any AE (%): 82.3 (PBO), 90.3 (telitacicept 80 mg), 92.1 (telitacicept 160 mg), 93.5 (telitacicept 240 mg) (p >0.05) SAEs (%): 16.1 (PBO), 12.9 (telitacicept 80 mg), 15.9 (telitacicept 160 mg), 12.9 (telitacicept 240 mg) (p >0.05) |
| Rituximab + Belimumab | |||
|---|---|---|---|
| Clinical trial name and/or identification | CALIBRATE (NCT02260934) | BEAT-LUPUS | Synbiose (NCT02284984) |
| Primary endpoint What was it? Was it achieved? | Safety: % of pts with AE (≥ grade 3) up to W48 23% RC vs. 9.5% RCB (p >0.05) | Serum IgG anti-dsDNA levels at W52 Anti-dsDNA levels, geometric mean (IU/mL) [95% CI], BEL vs. PBO: 47 [25–88] vs. 103 [49–213], p < 0.001 | Immunological effects of RTX + BEL at W24 -BlyS decreased to 0.15 ng/mL [0.05–0.4], p < 0.01 -anti-dsDNA Ab decreased to 57 IU/mL [10–374], p = 0.0004 -NETs formation reduced to 1.9-fold increase compared to controls [0.4–6.1] (p = 0.0006) |
| Eligibility criteria | -SLE ACR or SLICC criteria -ANA and/or anti-dsDNA Ab + -≥18 years -recurrent or refractory LN -prior therapy with CYC or MMF | -SLE -18–75 years -anti-dsDNA Ab + at least once in the 5 years before the screening -failure of conventional therapy | -SLE (ACR criteria 1997) -≥18 years -severe SLE flare or refractory disease -ANA ≥1:80, anti-dsDNA Ab ≥30 IU/mL, hypocomplementemia |
| Key exclusion criteria | -prior therapy with RTX -prior therapy with another B-cell biologic therapy within the prior 12 months | -BILAG A flare in CNS -prior therapy with biological drugs (except RTX) -low IgG or IgA levels | -significant B-cell depletion -significant hypogammaglobulinemia |
| Treatment scheme | All pts: MP 100 mg + RTX 1000 mg + CYC IV 750 mg W0, W2 At W4 randomization: -RTX + CYC followed by BEL 10 mg/kg IV W4, 6, 8, and then every 4 weeks (RCB group) -RTX + CYC | All pts: RTX IV 1000 mg two weeks apart 4–8 W after RTX randomization: -BEL 10 mg/kg IV W0, 2, 4, and then every 4 weeks up to W52 -PBO | Single arm: RTX 1000 mg IV W0 and W2 + BEL 10 mg/kg W4, 6, 8, then monthly. |
| Number of patients who received the treatment | 21 RCB group, 22 RC group | 26 RTX + BEL, 26 RTX + PBO | 16 |
| Study duration | 96 W | 52 W | 24 W |
| Key secondary endpoints | -ORR W48, n (%) RCB vs. RC: 11 (52) vs. 9 (41), p = 0.452 -n of B-cells, geometric mean (cells/µL) [95% CI] W60, RCB vs. RC: 11 [6–20] vs. 53 [26–109], p = 0.0012 -pts with B-cell reconstitution W24, n RCB vs. RC: 0 vs. 5, p = 0.041 -median IgG (mg/dl), RCB vs. RC: 904.5 vs. 1410.0, p = 0.022 | -severe flares W52, n, BEL vs. PBO: 3 vs. 10, HR: 0.27 (95% CI, 0.07–0.98), unadjusted log-rank p = 0.003 -B-cell count W52, geometric mean [95% CI], BEL vs. PBO: 0.012 [0.006–0.014] vs. 0.037 [0.021–0.081], p = 0.031 | -median SLEDAI BL vs. W24: 18 vs. 2, p < 0.0001 -median GC dose (mg/day) BL vs. W24: 60 [5–60] vs. 7.5 [5–12.5], p = 0.001 |
| Safety | TEAEs ≥ grade 2, n (%) [95% CI], RCB vs. RC: 21 (100) [0.00–16.11] vs. 22 (100) [0.00–15.44] Serious TEAEs, n (%) [95% CI], RCB vs. RC: 4 (19) [5.45–41.91] vs. 11 (50) [28.22–71.78] Infectious TEAEs ≥grade 3, n (%) [95% CI], RCB vs. RC: 2 (10) [1.17–30.38] vs. 6 (27) [10.73–50.22] | All AEs, n of pts (%), BEL vs. PBO: 24 (92) vs. 24 (92) All AEs, n of events (%), BEL vs. PBO: 241 vs. 242 SAEs, n (%), BEL vs. PBO: 6 (23) vs. 6 (23) | Any AE, n = 41 Major infections, n (%) 1 (2) Minor infections, n (%) 15 (37%) |
| Rituximab + belimumab | |
|---|---|
| Clinical trial name and/or identification | BLISS-BELIEVE (NCT03312907) |
| Primary endpoint What was it? Was it achieved? | Disease control (SLEDAI-2K = 2) at W52 Not met. Proportion of pts with disease control, n (%) BEL/PBO, BEL/RTX, BEL/ST: 12 (16.7), 28 (19.4), 12 (25.5) -Disease control BEL/RTX vs BEL/PBO, observed difference [OR (95% CI)]: 2.78 [1.27 (0.60-2.71)], p = 0.5342 -Exploratory comparison: BEL/RTX vs BEL/ST, observed difference [OR (95% CI)]: -6.09 [0.71 (0.32–1.54)] |
| Eligibility criteria | -SLE (ACR criteria) -SLEDAI-2K ≥6 -ANA and/or anti-dsDNA Ab + -≥18 years |
| Key exclusion criteria | -severe lupus kidney disease -severe active CNS lupus -evidence of serious suicide risk -IgA <10 mg/dl -IgG <250 mg/dl |
| Treatment scheme | 3 arms: -BEL 200 mg sc QW for 52 weeks + PBO IV W4 and W6 (BEL/PBO) -BEL 200 mg sc QW for 52 weeks + RTX 1000 mg IV at W4 and W6 (BEL/RTX) -BEL 200 mg sc QW + ST for 104 weeks (BEL/ST). |
| Number of patients who received the treatment | 72 BEL/PBO, 144 BEL/RTX, 76 BEL/ST |
| Study duration | 104 W |
| Key secondary endpoints | -pts with clinical remission at W64, observed difference [OR (95% CI)]
|
| Safety | All AEs, n (%) BEL/PBO, BEL/RTX, BEL/ST: 63 (87.5), 127 (88.2), 62 (81.6) Serious AE -infections-, n (%) BEL/PBO, BEL/RTX, BEL/ST: 2 (2.8), 8 (5.6), 1 (1.3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venturelli, V.; Isenberg, D.A. Targeted Therapy for SLE—What Works, What Doesn’t, What’s Next. J. Clin. Med. 2023, 12, 3198. https://doi.org/10.3390/jcm12093198
Venturelli V, Isenberg DA. Targeted Therapy for SLE—What Works, What Doesn’t, What’s Next. Journal of Clinical Medicine. 2023; 12(9):3198. https://doi.org/10.3390/jcm12093198
Chicago/Turabian StyleVenturelli, Veronica, and David Alan Isenberg. 2023. "Targeted Therapy for SLE—What Works, What Doesn’t, What’s Next" Journal of Clinical Medicine 12, no. 9: 3198. https://doi.org/10.3390/jcm12093198
APA StyleVenturelli, V., & Isenberg, D. A. (2023). Targeted Therapy for SLE—What Works, What Doesn’t, What’s Next. Journal of Clinical Medicine, 12(9), 3198. https://doi.org/10.3390/jcm12093198