
A Closer Look at EGFR Inhibitor Resistance in Non-Small Cell Lung Cancer through the Lens of Precision Medicine

 , , , , and
, , , , and 
Abstract
1. EGFR Mutations in Cancer
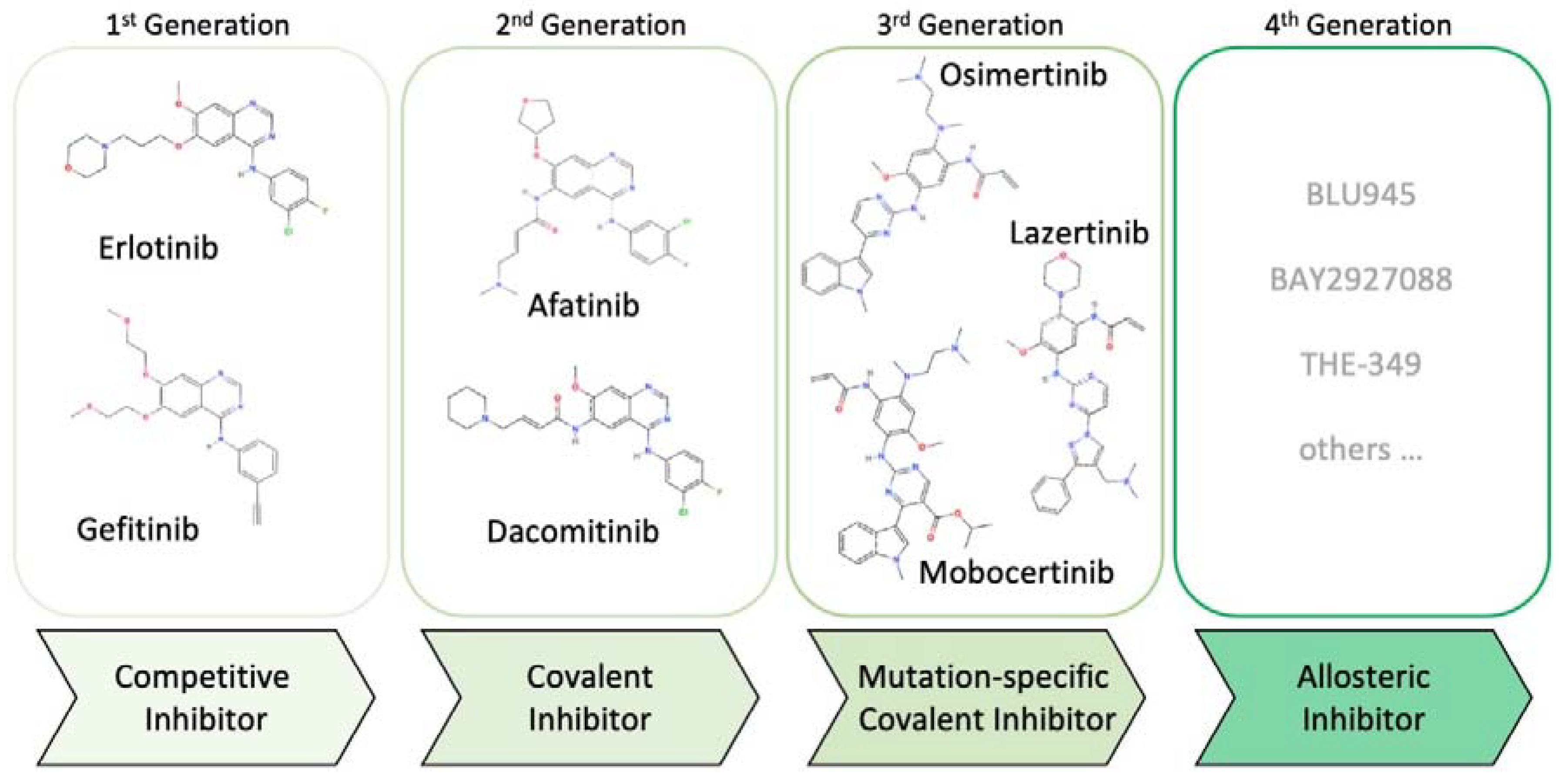
2. Evolution of EGFR Inhibitors
3. Genetic Mechanisms of EGFR Inhibitor Resistance beyond On-Target Mutations
4. The EGFR Inhibitor Genetic Resistance Gap
5. Clinical Strategies in the Treatment of EGFR Inhibitor Resistance
6. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramani, S.; Samant, S.; Manohar, S.M. The story of EGFR: From signaling pathways to a potent anticancer target. Future Med. Chem. 2022, 14, 1267–1288. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Kuwano, H.; Takahashi, T.; Mitsudomi, T. Mutations of the epidermal growth factor receptor gene in lung cancer: Biological and clinical implications. Cancer Res. 2004, 64, 8919–8923. [Google Scholar] [CrossRef] [PubMed]
- Midha, A.; Dearden, S.; McCormack, R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: A systematic review and global map by ethnicity (mutMapII). Am. J. Cancer Res. 2015, 5, 2892–2911. [Google Scholar]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Mok, T.; Peters, S.; Popat, S.; Ahn, M.J.; de Marinis, F. Recent Advances on the Role of EGFR Tyrosine Kinase Inhibitors in the Management of NSCLC With Uncommon, Non Exon 20 Insertions, EGFR Mutations. J. Thorac. Oncol. 2021, 16, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Mohanty, A.; Kulkarni, P.; Salgia, R. Precision oncology provides opportunities for targeting KRAS-inhibitor resistance. Trends Cancer 2023, 9, 42–54. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Zhao, Z.; Xie, L.; Bourne, P.E. Structural Insights into Characterizing Binding Sites in Epidermal Growth Factor Receptor Kinase Mutants. J. Chem. Inf. Model. 2019, 59, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Todsaporn, D.; Mahalapbutr, P.; Poo-Arporn, R.P.; Choowongkomon, K.; Rungrotmongkol, T. Structural dynamics and kinase inhibitory activity of three generations of tyrosine kinase inhibitors against wild-type, L858R/T790M, and L858R/T790M/C797S forms of EGFR. Comput. Biol. Med. 2022, 147, 105787. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef]
- Janne, P.A.; Boss, D.S.; Camidge, D.R.; Britten, C.D.; Engelman, J.A.; Garon, E.B.; Guo, F.; Wong, S.; Liang, J.; Letrent, S.; et al. Phase I dose-escalation study of the pan-HER inhibitor, PF299804, in patients with advanced malignant solid tumors. Clin. Cancer Res. 2011, 17, 1131–1139. [Google Scholar] [CrossRef]
- Yap, T.A.; Vidal, L.; Adam, J.; Stephens, P.; Spicer, J.; Shaw, H.; Ang, J.; Temple, G.; Bell, S.; Shahidi, M.; et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J. Clin. Oncol. 2010, 28, 3965–3972. [Google Scholar] [CrossRef]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Ahn, M.J.; Han, J.Y.; Lee, K.H.; Kim, S.W.; Kim, D.W.; Lee, Y.G.; Cho, E.K.; Kim, J.H.; Lee, G.W.; Lee, J.S.; et al. Lazertinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: Results from the dose escalation and dose expansion parts of a first-in-human, open-label, multicentre, phase 1–2 study. Lancet Oncol. 2019, 20, 1681–1690. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Erickson, A.W.; Brastianos, P.K.; Das, S. Assessment of Effectiveness and Safety of Osimertinib for Patients With Intracranial Metastatic Disease: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e201617. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Vincent, S.; Baker, T.E.; Gould, A.E.; Li, S.; Wardwell, S.D.; Nadworny, S.; Ning, Y.; Zhang, S.; Huang, W.S.; et al. Mobocertinib (TAK-788): A Targeted Inhibitor of EGFR Exon 20 Insertion Mutants in Non-Small Cell Lung Cancer. Cancer Discov. 2021, 11, 1672–1687. [Google Scholar] [CrossRef]
- Riely, G.J.; Neal, J.W.; Camidge, D.R.; Spira, A.I.; Piotrowska, Z.; Costa, D.B.; Tsao, A.S.; Patel, J.D.; Gadgeel, S.M.; Bazhenova, L.; et al. Activity and Safety of Mobocertinib (TAK-788) in Previously Treated Non-Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations from a Phase I/II Trial. Cancer Discov. 2021, 11, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Riess, J.W.; Gandara, D.R.; Frampton, G.M.; Madison, R.; Peled, N.; Bufill, J.A.; Dy, G.K.; Ou, S.I.; Stephens, P.J.; McPherson, J.D.; et al. Diverse EGFR Exon 20 Insertions and Co-Occurring Molecular Alterations Identified by Comprehensive Genomic Profiling of NSCLC. J. Thorac. Oncol. 2018, 13, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ramalingam, S.S.; Kim, T.M.; Kim, S.W.; Yang, J.C.; Riely, G.J.; Mekhail, T.; Nguyen, D.; Garcia Campelo, M.R.; Felip, E.; et al. Treatment Outcomes and Safety of Mobocertinib in Platinum-Pretreated Patients With EGFR Exon 20 Insertion-Positive Metastatic Non-Small Cell Lung Cancer: A Phase 1/2 Open-label Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, e214761. [Google Scholar] [CrossRef]
- Kian, W.; Christopoulos, P.; Remilah, A.A.; Levison, E.; Dudnik, E.; Shalata, W.; Krayim, B.; Marei, R.; Yakobson, A.; Faehling, M.; et al. Real-world efficacy and safety of mobocertinib in EGFR exon 20 insertion-mutated lung cancer. Front. Oncol. 2022, 12, 1010311. [Google Scholar] [CrossRef]
- Cho, B.C.; Han, J.Y.; Kim, S.W.; Lee, K.H.; Cho, E.K.; Lee, Y.G.; Kim, D.W.; Kim, J.H.; Lee, G.W.; Lee, J.S.; et al. A Phase 1/2 Study of Lazertinib 240 mg in Patients With Advanced EGFR T790M-Positive NSCLC After Previous EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2022, 17, 558–567. [Google Scholar] [CrossRef]
- Castellano, G.M.; Aisner, J.; Burley, S.K.; Vallat, B.; Yu, H.A.; Pine, S.R.; Ganesan, S. A Novel Acquired Exon 20 EGFR M766Q Mutation in Lung Adenocarcinoma Mediates Osimertinib Resistance but is Sensitive to Neratinib and Poziotinib. J. Thorac. Oncol. 2019, 14, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Tumbrink, H.L.; Heimsoeth, A.; Sos, M.L. The next tier of EGFR resistance mutations in lung cancer. Oncogene 2021, 40, 1–11. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Song, A.; Kim, D.W.; Kim, S.; Ahn, Y.O.; Keam, B.; Jeon, Y.K.; Lee, S.H.; Chung, D.H.; Heo, D.S. Mechanisms of Acquired Resistance to AZD9291: A Mutation-Selective, Irreversible EGFR Inhibitor. J. Thorac. Oncol. 2015, 10, 1736–1744. [Google Scholar] [CrossRef]
- Ou, S.I.; Horn, L.; Cruz, M.; Vafai, D.; Lovly, C.M.; Spradlin, A.; Williamson, M.J.; Dagogo-Jack, I.; Johnson, A.; Miller, V.A.; et al. Emergence of FGFR3-TACC3 fusions as a potential by-pass resistance mechanism to EGFR tyrosine kinase inhibitors in EGFR mutated NSCLC patients. Lung Cancer 2017, 111, 61–64. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef]
- von Buttlar, X.; Reuss, J.E.; Liu, S.V.; Kim, C. EML4-ALK Rearrangement as a Mechanism of Resistance to Osimertinib in Metastatic Lung Adenocarcinoma: A Case Report. JTO Clin. Res. Rep. 2021, 2, 100179. [Google Scholar] [CrossRef]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef]
- Del Re, M.; Tiseo, M.; Bordi, P.; D’Incecco, A.; Camerini, A.; Petrini, I.; Lucchesi, M.; Inno, A.; Spada, D.; Vasile, E.; et al. Contribution of KRAS mutations and c.2369C > T (p.T790M) EGFR to acquired resistance to EGFR-TKIs in EGFR mutant NSCLC: A study on circulating tumor DNA. Oncotarget 2017, 8, 13611–13619. [Google Scholar] [CrossRef]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef]
- Cheung, H.W.; Du, J.; Boehm, J.S.; He, F.; Weir, B.A.; Wang, X.; Butaney, M.; Sequist, L.V.; Luo, B.; Engelman, J.A.; et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011, 1, 608–625. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Yang, S.D.; Lim, S.; Shim, H.S.; Cho, B.C. Brief Report: Heterogeneity of Acquired Resistance Mechanisms to Osimertinib and Savolitinib. JTO Clin. Res. Rep. 2021, 2, 100180. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef]
- Pan, Y.; Yuan, C.; Cheng, C.; Zhang, Y.; Ma, Y.; Zheng, D.; Zheng, S.; Li, Y.; Jin, Y.; Sun, Y.; et al. Frequency and clinical significance of NF1 mutation in lung adenocarcinomas from East Asian patients. Int. J. Cancer 2019, 144, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Isobe, K.; Hata, Y.; Tochigi, N.; Kaburaki, K.; Kobayashi, H.; Makino, T.; Otsuka, H.; Sato, F.; Ishida, F.; Kikuchi, N.; et al. Clinical significance of BIM deletion polymorphism in non-small-cell lung cancer with epidermal growth factor receptor mutation. J. Thorac. Oncol. 2014, 9, 483–487. [Google Scholar] [CrossRef]
- He, J.; Huang, Z.; Han, L.; Gong, Y.; Xie, C. Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer (Review). Int. J. Oncol. 2021, 59, 90. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J. Clin. Oncol. 2019, 37, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Mambetsariev, I.; Arvanitis, L.; Fricke, J.; Pharaon, R.; Baroz, A.R.; Afkhami, M.; Koczywas, M.; Massarelli, E.; Salgia, R. Small Cell Lung Cancer Transformation following Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. J. Clin. Med. 2022, 11, 1429. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef]
- Yano, S.; Yamada, T.; Takeuchi, S.; Tachibana, K.; Minami, Y.; Yatabe, Y.; Mitsudomi, T.; Tanaka, H.; Kimura, T.; Kudoh, S.; et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J. Thorac. Oncol. 2011, 6, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Ware, K.E.; Hinz, T.K.; Kleczko, E.; Singleton, K.R.; Marek, L.A.; Helfrich, B.A.; Cummings, C.T.; Graham, D.K.; Astling, D.; Tan, A.C.; et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis 2013, 2, e39. [Google Scholar] [CrossRef]
- Ware, K.E.; Marshall, M.E.; Heasley, L.R.; Marek, L.; Hinz, T.K.; Hercule, P.; Helfrich, B.A.; Doebele, R.C.; Heasley, L.E. Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell lines through de-repression of FGFR2 and FGFR3 expression. PLoS ONE 2010, 5, e14117. [Google Scholar] [CrossRef]
- Cortot, A.B.; Repellin, C.E.; Shimamura, T.; Capelletti, M.; Zejnullahu, K.; Ercan, D.; Christensen, J.G.; Wong, K.K.; Gray, N.S.; Janne, P.A. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013, 73, 834–843. [Google Scholar] [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- Shah, K.N.; Bhatt, R.; Rotow, J.; Rohrberg, J.; Olivas, V.; Wang, V.E.; Hemmati, G.; Martins, M.M.; Maynard, A.; Kuhn, J.; et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 2019, 25, 111–118. [Google Scholar] [CrossRef]
- Tanaka, K.; Yu, H.A.; Yang, S.; Han, S.; Selcuklu, S.D.; Kim, K.; Ramani, S.; Ganesan, Y.T.; Moyer, A.; Sinha, S.; et al. Targeting Aurora B kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer by enhancing BIM- and PUMA-mediated apoptosis. Cancer Cell 2021, 39, 1245–1261.e6. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Guo, G.; Panchani, N.; Bender, M.E.; Gerber, D.E.; Minna, J.D.; Fattah, F.; Gao, B.; Peyton, M.; Kernstine, K.; et al. EGFR inhibition triggers an adaptive response by co-opting antiviral signaling pathways in lung cancer. Nat. Cancer 2020, 1, 394–409. [Google Scholar] [CrossRef]
- Blakely, C.M.; Pazarentzos, E.; Olivas, V.; Asthana, S.; Yan, J.J.; Tan, I.; Hrustanovic, G.; Chan, E.; Lin, L.; Neel, D.S.; et al. NF-kappaB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 2015, 11, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Eno, M.S.; Brubaker, J.D.; Campbell, J.E.; De Savi, C.; Guzi, T.J.; Williams, B.D.; Wilson, D.; Wilson, K.; Brooijmans, N.; Kim, J.; et al. Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65, 9662–9677. [Google Scholar] [CrossRef] [PubMed]
- Poh, A. BDTX-1535 Goes after Osimertinib Resistance. Cancer Discov. 2021, 11, 2952–2953. [Google Scholar] [CrossRef]
- Wiest, N.; Majeed, U.; Seegobin, K.; Zhao, Y.; Lou, Y.; Manochakian, R. Role of Immune Checkpoint Inhibitor Therapy in Advanced EGFR-Mutant Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 751209. [Google Scholar] [CrossRef]
- Gettinger, S.; Hellmann, M.D.; Chow, L.Q.M.; Borghaei, H.; Antonia, S.; Brahmer, J.R.; Goldman, J.W.; Gerber, D.E.; Juergens, R.A.; Shepherd, F.A.; et al. Nivolumab Plus Erlotinib in Patients With EGFR-Mutant Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1363–1372. [Google Scholar] [CrossRef]
- Yang, J.C.; Gadgeel, S.M.; Sequist, L.V.; Wu, C.L.; Papadimitrakopoulou, V.A.; Su, W.C.; Fiore, J.; Saraf, S.; Raftopoulos, H.; Patnaik, A. Pembrolizumab in Combination With Erlotinib or Gefitinib as First-Line Therapy for Advanced NSCLC With Sensitizing EGFR Mutation. J. Thorac. Oncol. 2019, 14, 553–559. [Google Scholar] [CrossRef]
- Ahn, M.J.; Cho, B.C.; Ou, X.; Walding, A.; Dymond, A.W.; Ren, S.; Cantarini, M.; Janne, P.A. Osimertinib Plus Durvalumab in Patients With EGFR-Mutated, Advanced NSCLC: A Phase 1b, Open-Label, Multicenter Trial. J. Thorac. Oncol. 2022, 17, 718–723. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wu, L.; Jian, H.; Chen, Y.; Wang, Q.; Fang, J.; Wang, Z.; Hu, Y.; Sun, M.; Han, L.; et al. Sintilimab plus bevacizumab biosimilar IBI305 and chemotherapy for patients with EGFR-mutated non-squamous non-small-cell lung cancer who progressed on EGFR tyrosine-kinase inhibitor therapy (ORIENT-31): First interim results from a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2022, 23, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Zhou, B.; Zhang, B.; Ren, H.; Zhu, L.; Zheng, G.; Ge, M.; Ge, J. Recent advances in the development of EGFR degraders: PROTACs and LYTACs. Eur. J. Med. Chem. 2022, 239, 114533. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Primary Target | Primary Therapeutic | Secondary Target | Secondary Therapeutic | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| EGFR | Osimertinib | CDK4/CDK6 | Abemaciclib | NCT04545710 |
| EGFR | Osimertinib | mTOR Aurora A | Sapanisertib Alisertib | NCT04479306 |
| EGFR | Osimertinib | Anti-EGFR | Necitumumab | NCT02496663 |
| EGFR | Osimertinib | MET | Tepotinib | NCT03940703 |
| EGFR | Osimertinib | MET | Tepotinib | NCT05120960 |
| EGFR | Osimertinib | COX1/COX2 (AKT/BIM) | Aspirin | NCT04184921 |
| EGFR | Osimertinib | MET | Savolitinib | NCT03944772 |
| EGFR | Gefitinib | |||
| Anti-EGFR | Necitumumab | |||
| Antifolate + Anti-PD1 | Pemetrexed + Durvalumab | |||
| ALK | Alectinib | |||
| RET | Selpercatinib | |||
| Antifolate + Platinum Chemotherapy | Pemetrexed + Carboplatin or Cisplatin | |||
| MEK1/MEK2 | Selumetinib | |||
| TROP2 ADC | Datopotamab-deruxtecan | |||
| - | - | Topoisomerase + Anti PD-L1 + Platinum Chemotherapy | Etoposide + Durvalumab + Carboplatin or Cisplatin | |
| EGFR | Osimertinib | BCL-2/BCL-xL | Pelcitoclax | NCT04001777 |
| EGFR | Osimertinib | BCL-2/BCL-xL | Navitoclax | NCT02520778 |
| EGFR | Osimertinib | SRC | Dasatinib | NCT02954523 |
| EGFR | Osimertinib | α/δ Phosphatidylinositol 3-kinase | TQ-B3525 | NCT05284994 |
| EGFR | Osimertinib | EGFR | Necitumumab + | NCT04285671 |
| HER2 | Trastuzumab | |||
| EGFR, HER2, HER4 | Dacomitinib | EGFR | Alone or + Osimertinib | NCT03755102 |
| EGFR-MET bispecific antibody | Amivantamab | EGFR | Lazertinib or | NCT05299125, |
| Antifolate | + Pemetrexed | NCT02609776, | ||
| Chemotherapy | + Carboplatin | NCT04077463 | ||
| EGFR-MET bispecific antibody | EMB-01 | NCT03797391 | ||
| Anti-HER3 ADC | Patritumab Deruxtecan | EGFR | Osimertinib | NCT04676477 |
| EGFR | Nazartinib (EGF816) | MEK1/MEK2 | Trametinib | NCT03516214 |
| PARP | Olaparib | Anti-PD-L1 | Durvalumab | NCT04538378 |
| Antifolate + Chemotherapy | Pemetrexed + Platinum Chemotherapy | Anti-PD-1 | Alone or + Pembrolizumab | NCT03515837 |
| EGFR (C797X) | BLU-701 | EGFR | Alone or + Osimertinib | NCT05153408 |
| Antifolate | + Pemetrexed | |||
| Chemotherapy | + Carboplatin | |||
| EGFR (C797X) | BLU-945 | EGFR | Alone or + Osimertinib | NCT04862780 |
| EGFR (C797X) | WJ13405 | NCT05662670 | ||
| EGFR (C797X) | BAY2927088 | NCT05099172 | ||
| EGFR (C797X) | JIN-A02 | NCT05394831 | ||
| EGFR (C797X) | HS-10375 | NCT05435248 | ||
| EGFR (C797X) | QLH11811 | NCT05555212 | ||
| EGFR (C797X) | BPI-361175 | NCT05393466 | ||
| EGFR (C797X) | BDTX-1535 | NCT05256290 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sattler, M.; Mambetsariev, I.; Fricke, J.; Tan, T.; Liu, S.; Vaidehi, N.; Pisick, E.; Mirzapoiazova, T.; Rock, A.G.; Merla, A.; et al. A Closer Look at EGFR Inhibitor Resistance in Non-Small Cell Lung Cancer through the Lens of Precision Medicine. J. Clin. Med. 2023, 12, 1936. https://doi.org/10.3390/jcm12051936
Sattler M, Mambetsariev I, Fricke J, Tan T, Liu S, Vaidehi N, Pisick E, Mirzapoiazova T, Rock AG, Merla A, et al. A Closer Look at EGFR Inhibitor Resistance in Non-Small Cell Lung Cancer through the Lens of Precision Medicine. Journal of Clinical Medicine. 2023; 12(5):1936. https://doi.org/10.3390/jcm12051936
Chicago/Turabian StyleSattler, Martin, Isa Mambetsariev, Jeremy Fricke, Tingting Tan, Sariah Liu, Nagarajan Vaidehi, Evan Pisick, Tamara Mirzapoiazova, Adam G. Rock, Amartej Merla, and et al. 2023. "A Closer Look at EGFR Inhibitor Resistance in Non-Small Cell Lung Cancer through the Lens of Precision Medicine" Journal of Clinical Medicine 12, no. 5: 1936. https://doi.org/10.3390/jcm12051936
APA StyleSattler, M., Mambetsariev, I., Fricke, J., Tan, T., Liu, S., Vaidehi, N., Pisick, E., Mirzapoiazova, T., Rock, A. G., Merla, A., Sharma, S., & Salgia, R. (2023). A Closer Look at EGFR Inhibitor Resistance in Non-Small Cell Lung Cancer through the Lens of Precision Medicine. Journal of Clinical Medicine, 12(5), 1936. https://doi.org/10.3390/jcm12051936