
Current Knowledge of the Molecular Pathogenesis of Cutaneous Lupus Erythematosus

Abstract
1. Introduction
2. Clinical Features of CLE
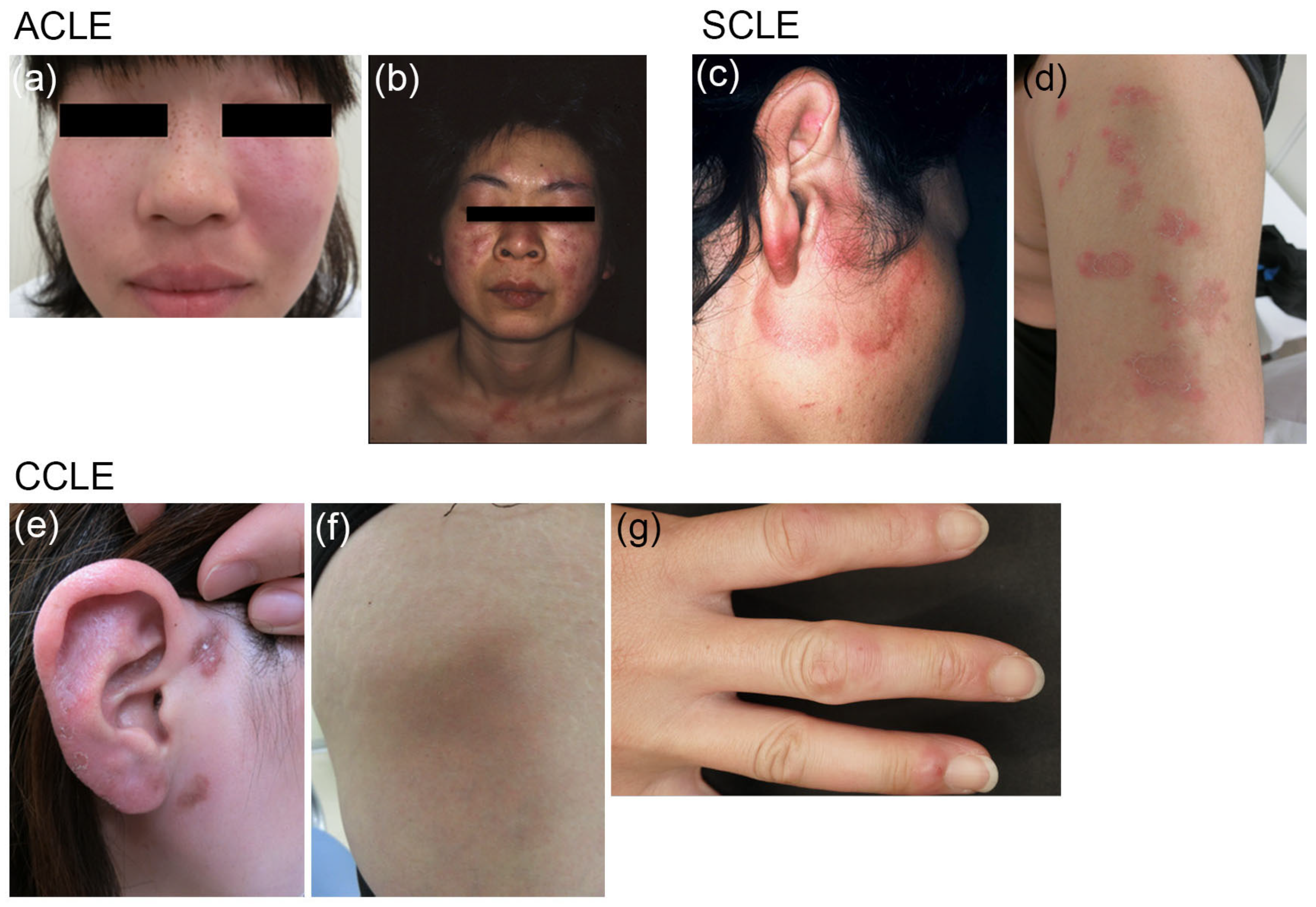
2.1. Classification
2.2. Epidemiology
3. Pathogenesis of CLE
3.1. Gene Expression Patterns
3.2. Autoantibodies
3.3. Toll-Like Receptors
3.4. Cytokines
3.4.1. Type I IFNs
3.4.2. Interleukin-6
3.5. Apoptosis
3.6. Immune Cells
3.6.1. Plasmacytoid Dendritic Cells
3.6.2. T-Helper 1 Cells
3.7. Environmental Factors
3.7.1. Ultraviolet
3.7.2. Cigarette Smoke
3.7.3. Drugs
3.8. Genetic Factors
3.8.1. Mutations
3.8.2. Polymorphisms
3.9. Sex Bias
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wenzel, J. Cutaneous lupus erythematosus: New insights into pathogenesis and therapeutic strategies. Nat. Rev. Rheumatol. 2019, 15, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M. Rethinking the Pathogenesis of Cutaneous Lupus. J. Investig. Dermatol. 2021, 141, 32–35. [Google Scholar] [CrossRef]
- Gilliam, J.N.; Sontheimer, R.D. Distinctive cutaneous subsets in the spectrum of lupus erythematosus. J. Am. Acad. Dermatol. 1981, 4, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Tsuchida, T. Classification of Lupus erythematosus Based upon Cutaneous Manifestations. Dermatological, systemic and laboratory findings in 191 patients. Dermatology 1995, 190, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Kunz, M.; Konig, I.R.; Schillert, A.; Kruppa, J.; Ziegler, A.; Grallert, H.; Müller-Nurasyid, M.; Lieb, W.; Franke, A.; Ranki, A.; et al. Genome-wide association study identifies new susceptibility loci for cutaneous lupus erythematosus. Exp. Dermatol. 2015, 24, 510–515. [Google Scholar] [CrossRef]
- Kuhn, A.; Wenzel, J.; Bijl, M. Lupus erythematosus revisited. Semin. Immunopathol. 2016, 38, 97–112. [Google Scholar] [CrossRef]
- Grönhagen, C.; Fored, C.; Granath, F.; Nyberg, F. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: A population-based cohort of 1088 patients in Sweden. Br. J. Dermatol. 2011, 164, 1335–1341. [Google Scholar] [CrossRef]
- Garelli, C.J.; Refat, M.A.; Nanaware, P.P.; Ramirez-Ortiz, Z.G.; Rashighi, M.; Richmond, J.M. Current Insights in Cutaneous Lupus Erythematosus Immunopathogenesis. Front. Immunol. 2020, 11, 1353. [Google Scholar] [CrossRef]
- Jarukitsopa, S.; Hoganson, D.D.; Crowson, C.S.; Sokumbi, O.; Davis, M.D.; Michet, C.J., Jr.; Matteson, E.L.; Kremers, H.M.; Chowdhary, V.R. Epidemiology of systemic lupus erythematosus and cutaneous lupus erythematosus in a predominantly white population in the United States. Arthritis Care Res. 2015, 67, 817–828. [Google Scholar] [CrossRef]
- Durosaro, O.; Davis, M.D.P.; Reed, K.B.; Rohlinger, A.L. Incidence of Cutaneous Lupus Erythematosus, 1965–2005: A Population-Based Study. Arch. Dermatol. 2009, 145, 249–253. [Google Scholar] [CrossRef]
- Scholtissek, B.; Zahn, S.; Maier, J.; Klaeschen, S.; Braegelmann, C.; Hoelzel, M.; Bieber, T.; Barchet, W.; Wenzel, J. Immunostimulatory Endogenous Nucleic Acids Drive the Lesional Inflammation in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2017, 137, 1484–1492. [Google Scholar] [CrossRef]
- Provost, T.T. Lupus band test. Int. J. Dermatol. 1981, 20, 475–481. [Google Scholar] [CrossRef]
- Wenzel, J.; Bauer, R.; Bieber, T.; Böhm, I. Autoantibodies in Patients with Lupus erythematosus: Spectrum and Frequencies. Dermatology 2000, 201, 283. [Google Scholar] [CrossRef]
- Lee, L.A.; Gaither, K.K.; Coulter, S.N.; Norris, D.A.; Harley, J.B. Pattern of cutaneous immunoglobulin G deposition in subacute cutaneous lupus erythematosus is reproduced by infusing purified anti-Ro (SSA) autoantibodies into human skin-grafted mice. J. Clin. Investig. 1989, 83, 1556–1562. [Google Scholar] [CrossRef]
- Greiling, T.M.; Dehner, C.; Chen, X.; Hughes, K.; Iñiguez, A.J.; Boccitto, M.; Ruiz, D.Z.; Renfroe, S.C.; Vieira, S.M.; Ruff, W.E.; et al. Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci. Transl. Med. 2018, 10, eaan2306. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef]
- Banchereau, J.; Pascual, V. Type I Interferon in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef]
- Miyagawa, F.; Tagaya, Y.; Ozato, K.; Asada, H. Essential Requirement for IFN Regulatory Factor 7 in Autoantibody Production but Not Development of Nephritis in Murine Lupus. J. Immunol. 2016, 197, 2167–2176. [Google Scholar] [CrossRef]
- Braunstein, I.; Klein, R.; Okawa, J.; Werth, V. The interferon-regulated gene signature is elevated in subacute cutaneous lupus erythematosus and discoid lupus erythematosus and correlates with the cutaneous lupus area and severity index score. Br. J. Dermatol. 2012, 166, 971–975. [Google Scholar] [CrossRef]
- Fäh, J.; Pavlovic, J.; Burg, G. Expression of MxA protein in inflammatory dermatoses. J. Histochem. Cytochem. 1995, 43, 47–52. [Google Scholar] [CrossRef]
- Wenzel, J.; Uerlich, M.; Worrenkamper, E.; Freutel, S.; Bieber, T.; Tuting, T. Scarring skin lesions of discoid lupus erythematosus are characterized by high numbers of skin-homing cytotoxic lymphocytes associated with strong expression of the type I interferon-induced protein MxA. Br. J. Dermatol. 2005, 153, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J.; Zahn, S.; Mikus, S.; Wiechert, A.; Bieber, T.; Tüting, T. The expression pattern of interferon-inducible proteins reflects the characteristic histological distribution of infiltrating immune cells in different cutaneous lupus erythematosus subsets. Br. J. Dermatol. 2007, 157, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gonzalez, J.; Cueto, I.; Franks, A.G.; Krueger, J.G. Dominant Th1 and Minimal Th17 Skewing in Discoid Lupus Revealed by Transcriptomic Comparison with Psoriasis. J. Investig. Dermatol. 2014, 134, 87–95. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Hile, G.A.; Berthier, C.C.; Sarkar, M.K.; Reed, T.J.; Liu, J.; Uppala, R.; Patrick, M.; Raja, K.; Xing, X.; et al. Hypersensitive IFN Responses in Lupus Keratinocytes Reveal Key Mechanistic Determinants in Cutaneous Lupus. J. Immunol. 2019, 202, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.K.; Hile, G.A.; Tsoi, L.C.; Xing, X.; Liu, J.; Liang, Y.; Berthier, C.C.; Swindell, W.R.; Patrick, M.T.; Shao, S.; et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann. Rheum. Dis. 2018, 77, 1653–1664. [Google Scholar] [CrossRef]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef]
- Morand, E.F.; Furie, R.A.; Bruce, I.N.; Vital, E.M.; Dall’Era, M.; Maho, E.; Pineda, L.; Tummala, R. Efficacy of anifrolumab across organ domains in patients with moderate-to-severe systemic lupus erythematosus: A post-hoc analysis of pooled data from the TULIP-1 and TULIP-2 trials. Lancet Rheumatol. 2022, 4, e282–e292. [Google Scholar] [CrossRef]
- Blum, F.R.; Sampath, A.J.; Foulke, G.T. Anifrolumab for treatment of refractory cutaneous lupus erythematosus. Clin. Exp. Dermatol. 2022, 47, 1998–2001. [Google Scholar] [CrossRef]
- Sabrautzki, S.; Janas, E.; Lorenz-Depiereux, B.; Calzada-Wack, J.; Aguilar-Pimentel, J.A.; Rathkolb, B.; Adler, T.; Cohrs, C.; Hans, W.; Diener, S.; et al. An ENU mutagenesis-derived mouse model with a dominant Jak1 mutation resembling phenotypes of systemic autoimmune disease. Am. J. Pathol. 2013, 183, 352–368. [Google Scholar] [CrossRef]
- Stannard, J.N.; Reed, T.J.; Myers, E.; Lowe, L.; Sarkar, M.K.; Xing, X.; Gudjonsson, J.E.; Kahlenberg, J.M. Lupus Skin Is Primed for IL-6 Inflammatory Responses through a Keratinocyte-Mediated Autocrine Type I Interferon Loop. J. Investig. Dermatol. 2017, 137, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Emlen, W.; Niebur, J.; Kadera, R. Accelerated in vitro apoptosis of lymphocytes from patients with systemic lupus erythematosus. J. Immunol. 1994, 152, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Amoura, Z.; Piette, J.-C.; Chabre, H.; Cacoub, P.; Papo, T.; Wechsler, B.; Bach, J.-F.; Koutouzov, S. Circulating plasma levels of nucleosomes in patients with systemic lupus erythematosus. Correlation with serum antinucleosome antibody titers and absence of clear association with disease activity. Arthritis Rheum. 1997, 40, 2217–2225. [Google Scholar] [CrossRef]
- Herrmann, M.; Voll, R.E.; Zoller, O.M.; Hagenhofer, M.; Ponner, B.B.; Kalden, J.R. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998, 41, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Lövgren, T.; Eloranta, M.-L.; Båve, U.; Alm, G.V.; Rönnblom, L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004, 50, 1861–1872. [Google Scholar] [CrossRef]
- Kuhn, A.; Herrmann, M.; Kleber, S.; Beckmann-Welle, M.; Fehsel, K.; Martin-Villalba, A.; Lehmann, P.; Ruzicka, T.; Krammer, P.H.; Kolb-Bachofen, V. Accumulation of apoptotic cells in the epidermis of patients with cutaneous lupus erythematosus after ultraviolet irradiation. Arthritis Rheum. 2006, 54, 939–950. [Google Scholar] [CrossRef]
- Katayama, S.; Panelius, J.; Koskenmies, S.; Skoog, T.; Mähönen, K.; Kisand, K.; Bondet, V.; Duffy, D.; Krjutškov, K.; Kere, J.; et al. Delineating the Healthy Human Skin UV Response and Early Induction of Interferon Pathway in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2019, 139, 2058–2061.e4. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, F.; Tanaka, H.; Sekita, K.; Nakamura, T.; Horiguchi, Y.; Hamashima, Y. Dermatopathological studies on skin lesions of MRL mice. Arch. Dermatol. Res. 1984, 276, 186–194. [Google Scholar] [CrossRef]
- Furukawa, F.; Kanauchi, H.; Wakita, H.; Tokura, Y.; Tachibana, T.; Horiguchi, Y.; Imamura, S.; Ozaki, S.; Takigawa, M. Spontaneous Autoimmune Skin Lesions of MRL/n Mice: Autoimmune Disease-Prone Genetic Background in Relation to Fas-Defect MRL/1pr Mice. J. Investig. Dermatol. 1996, 107, 95–100. [Google Scholar] [CrossRef]
- Mande, P.; Zirak, B.; Ko, W.-C.; Taravati, K.; Bride, K.L.; Brodeur, T.Y.; Deng, A.; Dresser, K.; Jiang, Z.; Ettinger, R.; et al. Fas ligand promotes an inducible TLR-dependent model of cutaneous lupus-like inflammation. J. Clin. Investig. 2018, 128, 2966–2978. [Google Scholar] [CrossRef]
- Furukawa, F.; Kashihara-Sawami, M.; Lyons, M.B.; Norris, D.A. Binding of antibodies to the extractable nuclear antigens SS-A/Ro and SS-B/La is induced on the surface of human keratinocytes by ultraviolet light (UVL): Implications for the pathogenesis of photosensitive cutaneous lupus. J. Investig. Dermatol. 1990, 94, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Casciola-Rosen, L.A.; Anhalt, G.; Rosen, A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 1994, 179, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Farkas, L.; Beiske, K.; Lund-Johansen, F.; Brandtzaeg, P.; Jahnsen, F.L. Plasmacytoid dendritic cells (natural interferon- α/β-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am. J. Pathol. 2001, 159, 237–243. [Google Scholar] [CrossRef]
- Guiducci, C.; Tripodo, C.; Gong, M.; Sangaletti, S.; Colombo, M.P.; Coffman, R.L.; Barrat, F.J. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J. Exp. Med. 2010, 207, 2931–2942. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J.; Wörenkämper, E.; Freutel, S.; Henze, S.; Haller, O.; Bieber, T.; Tüting, T. Enhanced type I interferon signalling promotes Th1-biased inflammation in cutaneous lupus erythematosus. J. Pathol. 2005, 205, 435–442. [Google Scholar] [CrossRef]
- Foering, K.; Chang, A.Y.; Piette, E.W.; Cucchiara, A.; Okawa, J.; Werth, V.P. Characterization of clinical photosensitivity in cutaneous lupus erythematosus. J. Am. Acad. Dermatol. 2013, 69, 205–213. [Google Scholar] [CrossRef]
- Biazar, C.; Sigges, J.; Patsinakidis, N.; Ruland, V.; Amler, S.; Bonsmann, G.; Kuhn, A. Cutaneous lupus erythematosus: First multicenter database analysis of 1002 patients from the European Society of Cutaneous Lupus Erythematosus (EUSCLE). Autoimmun. Rev. 2013, 12, 444–454. [Google Scholar] [CrossRef]
- Furukawa, F.; Itoh, T.; Wakita, H.; Yagi, H.; Tokura, Y.; Norris, D.A.; Takigawa, M. Keratinocytes from patients with lupus erythematosus show enhanced cytotoxicity to ultraviolet radiation and to antibody-mediated cytotoxicity. Clin. Exp. Immunol. 1999, 118, 164–170. [Google Scholar] [CrossRef]
- Gehrke, N.; Mertens, C.; Zillinger, T.; Wenzel, J.; Bald, T.; Zahn, S.; Tüting, T.; Hartmann, G.; Barchet, W. Oxidative Damage of DNA Confers Resistance to Cytosolic Nuclease TREX1 Degradation and Potentiates STING-Dependent Immune Sensing. Immunity 2013, 39, 482–495. [Google Scholar] [CrossRef]
- Piette, E.W.; Foering, K.P.; Chang, A.Y.; Okawa, J.; Ten Have, T.R.; Feng, R.; Werth, V.P. The Impact of smoking in cutaneous lupus erythematosus. Arch. Dermatol. 2012, 148, 317–322. [Google Scholar] [CrossRef]
- Bataille, P.; Chasset, F.; Monfort, J.-B.; De Risi-Pugliese, T.; Soria, A.; Francès, C.; Barbaud, A.; Senet, P. Cutaneous drug-induced lupus erythematosus: Clinical and immunological characteristics and update on new associated drugs. Ann. Dermatol. Vénéréologie 2021, 148, 211–220. [Google Scholar] [CrossRef]
- Vaglio, A.; Grayson, P.C.; Fenaroli, P.; Gianfreda, D.; Boccaletti, V.; Ghiggeri, G.M.; Moroni, G. Drug-induced lupus: Traditional and new concepts. Autoimmun. Rev. 2018, 17, 912–918. [Google Scholar] [CrossRef]
- Lowe, G.; Henderson, C.; Grau, R.; Hansen, C.; Sontheimer, R. A systematic review of drug-induced subacute cutaneous lupus erythematosus. Br. J. Dermatol. 2011, 164, 465–472. [Google Scholar] [CrossRef]
- Borucki, R.; Werth, V.P. Cutaneous lupus erythematosus induced by drugs-novel insights. Expert Rev. Clin. Pharmacol. 2020, 13, 35–42. [Google Scholar] [CrossRef]
- Michaelis, T.C.; Sontheimer, R.D.; Lowe, G.C. An update in drug-induced subacute cutaneous lupus erythematosus. Dermatol. Online J. 2017, 23. [Google Scholar] [CrossRef]
- Yoshimasu, T.; Nishide, T.; Seo, N.; Hiroi, A.; Ohtani, T.; Uede, K.; Furukawa, F. Susceptibility of T cell receptor-α chain knock-out mice to ultraviolet B light and fluorouracil: A novel model for drug-induced cutaneous lupus erythematosus. Clin. Exp. Immunol. 2004, 136, 245–254. [Google Scholar] [CrossRef]
- Rice, G.; Newman, W.G.; Dean, J.; Patrick, T.; Parmar, R.; Flintoff, K.; Robins, P.; Harvey, S.; Hollis, T.; O’Hara, A.; et al. Heterozygous Mutations in TREX1 Cause Familial Chilblain Lupus and Dominant Aicardi-Goutières Syndrome. Am. J. Hum. Genet. 2007, 80, 811–815. [Google Scholar] [CrossRef]
- Lee-Kirsch, M.A.; Chowdhury, D.; Harvey, S.; Gong, M.; Senenko, L.; Engel, K.; Pfeiffer, C.; Hollis, T.; Gahr, M.; Perrino, F.W.; et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J. Mol. Med. 2007, 85, 531–537. [Google Scholar] [CrossRef]
- Ravenscroft, J.C.; Suri, M.; Rice, G.I.; Szynkiewicz, M.; Crow, Y.J. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am. J. Med. Genet. Part A 2011, 155, 235–237. [Google Scholar] [CrossRef]
- Peschke, K.; Friebe, F.; Zimmermann, N.; Wahlicht, T.; Schumann, T.; Achleitner, M.; Berndt, N.; Luksch, H.; Behrendt, R.; Lee-Kirsch, M.A.; et al. Deregulated Type I IFN Response in TREX1-Associated Familial Chilblain Lupus. J. Investig. Dermatol. 2014, 134, 1456–1459. [Google Scholar] [CrossRef]
- Yang, Y.-G.; Lindahl, T.; Barnes, D.E. Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell 2007, 131, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Rapp, A.; Berndt, N.; Staroske, W.; Schuster, M.; Dobrick-Mattheuer, M.; Kretschmer, S.; König, N.; Kurth, T.; Wieczorek, D.; et al. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat. Commun. 2016, 7, 11752. [Google Scholar] [CrossRef] [PubMed]
- Konig, N.; Fiehn, C.; Wolf, C.; Schuster, M.; Cura Costa, E.; Tungler, V.; Ariel Alvarez, H.; Chara, O.; Engel, K.; Goldbach-Mansky, R.; et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann. Rheum. Dis. 2017, 76, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.B.; Pinnell, S.R.; Snyderman, R.; Ward, F.E. Hereditary C2 deficiency associated with cutaneous lupus erythematosus: Clinical, laboratory, and genetic studies. Arch. Dermatol. 1979, 115, 57–61. [Google Scholar] [CrossRef]
- Agnello, V.; Gell, J.; Tye, M.J. Partial genetic deficiency of the C4 component of complement in discoid lupus erythematosus and urticaria/angioedema. J. Am. Acad. Dermatol. 1983, 9, 894–898. [Google Scholar] [CrossRef]
- Lipsker, D.; Hauptmann, G. Cutaneous manifestations of complement deficiencies. Lupus 2010, 19, 1096–1106. [Google Scholar] [CrossRef]
- Jarvinen, T.M.; Hellquist, A.; Koskenmies, S.; Einarsdottir, E.; Koskinen, L.L.; Jeskanen, L.; Berglind, L.; Panelius, J.; Hasan, T.; Ranki, A.; et al. Tyrosine kinase 2 and interferon regulatory factor 5 polymorphisms are associated with discoid and subacute cutaneous lupus erythematosus. Exp. Dermatol. 2010, 19, 123–131. [Google Scholar] [CrossRef]
- Skonieczna, K.; Czajkowski, R.; Kaszewski, S.; Gawrych, M.; Jakubowska, A.; Grzybowski, T. Genetic similarities and differences between discoid and systemic lupus erythematosus patients within the Polish population. Postepy Dermatol. Alergol. 2017, 3, 228–232. [Google Scholar] [CrossRef]
- Millard, T.; Kondeatis, E.; Cox, A.; Wilson, A.; Grabczynska, S.; Carey, B.; Lewis, C.; Khamashta, M.; Duff, G.; Hughes, G.; et al. A candidate gene analysis of three related photosensitivity disorders: Cutaneous lupus erythematosus, polymorphic light eruption and actinic prurigo. Br. J. Dermatol. 2001, 145, 229–236. [Google Scholar] [CrossRef]
- Racila, D.M.; Sontheimer, C.J.; Sheffield, A.; Wisnieski, J.J.; Racila, E.; Sontheimer, R.D. Homozygous single nucleotide polymorphism of the complement C1QA gene is associated with decreased levels of C1q in patients with subacute cutaneous lupus erythematosus. Lupus 2003, 12, 124–132. [Google Scholar] [CrossRef]
- Jarvinen, T.M.; Hellquist, A.; Koskenmies, S.; Einarsdottir, E.; Panelius, J.; Hasan, T.; Julkunen, H.; Padyukov, L.; Kvarnström, M.; Wahren-Herlenius, M.; et al. Polymorphisms of the ITGAM Gene Confer Higher Risk of Discoid Cutaneous Than of Systemic Lupus Erythematosus. PLoS ONE 2010, 5, e14212. [Google Scholar] [CrossRef]
- Petersen, M.P.; Möller, S.; Bygum, A.; Voss, A.; Bliddal, M. Epidemiology of cutaneous lupus erythematosus and the associated risk of systemic lupus erythematosus: A nationwide cohort study in Denmark. Lupus 2018, 27, 1424–1430. [Google Scholar] [CrossRef]
- Liang, Y.; Tsoi, L.C.; Xing, X.; Beamer, M.A.; Swindell, W.R.; Sarkar, M.; Berthier, C.C.; Stuart, P.E.; Harms, P.W.; Nair, R.P.; et al. A gene network regulated by the transcription factor VGLL3 as a promoter of sex-biased autoimmune diseases. Nat. Immunol. 2017, 18, 152–160. [Google Scholar] [CrossRef]
- Billi, A.C.; Gharaee-Kermani, M.; Fullmer, J.; Tsoi, L.C.; Hill, B.D.; Gruszka, D.; Ludwig, J.; Xing, X.; Estadt, S.; Wolf, S.J.; et al. The female-biased factor VGLL3 drives cutaneous and systemic autoimmunity. JCI Insight 2019, 4, e127291. [Google Scholar] [CrossRef]


{kind=link}
| Subtype | Variant |
|---|---|
| ACLE | Localized (Malar rash) Generalized |
| SCLE | Annular Papulosquamous |
| ICLE | Lupus erythematosus tumidus |
| CCLE | Discoid lupus erythematosus (DLE) Localized Generalized Lupus erythematosus profundus Chilblain lupus erythematosus Hypertrophic CCLE Mucocutaneous CCLE |
| Class | Sub-Class | Drug |
|---|---|---|
| Antihypertensives | Thiazide diuretics | Hydrochlorothiazide, Hydrochlorothiazide + triamterene, Chlorothiazide |
| Calcium channel blockers | Diltiazem, Verapamil, Nifedipine, Nitrendipine | |
| ACE inhibitors | Captopril, Cilazapril, Enalapril, Lisinopril, Ramipril | |
| Beta blockers | Acebutolol, Oxprenolol | |
| Proton pump inhibitors | Lansoprazole, Esomeprazole, Omeprazole, Pantoprazole | |
| Antifungals | Terbinafine, Griseofulvin | |
| Antiepileptics | Carbamazepine, Phenytoin | |
| Statins | Pravastatin, Simvastatin | |
| Antihistamines | Ranitidine, Brompheniramine, Cinnarizine+thiethylperazine | |
| Antibiotics | Amoxicillin+clavulanic acid, Ciprofloxacin | |
| NSAIDs | Naproxen, Piroxicam | |
| Chemotherapeutics | Docetaxel, Paclitaxel, Tamoxifen, Capecitabine, Doxorubicin, Gemcitabine, Masitinib, Mitotane, Palbociclib, Uracil-tegafur, | |
| 5-Fluorouracil, Nivolumab, Pembrolizumab | ||
| Biologics | Anti-TNF | Etanercept, Infliximab, Adalimumab, Golimumab, Abatacept |
| Anti-CD11a | Efalizumab | |
| Anti-IL-12/23 | Ustekinumab | |
| Anti-IL-17 | Secukinumab | |
| Antidepressants | Bupropion | |
| Immunomodulators | Leflunomide, IFN-α and β | |
| Hormone-altering drugs | Leuprorelin, Anastrozole | |
| Others | Allopurinol, Ticlopidine, Tiotropium, IVIG |
| 5-Fluorouracil |
| NSAIDs |
| TNF-α inhibitors |
| IVIG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyagawa, F. Current Knowledge of the Molecular Pathogenesis of Cutaneous Lupus Erythematosus. J. Clin. Med. 2023, 12, 987. https://doi.org/10.3390/jcm12030987
Miyagawa F. Current Knowledge of the Molecular Pathogenesis of Cutaneous Lupus Erythematosus. Journal of Clinical Medicine. 2023; 12(3):987. https://doi.org/10.3390/jcm12030987
Chicago/Turabian StyleMiyagawa, Fumi. 2023. "Current Knowledge of the Molecular Pathogenesis of Cutaneous Lupus Erythematosus" Journal of Clinical Medicine 12, no. 3: 987. https://doi.org/10.3390/jcm12030987
APA StyleMiyagawa, F. (2023). Current Knowledge of the Molecular Pathogenesis of Cutaneous Lupus Erythematosus. Journal of Clinical Medicine, 12(3), 987. https://doi.org/10.3390/jcm12030987