


Pharmacological Modulation of β-Catenin Preserves Endothelial Barrier Integrity and Mitigates Retinal Vascular Permeability and Inflammation


 , , and
, , and
Abstract
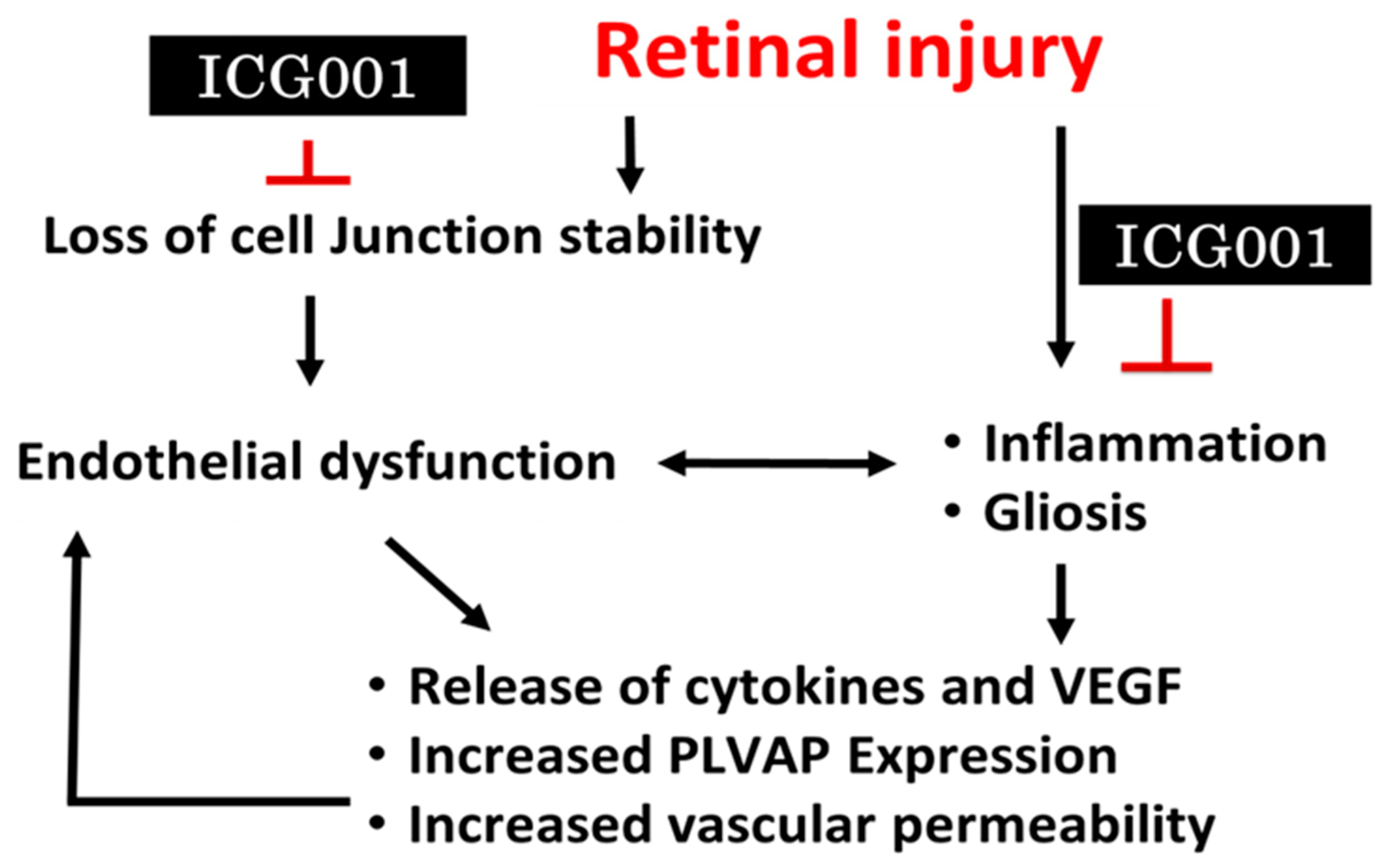
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Measurement of Endothelial-Barrier Resistance
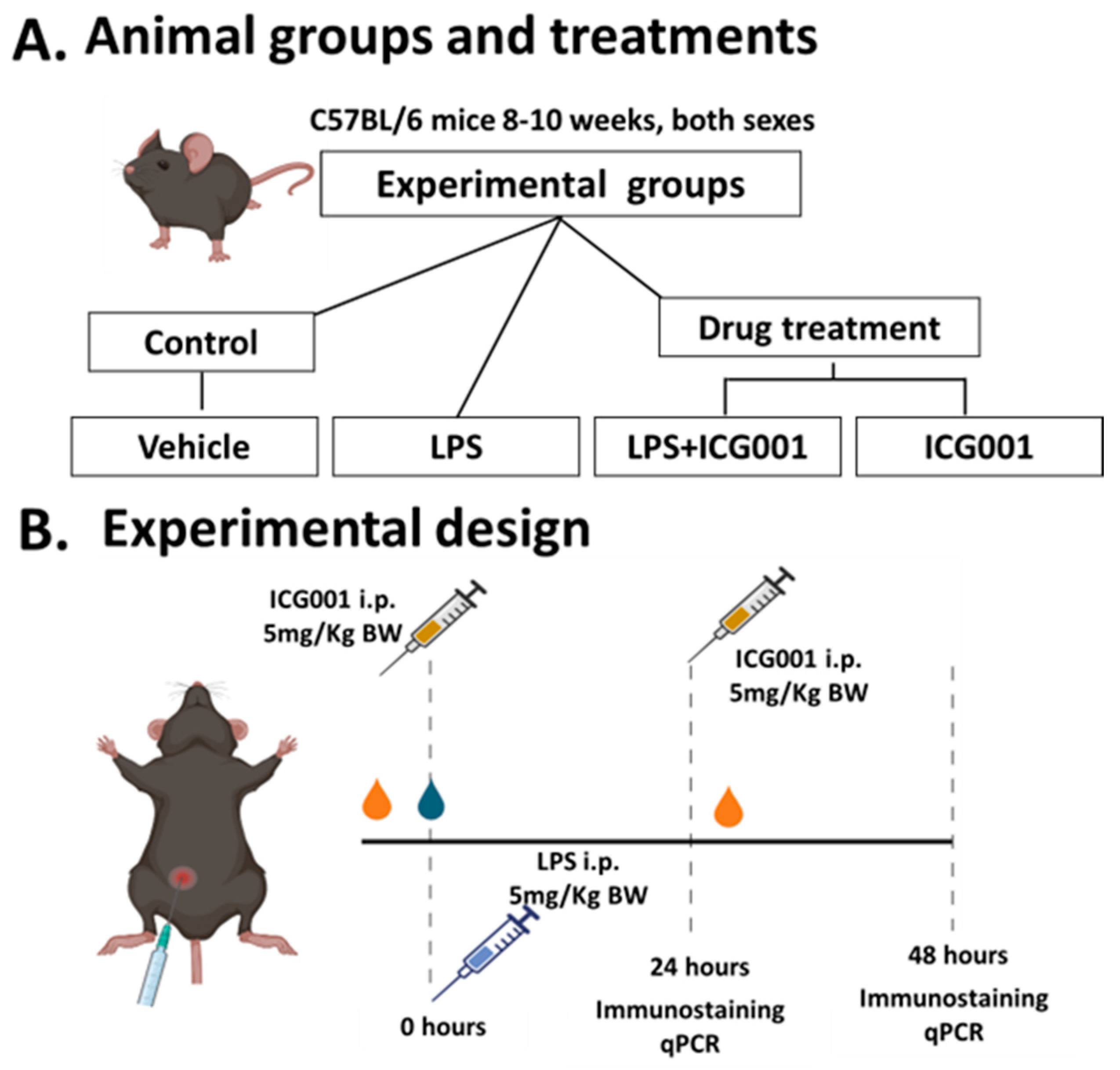
2.3. Animals
2.4. LPS-Induced Retinal Inflammation Model, ICG001 Treatment, and Sample Collection
2.5. Immunofluorescence Staining
2.6. Measurement of Vascular Permeability Using Albumin Immunostaining
2.7. Quantification of Immunofluorescence
2.8. RNA Isolation and Quantitative RT-PCR
2.9. Statistical Analysis
3. Results
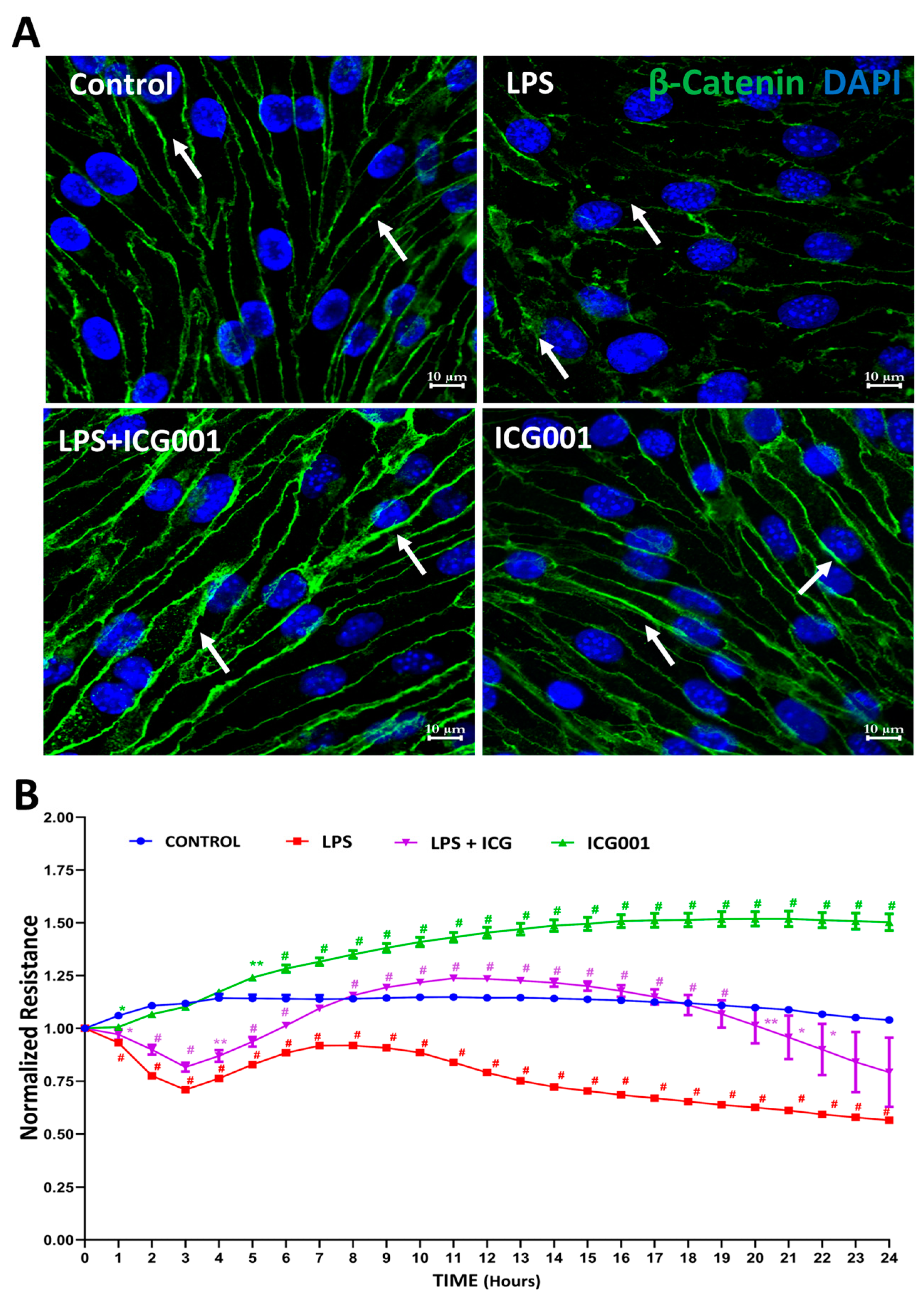
3.1. Treatment with ICG001 Strengthened the HREC Barrier by Improving β-Catenin Junctional Distribution
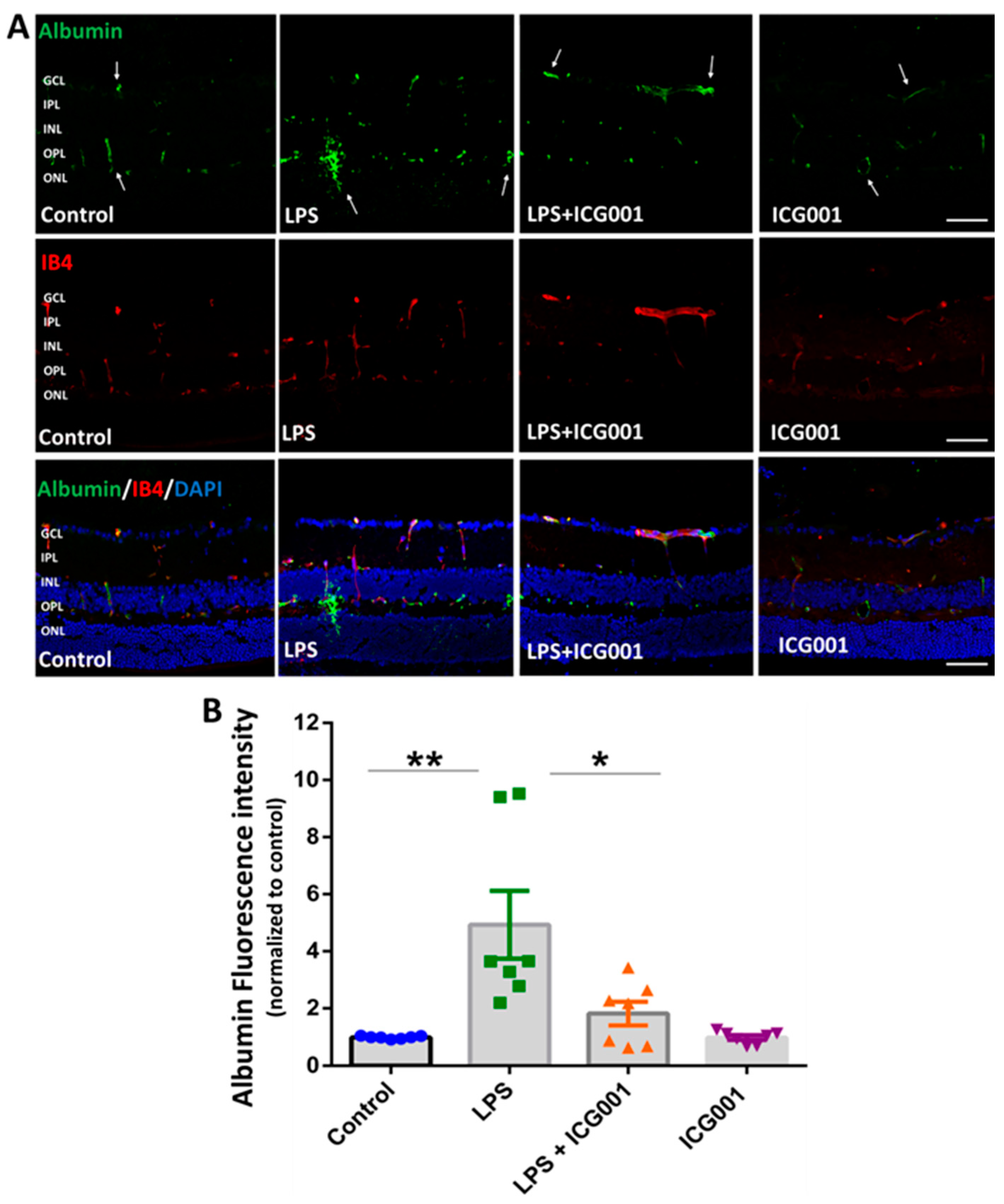
3.2. Treatment of Mice with ICG001 Blocked LPS-Induced Retinal Vascular Leakage
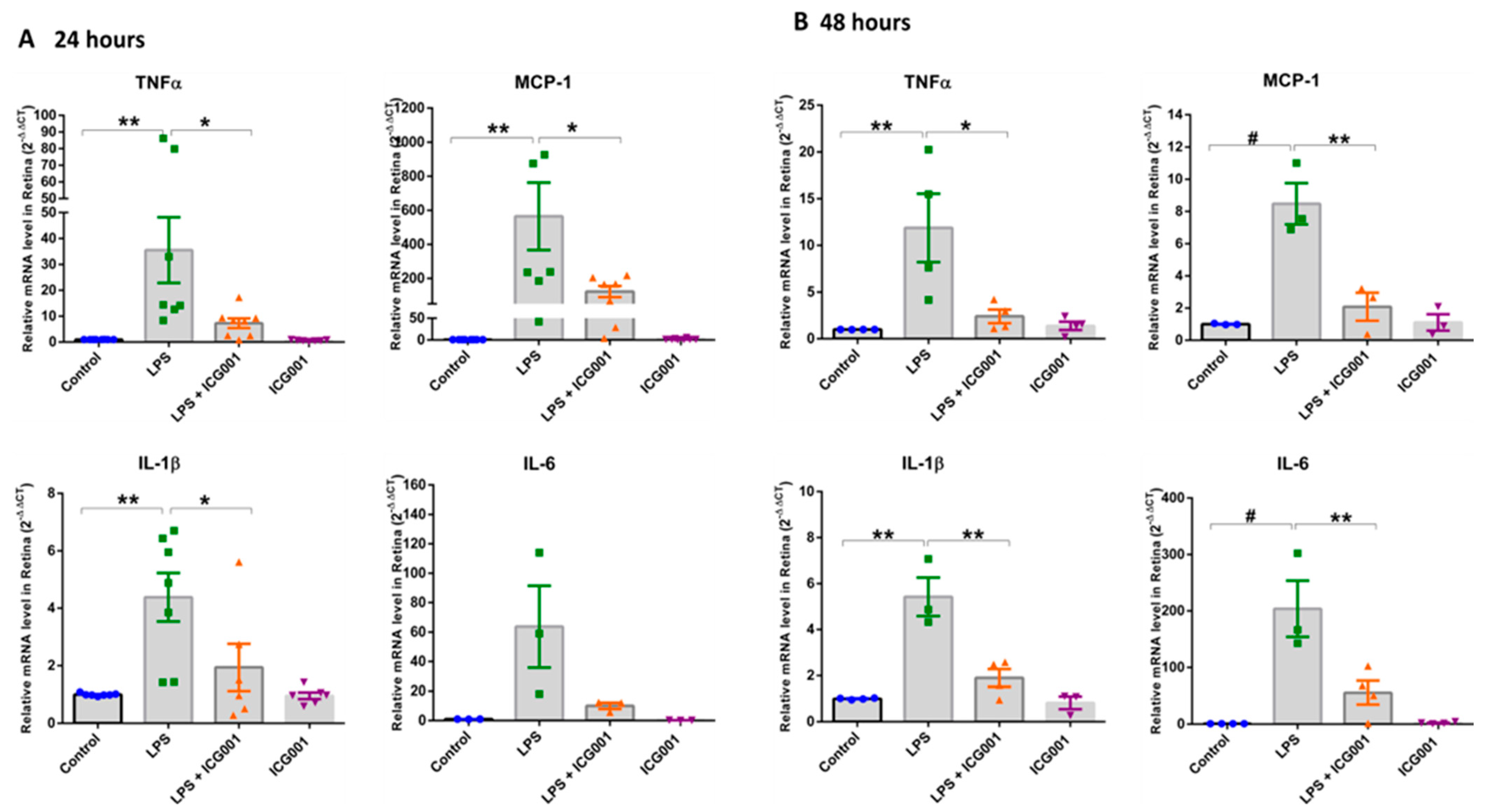
3.3. Treatment of Mice with ICG001 Blunted the LPS-Induced Increase in Retinal Pro-Inflammatory Cytokines and Chemokines
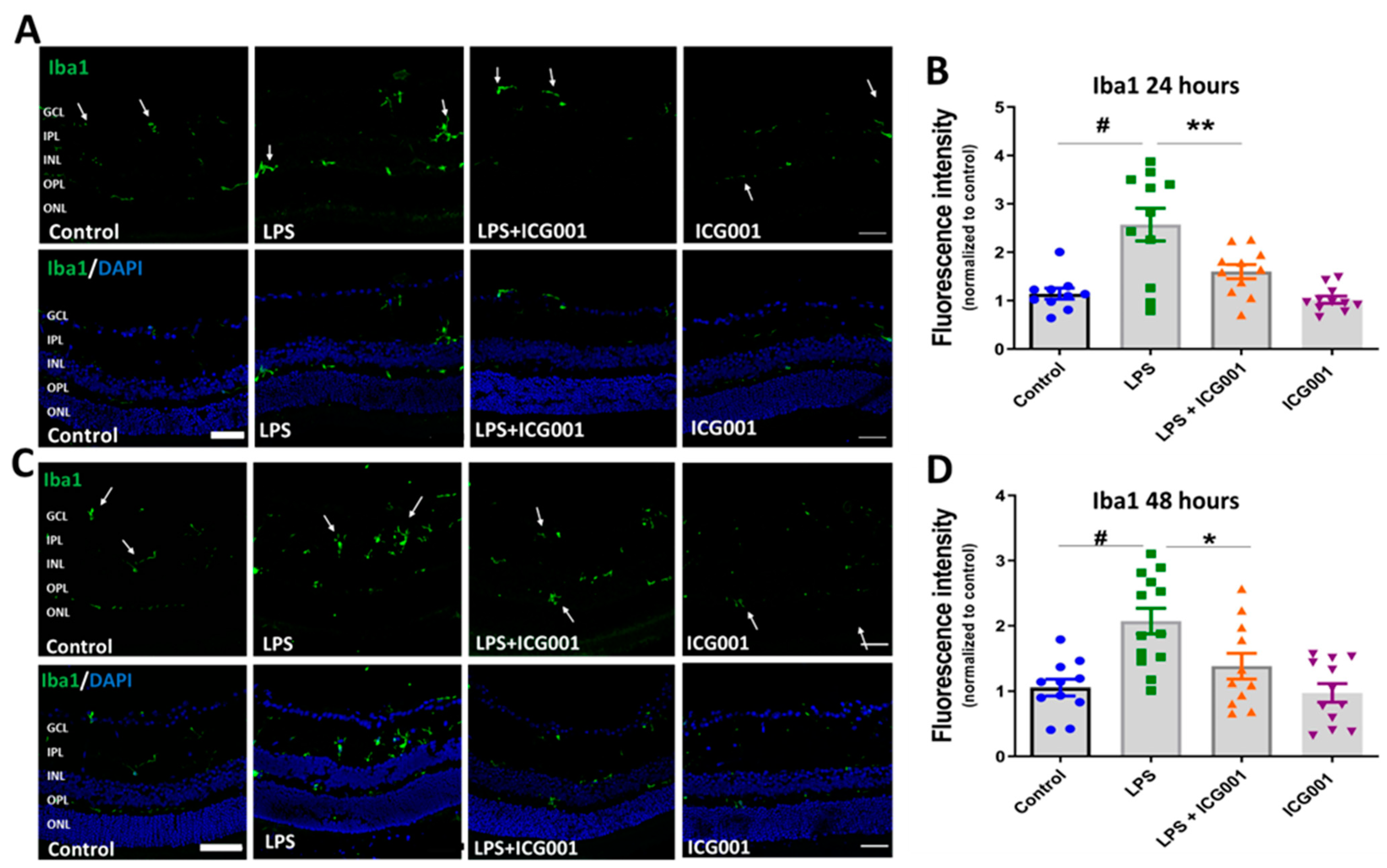
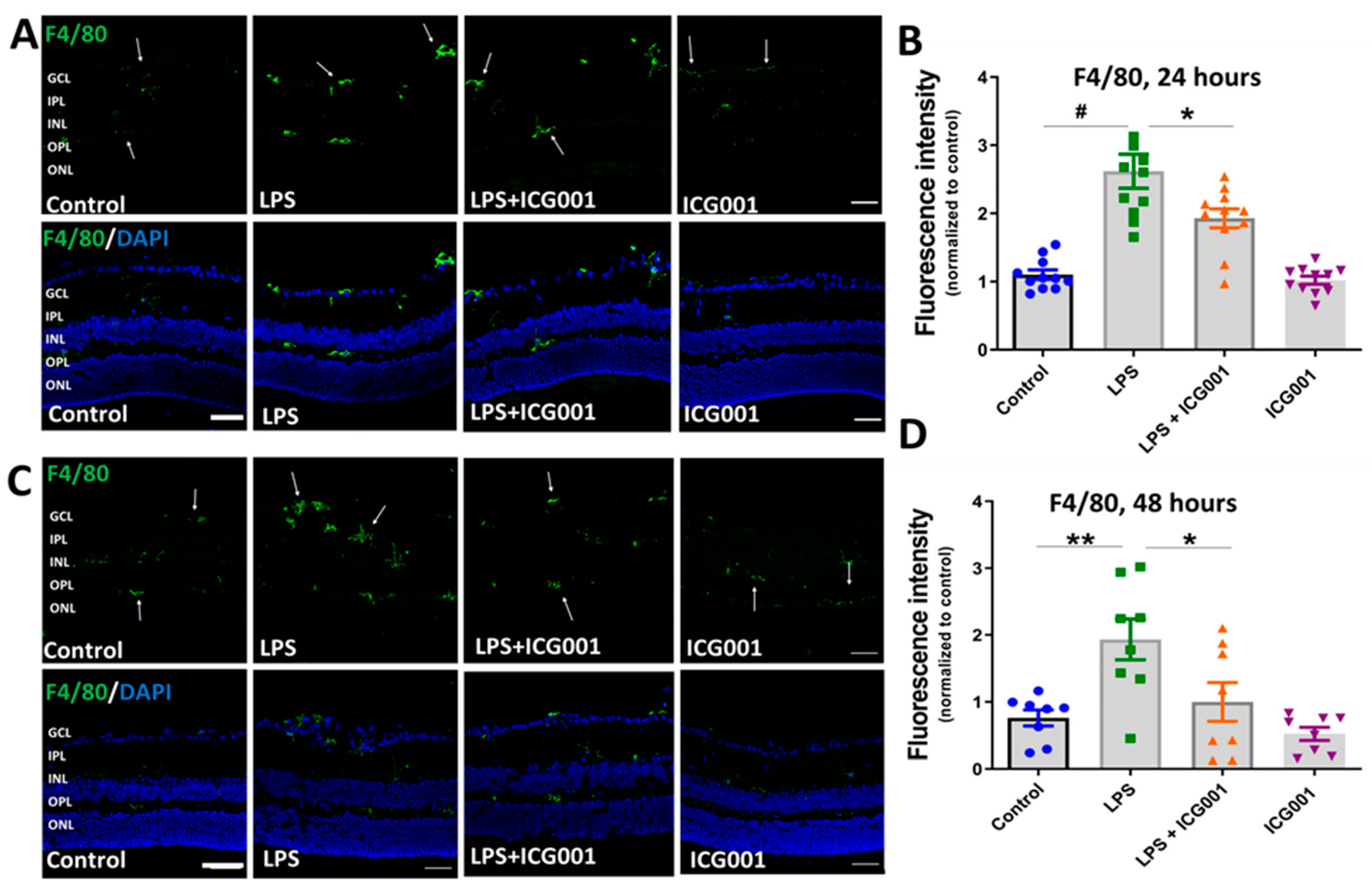
3.4. ICG001 Treatment Ameliorated LPS-Induced Microglia/Macrophage Activation
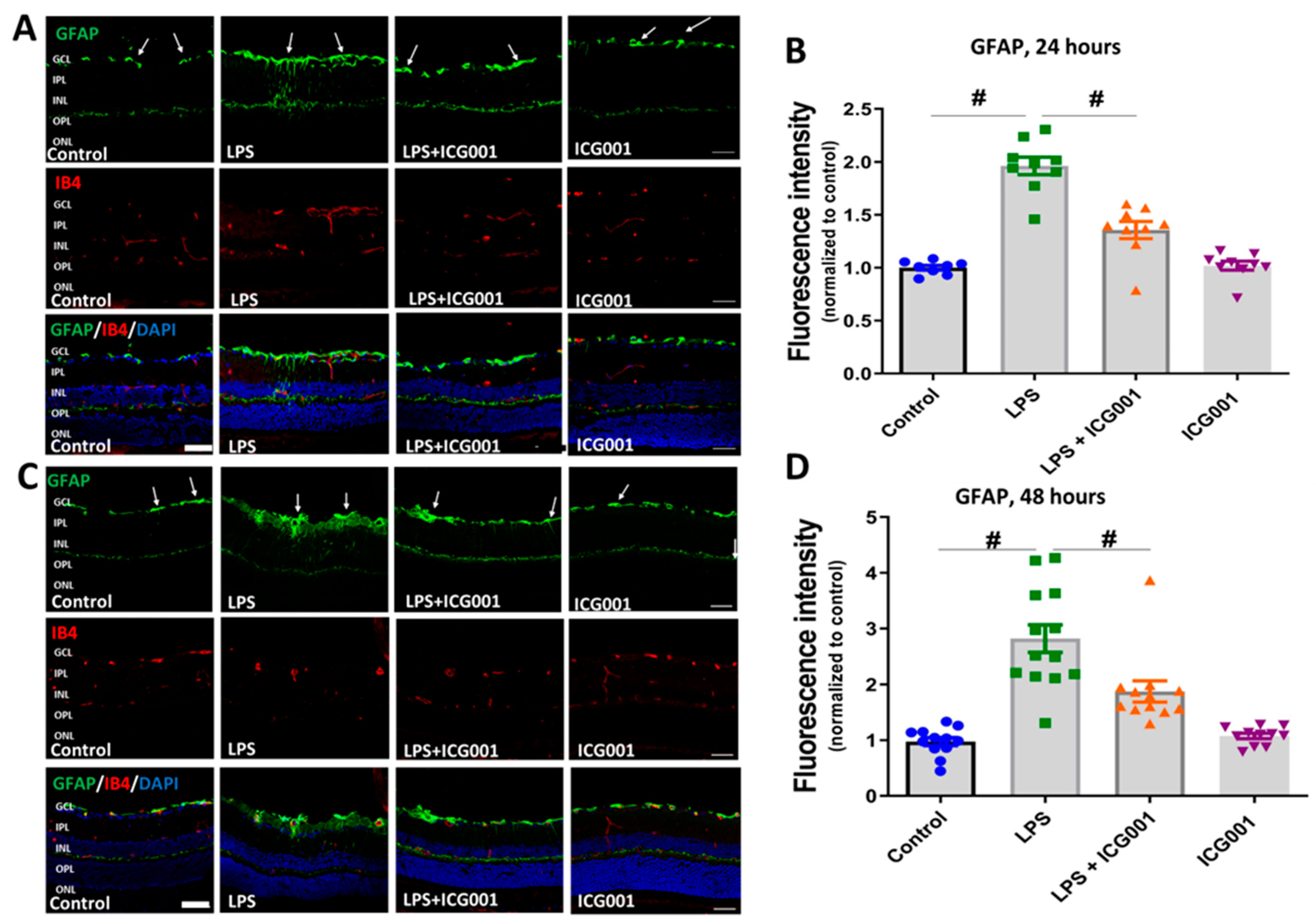
3.5. LPS-Induced Retinal Gliosis Was Attenuated by ICG001 Treatment
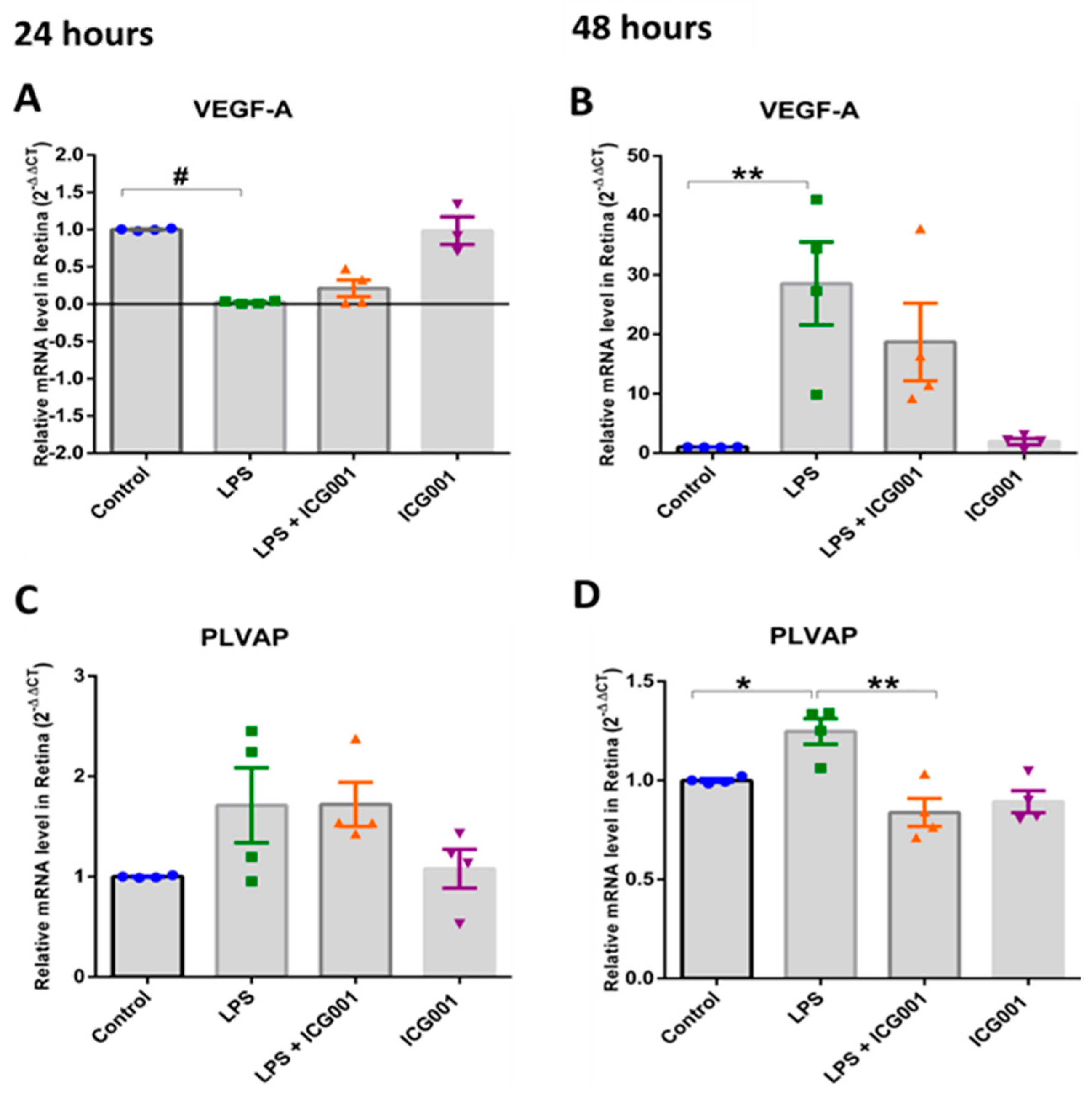
3.6. Treatment with ICG001-Modulated, LPS-Induced VEGF and PLVAP Expression in the Retina
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, J.R.; Hanumunthadu, D. Inflammatory eye disease: An overview of clinical presentation and management. Clin. Med. 2022, 22, 100–103. [Google Scholar] [CrossRef]
- Sherman, E.R.; Cafiero-Chin, M. Overcoming diagnostic and treatment challenges in uveitic glaucoma. Clin. Exp. Optom. 2019, 102, 109–115. [Google Scholar] [CrossRef]
- Deobhakta, A.; Chang, L.K. Inflammation in retinal vein occlusion. Int. J. Inflam. 2013, 2013, 438412. [Google Scholar] [CrossRef]
- Campa, C.; Costagliola, C.; Incorvaia, C.; Sheridan, C.; Semeraro, F.; De Nadai, K.; Sebastiani, A.; Parmeggiani, F. Inflammatory mediators and angiogenic factors in choroidal neovascularization: Pathogenetic interactions and therapeutic implications. Mediat. Inflamm. 2010, 2010, 546826. [Google Scholar] [CrossRef]
- Abcouwer, S.F. Angiogenic Factors and Cytokines in Diabetic Retinopathy. J. Clin. Cell Immunol. 2013, 11 (Suppl. S1), 1–12. [Google Scholar] [CrossRef]
- Gurung, R.L.; FitzGerald, L.M.; Liu, E.; McComish, B.J.; Kaidonis, G.; Ridge, B.; Hewitt, A.W.; Vote, B.J.; Verma, N.; Craig, J.E.; et al. Predictive factors for treatment outcomes with intravitreal anti-vascular endothelial growth factor injections in diabetic macular edema in clinical practice. Int. J. Retin. Vitr. 2023, 9, 23. [Google Scholar] [CrossRef]
- Abcouwer, S.F.; Shanmugam, S.; Muthusamy, A.; Lin, C.-m.; Kong, D.; Hager, H.; Liu, X.; Antonetti, D.A. Inflammatory resolution and vascular barrier restoration after retinal ischemia reperfusion injury. J. Neuroinflamm. 2021, 18, 186. [Google Scholar] [CrossRef]
- Bromberg-White, J.L.; Glazer, L.; Downer, R.; Furge, K.; Boguslawski, E.; Duesbery, N.S. Identification of VEGF-Independent Cytokines in Proliferative Diabetic Retinopathy Vitreous. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6472–6480. [Google Scholar] [CrossRef]
- Smith, R.O.; Ninchoji, T.; Gordon, E.; André, H.; Dejana, E.; Vestweber, D.; Kvanta, A.; Claesson-Welsh, L. Vascular permeability in retinopathy is regulated by VEGFR2 Y949 signaling to VE-cadherin. eLife 2020, 9, e54056. [Google Scholar] [CrossRef]
- Daruich, A.; Matet, A.; Moulin, A.; Kowalczuk, L.; Nicolas, M.; Sellam, A.; Rothschild, P.-R.; Omri, S.; Gélizé, E.; Jonet, L.; et al. Mechanisms of macular edema: Beyond the surface. Progress Retin. Eye Res. 2018, 63, 20–68. [Google Scholar] [CrossRef]
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.; Bach, A.; Obermüller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G216–G228. [Google Scholar] [CrossRef]
- Yang, J.M.; Yun, K.; Jeon, J.; Yang, H.Y.; Kim, B.; Jeong, S.; Lee, J.; Oh, W.-Y.; Uemura, A.; Song, J.S.; et al. Multimodal evaluation of an interphotoreceptor retinoid-binding protein-induced mouse model of experimental autoimmune uveitis. Exp. Mol. Med. 2022, 54, 252–262. [Google Scholar] [CrossRef]
- Goddard, L.M.; Iruela-Arispe, M.L. Cellular and molecular regulation of vascular permeability. Thromb. Haemost. 2013, 109, 407–415. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Lampugnani, M.G.; Dejana, E.; Giampietro, C. Vascular Endothelial (VE)-Cadherin, Endothelial Adherens Junctions, and Vascular Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029322. [Google Scholar] [CrossRef]
- Harris, E.S.; Nelson, W.J. VE-cadherin: At the front, center, and sides of endothelial cell organization and function. Curr. Opin. Cell Biol. 2010, 22, 651–658. [Google Scholar] [CrossRef]
- Lampugnani, M.G.; Orsenigo, F.; Gagliani, M.C.; Tacchetti, C.; Dejana, E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J. Cell Biol. 2006, 174, 593–604. [Google Scholar] [CrossRef]
- Braga, V.M.; Del Maschio, A.; Machesky, L.; Dejana, E. Regulation of cadherin function by Rho and Rac: Modulation by junction maturation and cellular context. Mol. Biol. Cell 1999, 10, 9–22. [Google Scholar] [CrossRef]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, J.S. VE-cadherin and claudin-5: It takes two to tango. Nat. Cell Biol. 2008, 10, 883–885. [Google Scholar] [CrossRef]
- Li, X.; Padhan, N.; Sjöström, E.O.; Roche, F.P.; Testini, C.; Honkura, N.; Sáinz-Jaspeado, M.; Gordon, E.; Bentley, K.; Philippides, A.; et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat. Commun. 2016, 7, 11017. [Google Scholar] [CrossRef] [PubMed]
- Akla, N.; Viallard, C.; Popovic, N.; Lora Gil, C.; Sapieha, P.; Larrivée, B. BMP9 (Bone Morphogenetic Protein-9)/Alk1 (Activin-Like Kinase Receptor Type I) Signaling Prevents Hyperglycemia-Induced Vascular Permeability. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1821–1836. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Breslin, J.W.; Wu, M.H.; Gottardi, C.J.; Yuan, S.Y. VE-cadherin and beta-catenin binding dynamics during histamine-induced endothelial hyperpermeability. Am. J. Physiol. Cell Physiol. 2008, 294, C977–C984. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Nyqvist, D.; Orsenigo, F.; Caprini, A.; Giampietro, C.; Taketo, M.M.; Iruela-Arispe, M.L.; Adams, R.H.; Dejana, E. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev. Cell 2010, 18, 938–949. [Google Scholar] [CrossRef]
- Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Distinct Mechanisms of Human Retinal Endothelial Barrier Modulation In Vitro by Mediators of Diabetes and Uveitis. Life 2022, 12, 33. [Google Scholar]
- Jang, J.; Jung, Y.; Kim, Y.; Jho, E.H.; Yoon, Y. LPS-induced inflammatory response is suppressed by Wnt inhibitors, Dickkopf-1 and LGK974. Sci. Rep. 2017, 7, 41612. [Google Scholar] [CrossRef]
- Troyanovsky, R.B.; Sokolov, E.P.; Troyanovsky, S.M. Endocytosis of cadherin from intracellular junctions is the driving force for cadherin adhesive dimer disassembly. Mol. Biol. Cell 2006, 17, 3484–3493. [Google Scholar] [CrossRef]
- Jones, C.A.; London, N.R.; Chen, H.; Park, K.W.; Sauvaget, D.; Stockton, R.A.; Wythe, J.D.; Suh, W.; Larrieu-Lahargue, F.; Mukouyama, Y.S.; et al. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat. Med. 2008, 14, 448–453. [Google Scholar] [CrossRef]
- Cowan, C.E.; Kohler, E.E.; Dugan, T.A.; Mirza, M.K.; Malik, A.B.; Wary, K.K. Kruppel-like factor-4 transcriptionally regulates VE-cadherin expression and endothelial barrier function. Circ. Res. 2010, 107, 959–966. [Google Scholar] [CrossRef]
- Gavard, J.; Patel, V.; Gutkind, J.S. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev. Cell 2008, 14, 25–36. [Google Scholar] [CrossRef]
- Lin, M.T.; Yen, M.L.; Lin, C.Y.; Kuo, M.L. Inhibition of vascular endothelial growth factor-induced angiogenesis by resveratrol through interruption of Src-dependent vascular endothelial cadherin tyrosine phosphorylation. Mol. Pharmacol. 2003, 64, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.L.; Kim, H.Y.; Moon, S.H.; et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Alwhaibi, A.; Artham, S.; Verma, A.; Somanath, P.R. Endothelial Akt1 loss promotes prostate cancer metastasis via β-catenin-regulated tight-junction protein turnover. Br. J. Cancer 2018, 118, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Sabbineni, H.; Verma, A.; Artham, S.; Anderson, D.; Amaka, O.; Liu, F.; Narayanan, S.P.; Somanath, P.R. Pharmacological inhibition of β-catenin prevents EndMT in vitro and vascular remodeling in vivo resulting from endothelial Akt1 suppression. Biochem. Pharmacol. 2019, 164, 205–215. [Google Scholar] [CrossRef]
- Gao, F.; Artham, S.; Sabbineni, H.; Al-Azayzih, A.; Peng, X.-D.; Hay, N.; Adams, R.H.; Byzova, T.V.; Somanath, P.R. Akt1 promotes stimuli-induced endothelial-barrier protection through FoxO-mediated tight-junction protein turnover. Cell. Mol. Life Sci. 2016, 73, 3917–3933. [Google Scholar] [CrossRef]
- Rosenbaum, J.T.; McDevitt, H.O.; Guss, R.B.; Egbert, P.R. Endotoxin-induced uveitis in rats as a model for human disease. Nature 1980, 286, 611–613. [Google Scholar] [CrossRef]
- Burkholder, T.H.; Linton, G.; Hoyt, R.F.; Young, R. Chapter 18—The Rabbit as an Experimental Model. In The Laboratory Rabbit, Guinea Pig, Hamster, and Other Rodents; Suckow, M.A., Stevens, K.A., Wilson, R.P., Eds.; Academic Press: Boston, MA, USA, 2012; pp. 529–560. [Google Scholar]
- Liu, F.; Saul, A.B.; Pichavaram, P.; Xu, Z.; Rudraraju, M.; Somanath, P.R.; Smith, S.B.; Caldwell, R.B.; Narayanan, S.P. Pharmacological Inhibition of Spermine Oxidase Reduces Neurodegeneration and Improves Retinal Function in Diabetic Mice. J. Clin. Med. 2020, 9, 340. [Google Scholar] [CrossRef]
- Alfarhan, M.; Liu, F.; Shan, S.; Pichavaram, P.; Somanath, P.R.; Narayanan, S.P. Pharmacological Inhibition of Spermine Oxidase Suppresses Excitotoxicity Induced Neuroinflammation in Mouse Retina. Int. J. Mol. Sci. 2022, 23, 2133. [Google Scholar] [CrossRef]
- Shan, S.; Liu, F.; Ford, E.; Caldwell, R.B.; Narayanan, S.P.; Somanath, P.R. Triciribine attenuates pathological neovascularization and vascular permeability in a mouse model of proliferative retinopathy. Biomed. Pharmacother. 2023, 162, 114714. [Google Scholar] [CrossRef]
- Radu, M.; Chernoff, J. An in vivo assay to test blood vessel permeability. JoVE (J. Vis. Exp.) 2013, 73, e50062. [Google Scholar]
- Wang, A.L.; Yu, A.C.; Lau, L.T.; Lee, C.; Wu, L.M.; Zhu, X.; Tso, M.O. Minocycline inhibits LPS-induced retinal microglia activation. Neurochem. Int. 2005, 47, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Regulation of blood-retinal barrier cell-junctions in diabetic retinopathy. Pharmacol. Res. 2020, 161, 105115. [Google Scholar] [CrossRef] [PubMed]
- Mrugacz, M.; Bryl, A.; Zorena, K. Retinal Vascular Endothelial Cell Dysfunction and Neuroretinal Degeneration in Diabetic Patients. J. Clin. Med. 2021, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, Y.; Stock, O.; Pfister, F.; Tanimoto, N.; Seeliger, M.W.; Hillebrands, J.L.; Hoffmann, S.; Wolburg, H.; Gretz, N.; et al. Vasoregression linked to neuronal damage in the rat with defect of polycystin-2. PLoS ONE 2009, 4, e7328. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.H.; Frank, R.N.; Kennedy, A.; Eliott, D.; Puklin, J.E.; Abrams, G.W. Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 1997, 38, 36–47. [Google Scholar]
- Günzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef]
- Furuse, M. Molecular basis of the core structure of tight junctions. Cold Spring Harb. Perspect. Biol. 2010, 2, a002907. [Google Scholar] [CrossRef]
- Niessen, C.M. Tight junctions/adherens junctions: Basic structure and function. J. Investig. Dermatol. 2007, 127, 2525–2532. [Google Scholar] [CrossRef]
- Adil, M.S.; Narayanan, S.P.; Somanath, P.R. Cell-cell junctions: Structure and regulation in physiology and pathology. Tissue Barriers 2021, 9, 1848212. [Google Scholar] [CrossRef]
- Kong, Y.; Naggert, J.K.; Nishina, P.M. The Impact of Adherens and Tight Junctions on Physiological Function and Pathological Changes in the Retina. Adv. Exp. Med. Biol. 2018, 1074, 545–551. [Google Scholar] [CrossRef]
- Erickson, K.K.; Sundstrom, J.M.; Antonetti, D.A. Vascular permeability in ocular disease and the role of tight junctions. Angiogenesis 2007, 10, 103–117. [Google Scholar] [CrossRef]
- Beckers, C.M.L.; García-Vallejo, J.J.; van Hinsbergh, V.W.M.; van Nieuw Amerongen, G.P. Nuclear targeting of β-catenin and p120ctn during thrombin-induced endothelial barrier dysfunction. Cardiovasc. Res. 2008, 79, 679–688. [Google Scholar] [CrossRef]
- Tran, K.A.; Zhang, X.; Predescu, D.; Huang, X.; Machado, R.F.; Göthert, J.R.; Malik, A.B.; Valyi-Nagy, T.; Zhao, Y.Y. Endothelial β-Catenin Signaling Is Required for Maintaining Adult Blood-Brain Barrier Integrity and Central Nervous System Homeostasis. Circulation 2016, 133, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sabbagh, M.F.; Gu, X.; Rattner, A.; Williams, J.; Nathans, J. Beta-catenin signaling regulates barrier-specific gene expression in circumventricular organ and ocular vasculatures. eLife 2019, 8, e43257. [Google Scholar] [CrossRef]
- Shang, S.; Hua, F.; Hu, Z.W. The regulation of β-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [PubMed]
- Drenser, K.A. Wnt signaling pathway in retinal vascularization. Eye Brain 2016, 8, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Sabbineni, H.; Artham, S.; Somanath, P. Modulation of Long-Term Endothelial-Barrier Integrity Is Conditional to the Cross-Talk between Akt and Src Signaling. J. Cell. Physiol. 2017, 232, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Zhou, T.; Zhou, K.K.; Mott, R.; Wu, M.; Boulton, M.; Lyons, T.J.; Gao, G.; Ma, J.-x. Activation of the Wnt Pathway Plays a Pathogenic Role in Diabetic Retinopathy in Humans and Animal Models. Am. J. Pathol. 2009, 175, 2676–2685. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, X.; Cheng, R.; Ma, J.-x.; Yi, J.; Li, J. Salutary effect of fenofibrate on type 1 diabetic retinopathy via inhibiting oxidative stress–mediated Wnt/β-catenin pathway activation. Cell Tissue Res. 2019, 376, 165–177. [Google Scholar] [CrossRef]
- Chen, Q.; Ma, J.-x. Canonical Wnt signaling in diabetic retinopathy. Vision Res. 2017, 139, 47–58. [Google Scholar] [CrossRef]
- Arensman, M.D.; Telesca, D.; Lay, A.R.; Kershaw, K.M.; Wu, N.; Donahue, T.R.; Dawson, D.W. The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol. Cancer Ther. 2014, 13, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Schlingemann, R.O.; Hofman, P.; Vrensen, G.F.; Blaauwgeers, H.G. Increased expression of endothelial antigen PAL-E in human diabetic retinopathy correlates with microvascular leakage. Diabetologia 1999, 42, 596–602. [Google Scholar] [CrossRef]
- Schlingemann, R.O.; Hofman, P.; Anderson, L.; Troost, D.; van der Gaag, R. Vascular expression of endothelial antigen PAL-E indicates absence of blood-ocular barriers in the normal eye. Ophthalmic Res. 1997, 29, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Bosma, E.K.; van Noorden, C.J.F.; Schlingemann, R.O.; Klaassen, I. The role of plasmalemma vesicle-associated protein in pathological breakdown of blood-brain and blood-retinal barriers: Potential novel therapeutic target for cerebral edema and diabetic macular edema. Fluids Barriers CNS 2018, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Stan, R.V.; Tkachenko, E.; Niesman, I.R. PV1 is a key structural component for the formation of the stomatal and fenestral diaphragms. Mol. Biol. Cell 2004, 15, 3615–3630. [Google Scholar] [CrossRef]
- Strickland, L.A.; Jubb, A.M.; Hongo, J.A.; Zhong, F.; Burwick, J.; Fu, L.; Frantz, G.D.; Koeppen, H. Plasmalemmal vesicle-associated protein (PLVAP) is expressed by tumour endothelium and is upregulated by vascular endothelial growth factor-A (VEGF). J. Pathol. 2005, 206, 466–475. [Google Scholar] [CrossRef]
- Zhao, L.; Zabel, M.K.; Wang, X.; Ma, W.; Shah, P.; Fariss, R.N.; Qian, H.; Parkhurst, C.N.; Gan, W.B.; Wong, W.T. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 2015, 7, 1179–1197. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody/Probe | Dilution | Company | Catalog No. | Experiment |
|---|---|---|---|---|
| Primary Antibodies | ||||
| β-Catenin | 1:200 | Cell Signaling, (Danvers, MA, USA) | 9562 | Immunostaining |
| Albumin | 1:200 | Bethyl Laboratories, (Montgomery, TX, USA) | A110-134 | Immunostaining |
| Iba-1 | 1:500 | Wako, (Neuss, Germany) | 019-19741 | Immunostaining |
| F4/80 | 1:100 | Abcam, (Waltham MA USA) | ab6640 | Immunostaining |
| GFAP | 1:500 | Agilent Technologies, (Santa Clara, CA, USA) | Z033429–2 | Immunostaining |
| Isolectin, GS-IB4 Alexa Fluor 594 | 1:200 | Thermo Fisher Scientific | I21413 | Immunostaining |
| Secondary Antibodies | ||||
| Donkey anti-Sheep IgG Alexa Fluor 488 | 1:500 | Jaxson Immune Research, (West Grove, PA, USA) | 713-545-147 | Immunostaining |
| Donkey anti-Rabbit IgG Alexa Fluor 488 | 1:500 | Invitrogen, (Waltham, MA, USA) | A21206 | Immunostaining |
| Donkey anti-Rat IgG Alexa Fluor 488 | 1:500 | Invitrogen | A21209 | Immunostaining |
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| TNFα | GGTCCCCAAAGGGATGAGAA | TGAGGGTCTGGGCCATAGAA |
| IL1β | CCAAGCAACGACAAAATACC | GTTGAAGACAAACCGTTTTTCC |
| MCP-1 | GGCTCAGCCAGATGCAGTTAA | CCTACTCATTGGGATCATCTTGCT |
| IL6 | AGACAAAGCCAGAGTCCTTCAG | TGCCGAGTAGATCTCAAAGTGA |
| VEGF-A | AGCTCATGGACGGGTGAGG | CCTGGGACCACTTGGCAT |
| PLVAP | CGTCAAGGCCAAGTCGCT | CAGCAGGGTTGACTACAGGG |
| GAPDH | TGGTGAAGGTCGGTGTGAAC | CCATGTAGTTGAGGTCAATGAAGG |
| HPRT | GAAAGACTTGCTCGAGATGTCATG | CACACAGAGGGCCACAATGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudraraju, M.; Shan, S.; Liu, F.; Tyler, J.; Caldwell, R.B.; Somanath, P.R.; Narayanan, S.P. Pharmacological Modulation of β-Catenin Preserves Endothelial Barrier Integrity and Mitigates Retinal Vascular Permeability and Inflammation. J. Clin. Med. 2023, 12, 7145. https://doi.org/10.3390/jcm12227145
Rudraraju M, Shan S, Liu F, Tyler J, Caldwell RB, Somanath PR, Narayanan SP. Pharmacological Modulation of β-Catenin Preserves Endothelial Barrier Integrity and Mitigates Retinal Vascular Permeability and Inflammation. Journal of Clinical Medicine. 2023; 12(22):7145. https://doi.org/10.3390/jcm12227145
Chicago/Turabian StyleRudraraju, Madhuri, Shengshuai Shan, Fang Liu, Jennifer Tyler, Ruth B. Caldwell, Payaningal R. Somanath, and S. Priya Narayanan. 2023. "Pharmacological Modulation of β-Catenin Preserves Endothelial Barrier Integrity and Mitigates Retinal Vascular Permeability and Inflammation" Journal of Clinical Medicine 12, no. 22: 7145. https://doi.org/10.3390/jcm12227145
APA StyleRudraraju, M., Shan, S., Liu, F., Tyler, J., Caldwell, R. B., Somanath, P. R., & Narayanan, S. P. (2023). Pharmacological Modulation of β-Catenin Preserves Endothelial Barrier Integrity and Mitigates Retinal Vascular Permeability and Inflammation. Journal of Clinical Medicine, 12(22), 7145. https://doi.org/10.3390/jcm12227145