

Anti-Amyloid Therapy, AD, and ARIA: Untangling the Role of CAA


Abstract
1. Introduction
2. Amyloid β Plays a Key Role in the Pathogenesis of AD and CAA
3. Anti-Amyloid Therapies Are Emerging as Treatments for AD
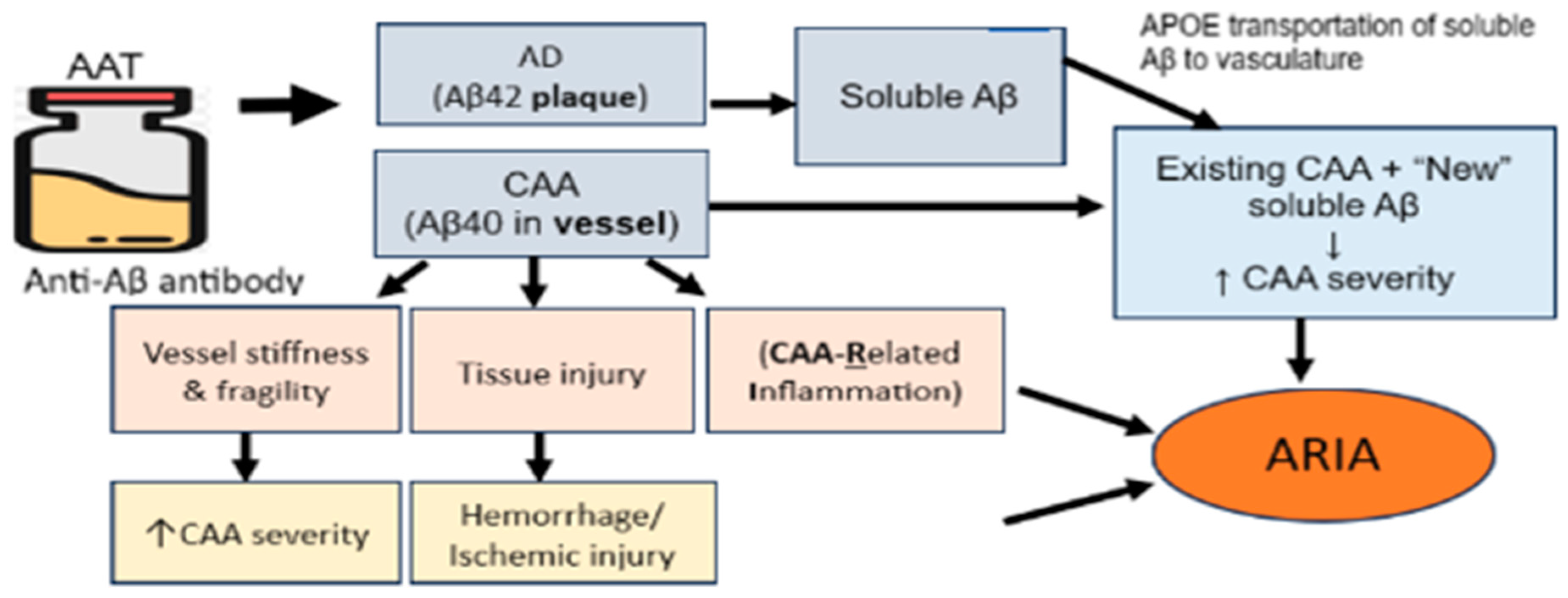
4. Amyloid-Related Imaging Abnormalities (ARIA) Are Significant Side Effects of AATs
5. CAA Is a Major Risk Factor for ARIA
6. CAA Is an Independent Risk Factor for Cognitive Impairment
7. Age and ApoE e4 Are Important Risk Factors for CAA
8. Transient Focal Neurological Episodes Are the Earliest Clinical Symptoms in CAA
9. A Sensitive and Reliable Diagnostic Biomarker for CAA Is Needed
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Disease Facts and Figures. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 19 September 2023).
- Sveikata, L.; Charidimou, A.; Viswanathan, A. Vessels sing their ARIAs: The role of vascular amyloid in the age of aducanumab. Stroke 2022, 53, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease Fact Sheet. Available online: https://www.nia.nih.gov/health/alzheimers-disease-fact-sheet (accessed on 19 September 2023).
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Ono, K.; Watanabe-Nakayama, T. Aggregation and structure of amyloid β-protein. Neurochem. Int. 2021, 151, 105208. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, H. Molecular and cellular mechanisms for Alzheimer’s disease: Understanding APP metabolism. Curr. Mol. Med. 2007, 7, 687–696. [Google Scholar] [CrossRef]
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 2013, 126, 305–311. [Google Scholar] [CrossRef]
- Han, B.H.; Zhou, M.; Vellimana, A.K.; Milner, E.; Kim, D.H.; Greenberg, J.K.; Chu, W.; Mach, R.H.; Zipfel, G.J. Resorufin analogs preferentially bind cerebrovascular amyloid: Potential use as imaging ligands for cerebral amyloid angiopathy. Mol. Neurodegener. 2011, 6, 86. [Google Scholar] [CrossRef]
- Qiu, T.; Liu, Q.; Chen, Y.; Zhao, Y.; Li, Y. Aβ42 and Aβ40: Similarities and differences. J. Pept. Sci. 2015, 21, 522–529. [Google Scholar] [CrossRef]
- DeSimone, C.V.; Graff-Radford, J.; El-Harasis, M.A.; Rabinstein, A.A.; Asirvatham, S.J.; Holmes, D.R.J. Cerebral amyloid angiopathy: Diagnosis, clinical implications, and management strategies in atrial fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Gordon, M.N.; Morgan, D. Quantification of cerebral amyloid angiopathy and parenchymal amyloid plaques with congo red histochemical stain. Nat. Protoc. 2006, 1, 1591–1595. [Google Scholar] [CrossRef]
- Kakuda, N.; Miyasaka, T.; Iwasaki, N.; Nirasawa, T.; Wade-Kakuda, S.; Takahashi-Fujigasaki, J.; Murayama, S.; Ihara, Y.; Ikegawa, M. Distinct deposition of amyloid-β species in brains with Alzheimer’s disease pathology visualized with MALDI imaging mass spectrometry. Acta Neuropathol. Commun. 2017, 5, 73. [Google Scholar] [CrossRef]
- Gravina, S.A.; Ho, L.; Eckman, C.B.; Long, K.E.; Otvos, L.; Younkin, L.H.; Suzuki, N.; Younkin, S.G. Amyloid beta protein (A beta) in Alzheimer’s disease brain. biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J. Biol. Chem. 1995, 270, 7013–7016. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Papayannopoulos, I.A.; Styles, J.; Bobin, S.A.; Biemann, L.K.; Iqbal, K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch. Biochem. Biophys. 1993, 301, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.; Sharman, T. Cerebral amyloid angiopathy. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2023. [Google Scholar]
- Verbeek, M.M.; Kremer, B.P.H.; Rikkert, M.O.; Van Domburg, P.H.M.F.; Skehan, M.E.; Greenberg, S.M. Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Ann. Neurol. 2009, 66, 245–249. [Google Scholar] [CrossRef]
- Norfray, J.F.; Provenzale, J.M. Alzheimer’s disease: Neuropathologic findings and recent advances in imaging. AJR Am. J. Roentgenol. 2004, 182, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Margraf, N.G.; Jensen-Kondering, U.; Weiler, C.; Leypoldt, F.; Maetzler, W.; Philippen, S.; Bartsch, T.; Fluh, C.; Rocken, C.; Moller, B.; et al. Cerebrospinal fluid biomarkers in cerebral amyloid angiopathy: New data and quantitative meta-analysis. Front. Aging Neurosci. 2022, 14, 783996. [Google Scholar] [CrossRef]
- Withington, C.G.; Turner, R.S. Amyloid-related imaging abnormalities with anti-amyloid antibodies for the treatment of dementia due to Alzheimer’s disease. Front. Neurol. 2022, 13, 862369. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- McDade, E.; Cummings, J.L.; Dhadda, S.; Swanson, C.J.; Reyderman, L.; Kanekiyo, M.; Koyama, A.; Irizarry, M.; Kramer, L.D.; Bateman, R.J. Lecanemab in patients with early Alzheimer’s disease: Detailed results on biomarker, cognitive, and clinical effects from the randomized and open-label extension of the phase 2 proof-of-concept study. Alzheimers Res. Ther. 2022, 14, 191–192. [Google Scholar] [CrossRef]
- Petersen, R.C.; Aisen, P.S.; Andrews, J.S.; Aatri, A.; Matthews, B.R.; Rentz, D.M.; Siemers, E.R.; Weber, C.J.; Carrillo, M.C. Expectations and clinical meaningfulness of randomized controlled trials. Alzheimers Dement. 2023, 19, 2730–2736. [Google Scholar] [CrossRef]
- Ramanan, V.K.; Day, G.S. Anti-amyloid therapies for alzheimer disease: Finally, good news for patients. Mol. Neurodegener. 2023, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Chamard, L.; Wallon, D.; Pijoff, A.; Berger, E.; Viennet, G.; Hannequin, D.; Magnin, E. Amyloid-related imaging abnormalities in AβPP duplication carriers. J. Alzheimers Dis. 2013, 37, 789–793. [Google Scholar] [CrossRef]
- Yang, J.; Chu, Y.; Tsai, H.; Jeng, J. Amyloid and tau PET in cerebral amyloid angiopathy-related inflammation two case reports and literature review. Front. Neurol. 2023, 14, 1153305. [Google Scholar] [CrossRef] [PubMed]
- Antolini, L.; Di Francesco, J.C.; Zedde, M.; Basso, G.; Arighi, A.; Shima, A.; Cagnin, A.; Caulo, M.; Carare, R.O.; Charidimou, A.; et al. Spontaneous ARIA-like events in cerebral amyloid angiopathy-related inflammation: A multicenter prospective longitudinal cohort study. Neurology 2021, 97, e1809–e1822. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.K.Y.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-aβ protofibril antibody. Alzheimers Res. Ther. 2021, 13, 80–88. [Google Scholar] [CrossRef]
- Honig, L.S.; Barakos, J.; Dhadda, S.; Kanekiyo, M.; Reyderman, L.; Irizarry, M.; Kramer, L.D.; Swanson, C.J.; Sabbagh, M. ARIA in patients treated with lecanemab (BAN2401) in a phase 2 study in early Alzheimer’s disease. Alzheimers Dement. 2023, 9, e12377. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Elhage, A.; Cho, M.; Apostolova, L.G.; Nicoll, J.A.R.; Atri, A. Amyloid-related imaging abnormalities (ARIA): Radiological, biological and clinical characteristics. Brain 2023, awad188. [Google Scholar] [CrossRef]
- Cogswell, P.M.; Barakos, J.A.; Barkhof, F.; Benzinger, T.S.; Jack, C.R.; Poussaint, T.Y.; Raji, C.A.; Whitlow, C.T. Amyloid-related imaging abnormalities with emerging alzheimer disease therapeutics: Detection and reporting recommendations for clinical practice. AJNR Am. J. Neuroradiol. 2022, 43, E19–E35. [Google Scholar] [CrossRef]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early alzheimer disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- Racke, M.M.; Boone, L.I.; Hepburn, D.L.; Parsadainian, M.; Bryan, M.; Ness, D.K.; Piroozi, K.S.; Jordan, W.H.; Brown, D.D.; Hoffman, W.P.; et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J. Neurosci. 2005, 25, 629–636. [Google Scholar]
- Piazza, F.; Greenberg, S.M.; Savoiardo, M.; Gardinetti, M.; Chiapparini, L.; Raicher, I.; Nitrini, R.; Sakaguchi, H.; Brioschi, M.; Billo, G.; et al. Anti-amyloid β autoantibodies in cerebral amyloid angiopathy-related inflammation: Implications for amyloid-modifying therapies. Ann. Neurol. 2013, 73, 449–458. [Google Scholar] [CrossRef]
- Sperling, R.A.; Jack, C.R.J.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s association research roundtable workgroup. Alzheimers Dement. 2011, 7, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Roytman, M.; Mashriqi, F.; Al-Tawil, K.; Schulz, P.E.; Zaharchuk, G.; Benzinger, T.L.S.; Franceschi, A.M. Amyloid-related imaging abnormalities: An update. AJR Am. J. Roentgenol. 2023, 220, 562–574. [Google Scholar] [CrossRef]
- Decourt, B.; Boumelhem, F.; Pope, E.D.; Shi, J.; Mari, Z.; Sabbagh, M.N. Critical appraisal of amyloid lowering agents in AD. Curr. Neurol. Neurosci. Rep. 2021, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Rabinovici, G.D.; Atri, A.; Aisen, P.; Apostolova, L.G.; Hendrix, S.; Sabbagh, M.; Selkoe, D.; Weiner, M.; Salloway, S. Aducanumab: Appropriate use recommendations update. J. Prev. Alzheimers Dis. 2022, 9, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Chiong, W.; Tolchin, B.D.; Bonnie, R.J.; Busl, K.; Cruz-Florea, S.; Epstein, L.G.; Greene, E.P.; Illes, J.; Kirschen, M.; Larriviere, D.G.; et al. Decisions with patients and families regarding aducanumab in alzheimer disease, with recommendations for consent: AAN position statement. Neurology. 2021, 98, 154–159. [Google Scholar] [CrossRef]
- Lyons, M.; Neve, A.; Huang, Z.; Das, B.; Wojtowicz, J.; Bullain, S. Baseline risk factors for developing ARIA-E from the SCarlet RoAD and marguerite RoAD open-label extension studies. Alzheimers Dement. 2022, 18, e065856. [Google Scholar] [CrossRef]
- Barakos, J.; Purcell, D.; Suhy, J.; Chalkias, S.; Burkett, P.; Marsica, C.; Grassi, C.M.; Castrillo-Viguera, C.; Rubino, I.; Vijverberg, E. Detection and management of amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with anti-amyloid beta therapy. J. Prev. Alzheimers Dis. 2022, 9, 211–220. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef]
- Gurol, M.E.; Becker, J.A.; Fotiadis, P.; Riley, G.; Schwab, K.; Johnson, K.A.; Greenberg, S.M. Florbetapir-PET to diagnose cerebral amyloid angiopathy: A prospective study. Neurology 2016, 87, 2043–2049. [Google Scholar] [CrossRef]
- Scolding, N.J.; Joseph, F.; Kirby, P.A.; Mazanti, I.; Gray, F.; Mikol, J.; Ellison, D.; Hilton, D.A.; Williams, T.L.; Mackenzie, J.M.; et al. Abeta-related angiitis: Primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 2005, 128, 500–515. [Google Scholar] [CrossRef]
- Eng, J.A.; Frosch, M.P.; Choi, K.; Rebeck, G.W.; Greenberg, S.M. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann. Neurol. 2004, 55, 250–256. [Google Scholar] [CrossRef]
- Carlson, C.; Siemers, E.; Hake, A.; Case, M.; Hayduk, R.; Suhy, J.; Oh, J.; Barakos, J. Amyloid-related imaging abnormalities from trials of solanezumab for Alzheimer’s disease. Alzheimers Dement. 2016, 2, 75–85. [Google Scholar] [CrossRef]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate alzheimer disease. Neurology 2018, 90, e1889–e1897. [Google Scholar] [CrossRef]
- Leurent, C.; Goodman, J.A.; Zhang, Y.; He, P.; Polimeni, J.R.; Gurol, M.E.; Lindsay, M.; Frattura, L.; Sohur, U.S.; Viswanathan, A.; et al. Immunotherapy with ponezumab for probable cerebral amyloid angiopathy. Ann. Clin. Transl. Neurol. 2019, 6, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Budd Haeberlein, S.; O’Gorman, J.; Chiao, P.; Bussiere, T.; von Rosenstiel, P.; Tian, Y.; Zhu, Y.; von Hehn, C.; Gheuens, S.; Skordos, L.; et al. Clinical development of aducanumab, an anti-aβ human monoclonal antibody being investigated for the treatment of early Alzheimer’s disease. J. Prev. Alzheimers Dis. 2017, 4, 255–263. [Google Scholar] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Landen, J.W.; Andreasen, N.; Cronenberger, C.L.; Schwartz, P.F.; Borjesson-Hanson, A.; Ostlund, H.; Sattler, C.A.; Binneman, B.; Bednar, M.M. Ponezumab in mild-to-moderate Alzheimer’s disease: Randomized phase II PET-PIB study. Alzheimers Dement. 2017, 3, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Zotova, E.; Weller, R.O.; Love, S.; Neal, J.W.; Pickering, R.M.; Wilkinson, D.; Holmes, C.; Nicoll, J.A.R. Consequences of Abet immunization on the vasculature of human Alzheimer’s disease brain. Brain 2008, 131, 3299–3310. [Google Scholar] [CrossRef]
- Chalmers, K.; Wilcock, G.K.; Love, S. APOE epsilon 4 influences the pathological phenotype of Alzheimer’s disease by favouring cerebrovascular over parenchymal accumulation of A beta protein. Neuropathol. Appl. Neurobiol. 2003, 29, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Love, S.; Miners, S.; Palmer, J.; Chalmers, K.; Kehoe, P. Insights into the pathogenesis and pathogenicity of cerebral amyloid angiopathy. Front. Biosci. (Landmark Ed.) 2009, 14, 4778–4792. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M.M.; Wilcock, G.K. Cerebral amyloid angiopathy in dementia and old age. J. Neurol. Neurosurg. Psychiatry 1986, 49, 1221–1226. [Google Scholar] [CrossRef]
- Sin, M.; Cheng, Y.; Roseman, J.M.; Zamrini, E.; Ahmed, A. Relationships between cerebral vasculopathies and microinfarcts in a community-based cohort of older adults. J. Clin. Med. 2023, 12, 3807. [Google Scholar] [CrossRef]
- Love, S.; Nicoll, J.A.; Hughes, A.; Wilcock, G.K. APOE and cerebral amyloid angiopathy in the elderly. Neuroreport. 2003, 14, 1535–1536. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.A.; Yu, L.; Nag, S.; Leurgans, S.; Wilson, R.S.; Bennett, D.A.; Schneider, J.A. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 2015, 85, 1930–1936. [Google Scholar] [CrossRef]
- Grabowski, T.J.; Cho, H.S.; Vonsattel, J.P.; Rebeck, G.W.; Greenberg, S.M. Novel amyloid precursor protein mutation in an iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 2001, 49, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, G.; Carare, R.; Cordonnier, C.; Greenberg, S.M.; Schneider, J.A.; Smith, E.E.; van Buchem, M.; van der Grong, J.; Verbeek, M.M.; Werring, D.J. The increasing impact of cerebral amyloid angiopathy: Essential new insights for clinical practice. J. Neurol. Neurosurg. Psychiatry 2017, 88, 982–994. [Google Scholar] [CrossRef]
- Vidoni, E.D.; Yeh, H.; Morris, J.K.; Newell, K.L.; Alqahtani, A.; Burns, N.C.; Burns, J.M.; Billinger, S.A. Cerebral β-amyloid angiopathy is associated with earlier dementia onset in Alzheimer’s disease. Neurodegener. Dis. 2016, 16, 218–224. [Google Scholar] [CrossRef]
- Samarasekera, N.; Rodrigues, M.A.; Toh, P.S.; Al-Shahi, S. Imaging features of intracerebral hemorrhage with cerebral amyloid angiopathy: Systematic review and meta-analysis. PLoS ONE 2017, 12, e0180923. [Google Scholar]
- Alban, S.L.; Lynch, K.M.; Ringman, J.M.; Toga, A.W.; Chui, H.C.; Sepehrband, F.; Choupan, J.; Alzheimer’s Disease Neuroimaging Initiative. The association between white matter hyperintensities and amyloid and tau deposition. Neuroimage Clin. 2023, 38, 103383. [Google Scholar] [CrossRef] [PubMed]
- Walsh, P.; Sudre, C.H.; Fiford, C.M.; Ryan, N.S.; Lashley, T.; Frost, C.; Barnes, J.; ADNI Investigators. CSF amyloid is a consistent predictor of white matter hyperintensities across the disease course from aging to Alzheimer’s disease. Neurobiol. Aging 2020, 91, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Graff-Radford, J.; Arenaza-Urquijo, E.M.; Knopman, D.S.; Schwarz, C.G.; Brown, R.D.; Rabinstein, A.A.; Gunter, J.L.; Senjem, M.L.; Przybelski, S.A.; Lesnick, T.; et al. White matter hyperintensities: Relationship to amyloid and tau burden. Brain 2019, 142, 2483–2491. [Google Scholar] [CrossRef]
- de Leeuw, F.E.; de Groot, J.C.; Achten, E.; Oudkerk, M.; Ramos, L.M.; Heijboer, R.; Hofman, A.; Jolles, J.; van Gijn, J.; Breteler, M.M. Prevalence of cerebral white matter lesions in elderly people: A population based magnetic resonance imaging study. the rotterdam scan study. J. Neurol. Neurosurg. Psychiatry 2001, 70, 9–14. [Google Scholar] [CrossRef]
- Pfefferbaum, A.; Adalsteinsson, E.; Sullivan, E.V. Frontal circuitry degradation marks healthy adult aging: Evidence from diffusion tensor imaging. Neuroimage 2005, 26, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chabriat, H.; Godin, O.; Dufouil, C.; Rosan, J.; Greenberg, S.M.; Smith, E.E.; Tzourio, C.; Viswanathan, A. istribution of white matter hyperintensity in cerebral hemorrhage and healthy aging. J. Neurol. 2012, 259, 530–536. [Google Scholar] [CrossRef]
- Smith, E.E.; Gurol, M.E.; Eng, J.A.; Nguyen, T.N.; Rosand, J.; Greenberg, S.M. White matter lesions, cognition, and recurrent hemorrhage in lobar intracerebral hemorrhage. Neurology 2004, 63, 1606–1612. [Google Scholar] [CrossRef]
- Charidimou, A.; Baron, J.; Werring, D.J. Transient focal neurological episodes, cerebral amyloid angiopathy, and intracerebral hemorrhage risk: Looking beyond TIAs. Int. J. Stroke 2013, 8, 105–108. [Google Scholar] [CrossRef]
- Thanprasertsuk, S.; Martinez-Ramirez, S.; Pontes-Neto, O.M.; Ni, J.; Ayres, A.; Reed, A.; Swords, K.; Gurol, M.E.; Greenberg, S.M.; Viswanathan, A. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology 2014, 83, 794–800. [Google Scholar] [CrossRef]
- Puy, L.; Cordonnier, C. Sporadic cerebral amyloid angiopathy. Geriatr. Psychol. Neuropsychiatr. Vieil. 2019, 17, 73–82. [Google Scholar]
- Chui, H.C.; Ramirez-Gomez, L. Clinical and imaging features of mixed alzheimer and vascular pathologies. Alzheimers Res. Ther. 2015, 7, 21–27. [Google Scholar] [CrossRef]
- Yu, L.; Boyle, P.A.; Nag, S.; Leurgans, S.; Buchman, A.S.; Wilson, R.S.; Arvanitakis, Z.; Farfel, J.M.; De Jager, P.L.; Bennett, D.A.; et al. APOE and cerebral amyloid angiopathy in community-dwelling older persons. Neurobiol. Aging 2015, 36, 2946–2953. [Google Scholar] [CrossRef]
- Premkumar, D.R.; Cohen, D.L.; Hedera, P.; Friedland, R.P.; Kalaria, R.N. Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am. J. Pathol. 1996, 148, 2083–2095. [Google Scholar]
- Olichney, J.M.; Hansen, L.A.; Galasko, D.; Saitoh, T.; Hofstetter, C.R.; Katzman, R.; Thal, L.J. The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer’s disease and lewy body variant. Neurology 1996, 47, 190–196. [Google Scholar] [CrossRef]
- Thal, D.R.; Griffin, W.S.T.; de Vos, R.A.I.; Ghebremedhin, E. Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol. 2008, 115, 599–609. [Google Scholar] [CrossRef]
- Smith, E.E.; Charidimou, A.; Ayata, C.; Werring, D.J.; Greenberg, S.M. Cerebral amyloid angiopathy-related transient focal neurologic episodes. Neurology 2021, 97, 231–238. [Google Scholar] [CrossRef]
- Weller, R.O.; Preston, S.D.; Subash, M.; Carare, R.O. Cerebral amyloid angiopathy in the aetiology and immunotherapy of alzheimer disease. Alzheimers Res. Ther. 2009, 1, 6. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Frosch, M.P.; Baron, J.C.; Pasi, M.; Albucher, J.F.; Banerjee, G.; Barbato, C.; Bonneville, F.; Brandner, S.; et al. The boston criteria version 2.0 for cerebral amyloid angiopathy: A multicentre, retrospective, MRI-neuropathology diagnostic accuracy study. Lancet Neurol. 2022, 21, 714–725. [Google Scholar] [CrossRef]
- Knudsen, K.A.; Rosand, J.; Karluk, D.; Greenberg, S.M. Clinical diagnosis of cerebral amyloid angiopathy: Validation of the boston criteria. Neurology 2001, 56, 537–539. [Google Scholar] [CrossRef]
- De Kort, A.M.; Kuiperij, H.B.; Marques, T.M.; Jakel, L.; Van den Berg, E.; Kersten, I.; van Berckel-Smit, H.E.P.; Duering, M.; Stoops, E.; Abdo, W.; et al. Decreased cerebrospinal fluid amyloid β 38, 40, 42, and 43 levels in sporadic and hereditary cerebral amyloid angiopathy. Ann. Neurol. 2023, 93, 1173–1186. [Google Scholar] [CrossRef]
- Zhu, X.; Xu, F.; Hoos, M.D.; Lee, H.; Benveniste, H.; Nostrand, W.E.V. Reduced levels of cerebrospinal fluid/plasma Aβ40 as an early biomarker for cerebral amyloid angiopathy in RTg-DI rats. Int. J. Mol. Sci. 2020, 21, 303. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, M.M.; Eikelenboom, P.; de Waal, R.M. Differences between the pathogenesis of senile plaques and congophilic angiopathy in alzheimer disease. J. Neuropathol. Exp. Neurol. 1997, 56, 751–761. [Google Scholar] [CrossRef] [PubMed][Green Version]
- van Etten, E.S.; Verbeek, M.M.; van der Grond, J.; Zielman, R.; van Rooden, S.; van Zwet, E.W.; van Opstal, A.M.; Haan, J.J.; Greenberg, S.M.; van Buchem, M.A.; et al. Β-amyloid in CSF: Biomarker for preclinical cerebral amyloid angiopathy. Neurology 2017, 88, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Viqar, F.; Zimmerman, M.E.; Narkhede, A.; Tosto, G.; Benzinger, T.L.S.; Marcus, D.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; et al. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited alzheimer network. Ann. Neurol. 2016, 79, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Weaver, N.A.; Doeven, T.; Barkhof, F.; Biesbroek, J.M.; Groeneveld, O.N.; Kuijf, H.J.; Prins, N.D.; Scheltens, P.; Teunissen, C.E.; van der Flier, M.M.; et al. Cerebral amyloid burden is associated with white matter hyperintensity location in specific posterior white matter regions. Neurobiol. Aging 2019, 84, 225–234. [Google Scholar] [CrossRef]
- Schoemaker, D.; Zanon Zotin, M.C.; Chen, K.; Igwe, K.C.; Vila-Castelar, C.; Martinez, J.; Baena, A.; Fox-Fuller, J.T.; Lopera, F.; Reiman, E.M.; et al. White matter hyperintensities are a prominent feature of autosomal dominant Alzheimer’s disease that emerge prior to dementia. Alzheimers Res. Ther. 2022, 14, 89. [Google Scholar] [CrossRef]
- Charidimou, A.; Gang, Q.; Werring, D.J. Sporadic cerebral amyloid angiopathy revisited: Recent insights into pathophysiology and clinical spectrum. J. Neurol. Neurosurg. Psychiatry 2012, 83, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, G.; Ambler, G.; Keshavan, A.; Paterson, R.W.; Foiani, M.S.; Toombs, J.; Heslegrave, A.; Dickson, J.C.; Fraioli, F.; Groves, A.M.; et al. Cerebrospinal fluid biomarkers in cerebral amyloid angiopathy. J. Alzheimers Dis. 2020, 74, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Grangeon, L.; Paquet, C.; Guey, S.; Zarea, A.; Martinaud, O.; Rotharmel, M.; Maltete, D.; Quillard-Muraine, M.; Nicolas, G.; Charbonnier, C.; et al. Cerebrospinal fluid profile of tau, phosphorylated tau, Aβ42, and Aβ40 in probable cerebral amyloid angiopathy. J. Alzheimers Dis. 2022, 87, 791–802. [Google Scholar] [CrossRef]
- Sembill, J.A.; Lusse, C.; Linnerbauer, M.; Sprugel, M.I.; Mrochen, A.; Knott, M.; Engelhorn, T.; Schmidt, M.A.; Doerfler, A.; Oberstein, T.J.; et al. Cerebrospinal fluid biomarkers for cerebral amyloid angiopathy. Brain Commun. 2023, 5, fcad159. [Google Scholar] [CrossRef] [PubMed]
- Gurol, M.E.; Irizarry, M.C.; Smith, E.E.; Raju, S.; Diaz-Arrastia, R.; Bottiglieri, T.; Rosand, J.; Growdon, J.H.; Greenberg, S.M. Plasma beta-amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology 2006, 66, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.L.; Lawler, P.E.; Bollinger, J.G.; Li, Y.; Schindler, S.E.; Li, M.; Lopez, S.; Ovod, V.; Nakamura, A.; Shaw, L.M.; et al. The performance of plasma amyloid beta measurements in identifying amyloid plaques in Alzheimer’s disease: A literature review. Alzheimers Res. Ther. 2022, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.M.; Li, Y.; Gorden, B.A.; Holtzman, D.M.; Moris, J.C.; Benzinger, T.L.S.; Xiong, C.; et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019, 93, e1647–e1659. [Google Scholar] [CrossRef]
- Verberk, I.M.W.; Slot, R.E.; Verfaillie, S.C.J.; Heijst, H.; Prins, N.D.; van Berckel, B.N.M.; Scheltens, P.; Teunissen, C.E.; van der Flier, W.M. Plasma amyloid as prescreener for the earliest alzheimer pathological changes. Ann. Neurol. 2018, 84, 648–658. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Overview | AD | CAA |
|---|---|---|
| Common clinical symptom | Memory disorders | Intracranial hemorrhage |
| Imaging finding | Hippocampal atrophy | Hemorrhage (macrobleed in very severe cases; multiple microbleeds in chronic, late-stage); cortical superficial siderosis |
| Biomarkers findings (CSF, plasma) | ↓ Amyloid β42, ↑ amyloid β40, ↓ amyloid β42/40, ↑ total tau (t-tau), ↑ phosphorylated tau (p-tau181) | ↓ Amyloid β40, ↑ amyloid β42, ↓ amyloid β40/42, ↓ total tau, ↓ phosphorylated tau 181 (p-tau181) |
| Neuropathological findings | Amyloid β accumulation in parenchyma as plaques | Amyloid β accumulation in the leptomeninges and small to medium-sized cerebral vessels |
| ARIA Type | Radiological Severity | ||
|---|---|---|---|
| Mild | Moderate | Severe | |
| ARIA-E | |||
| Size | <5 cm | 5–10 cm | >10 cm |
| Location | Limited to a single site within sulcus or cortex/subcortical white matter | One or multiple brain locations | Significant involvement in the sulcus or subcortical white matter in one or more distinct sites |
| ARIA-H | |||
| New incident of microhemorrhages | ≤4 | 5–9 | ≥10 |
| Focal areas of superficial siderosis | 1 | 2 | >2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sin, M.-K.; Zamrini, E.; Ahmed, A.; Nho, K.; Hajjar, I. Anti-Amyloid Therapy, AD, and ARIA: Untangling the Role of CAA. J. Clin. Med. 2023, 12, 6792. https://doi.org/10.3390/jcm12216792
Sin M-K, Zamrini E, Ahmed A, Nho K, Hajjar I. Anti-Amyloid Therapy, AD, and ARIA: Untangling the Role of CAA. Journal of Clinical Medicine. 2023; 12(21):6792. https://doi.org/10.3390/jcm12216792
Chicago/Turabian StyleSin, Mo-Kyung, Edward Zamrini, Ali Ahmed, Kwangsik Nho, and Ihab Hajjar. 2023. "Anti-Amyloid Therapy, AD, and ARIA: Untangling the Role of CAA" Journal of Clinical Medicine 12, no. 21: 6792. https://doi.org/10.3390/jcm12216792
APA StyleSin, M.-K., Zamrini, E., Ahmed, A., Nho, K., & Hajjar, I. (2023). Anti-Amyloid Therapy, AD, and ARIA: Untangling the Role of CAA. Journal of Clinical Medicine, 12(21), 6792. https://doi.org/10.3390/jcm12216792