
Sugar and Dyslipidemia: A Double-Hit, Perfect Storm

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
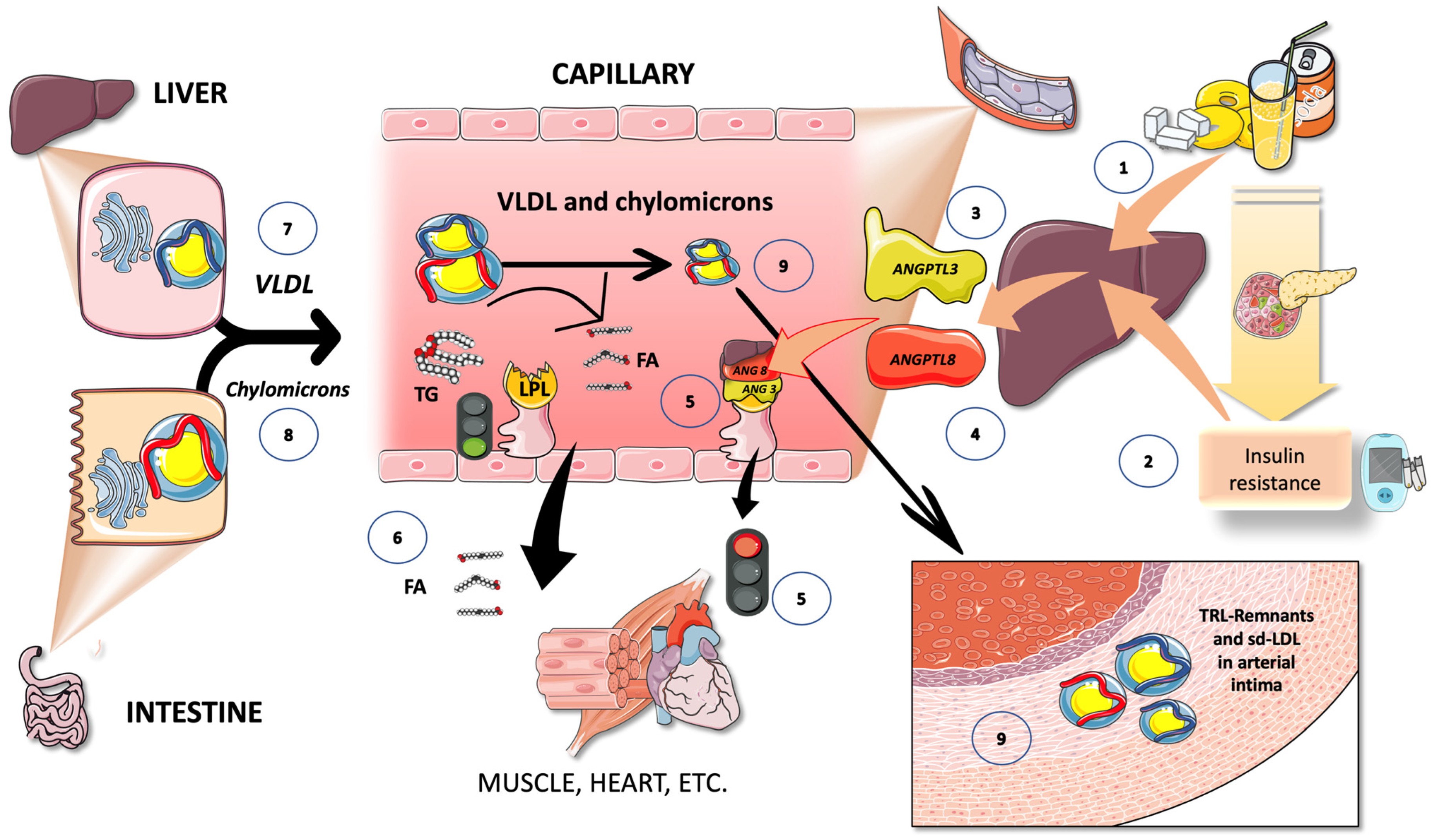{kind=link}
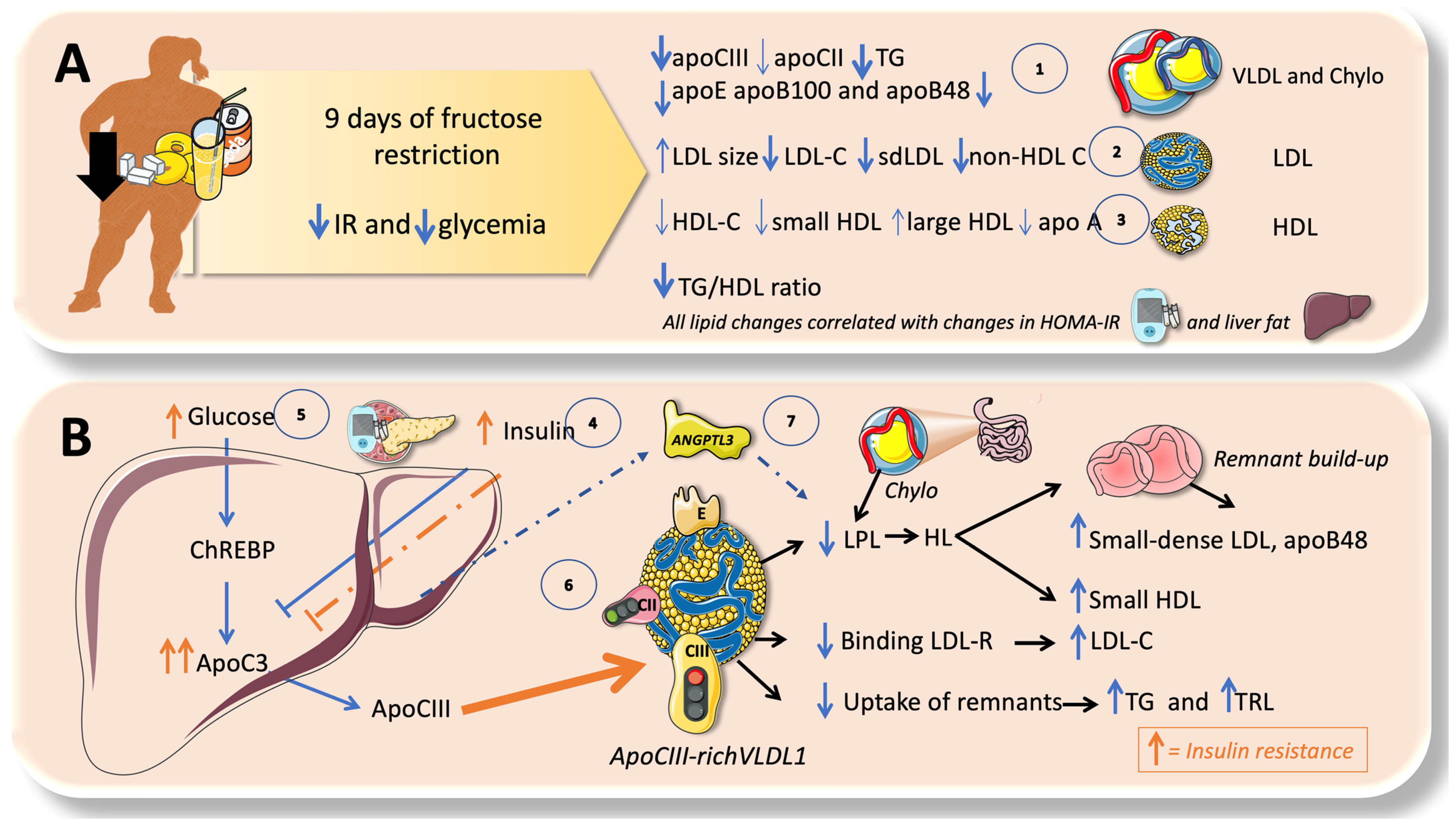{kind=link}
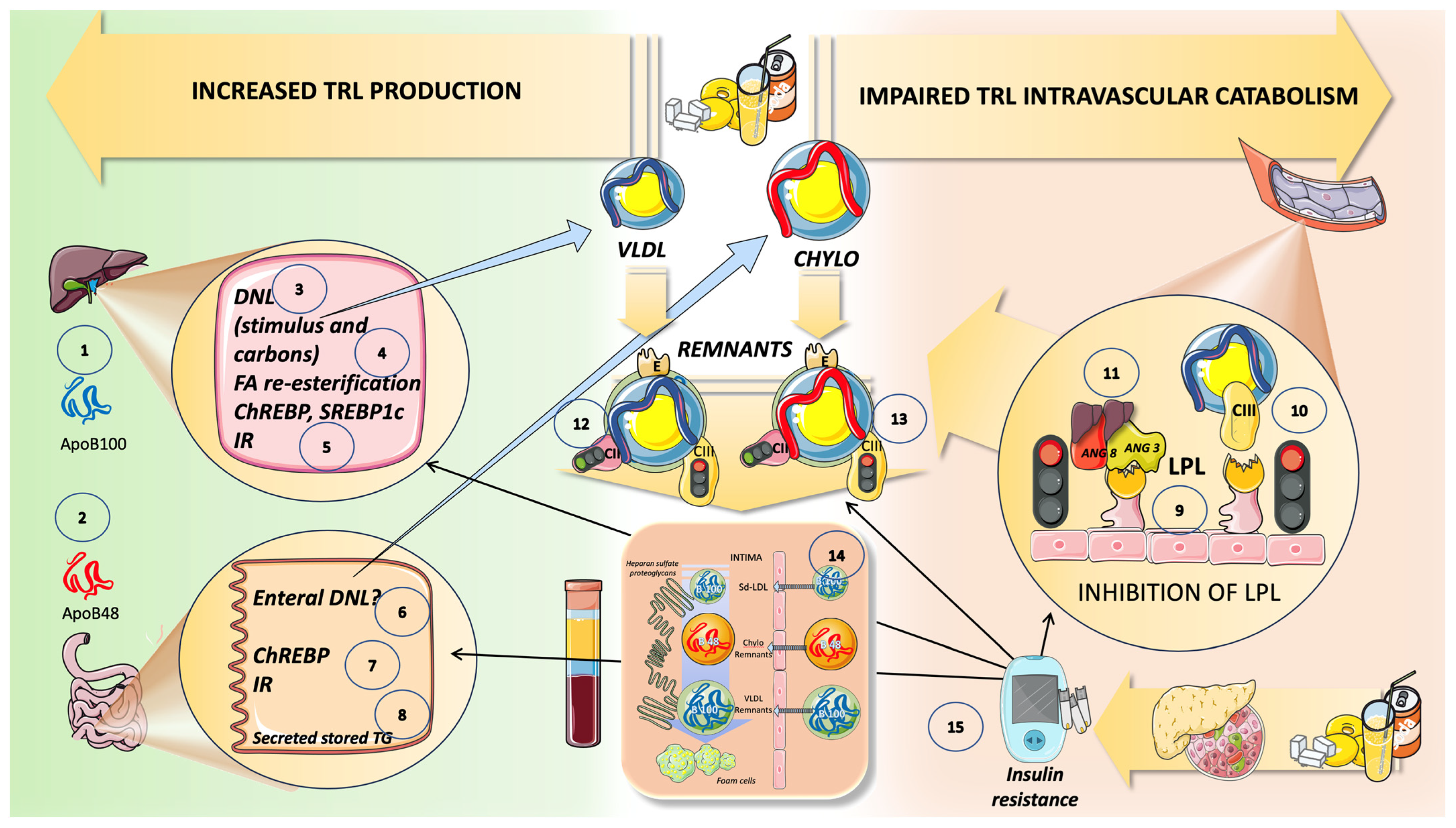{kind=link}
Abstract
1. Introduction
2. Sugar in Excess Produces Unwanted Metabolic Consequences
2.1. Sugar in Food, and More So in a Liquid Form, Is Hazardous; Metabolic Consequences Are Far Worse When It Is Provided in Liquid Form and on an Empty Stomach
2.2. “Healthy and Natural” Fruit Juice Is Also a Culprit
3. Triglyceride-Rich Lipoproteins (TRL): Energy-Rich Healthy Particles in Healthy Quantities, Villains When in Excess
3.1. Production of Circulating TRL
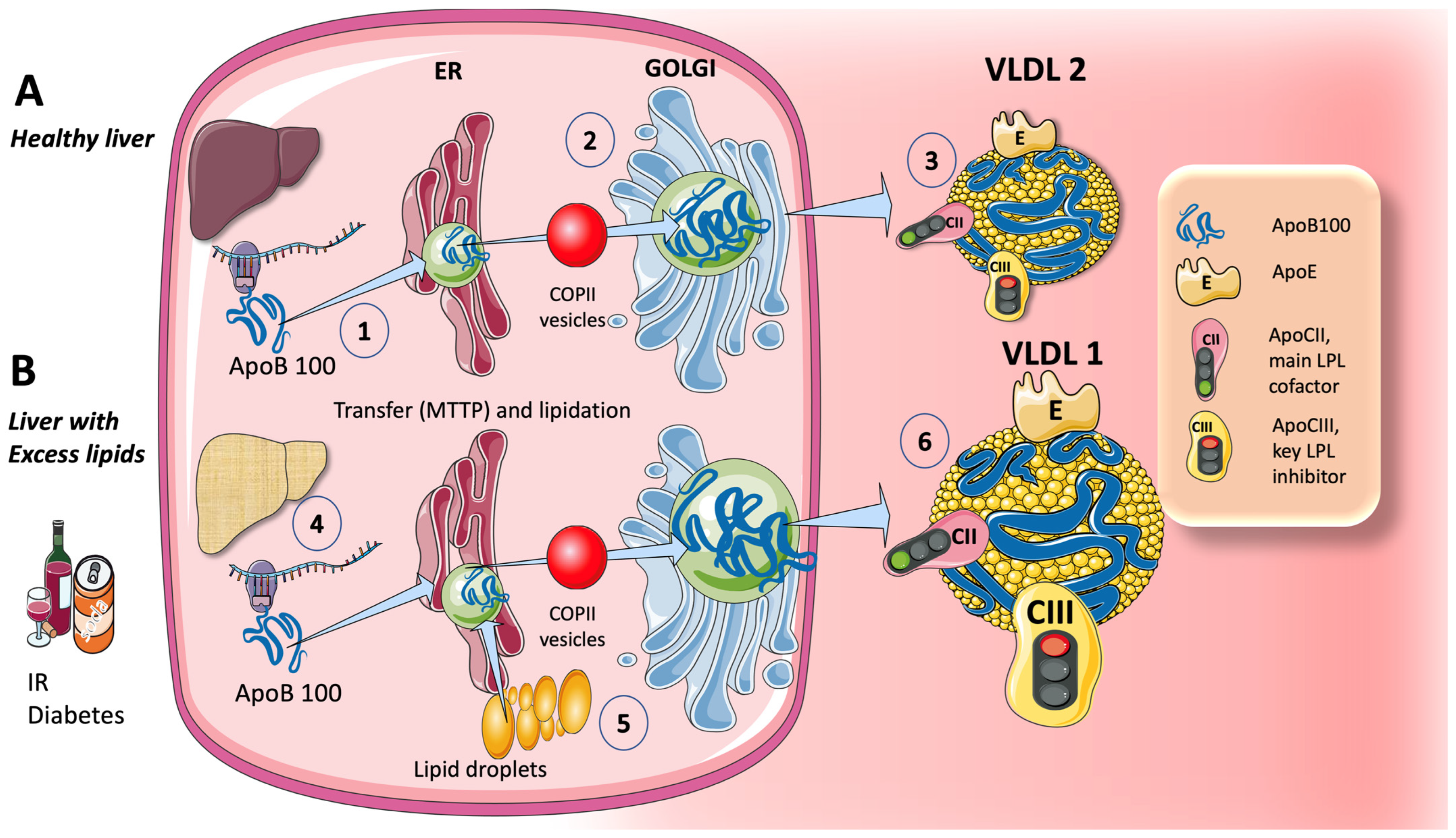
3.1.1. Endogenous Routes: The Liver Produces VLDL throughout the Day, Which Is Crucial during Fasting
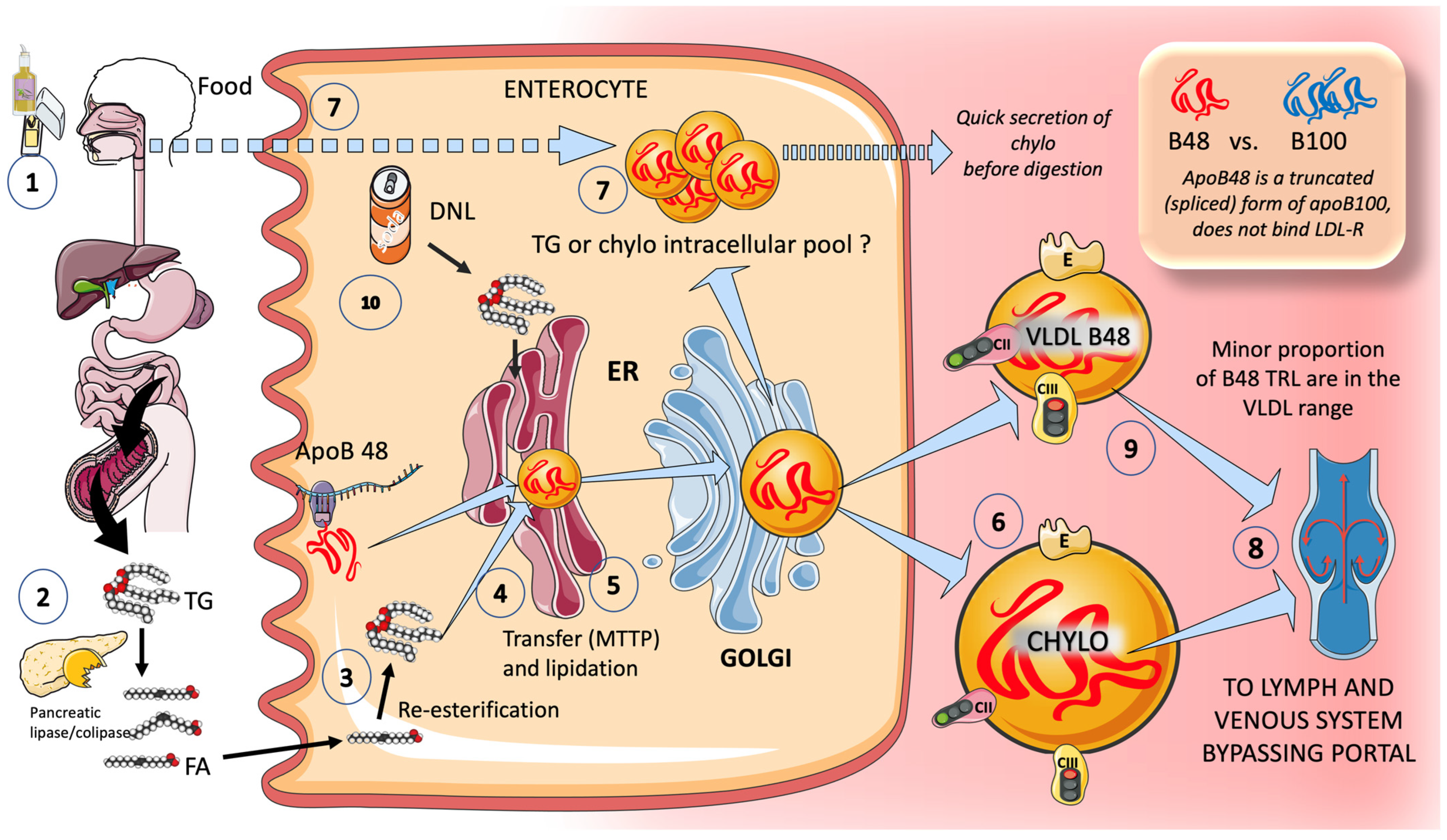
3.1.2. Exogenous Routes: In the Postprandial Period, Enterocytes Produce Chylomicrons
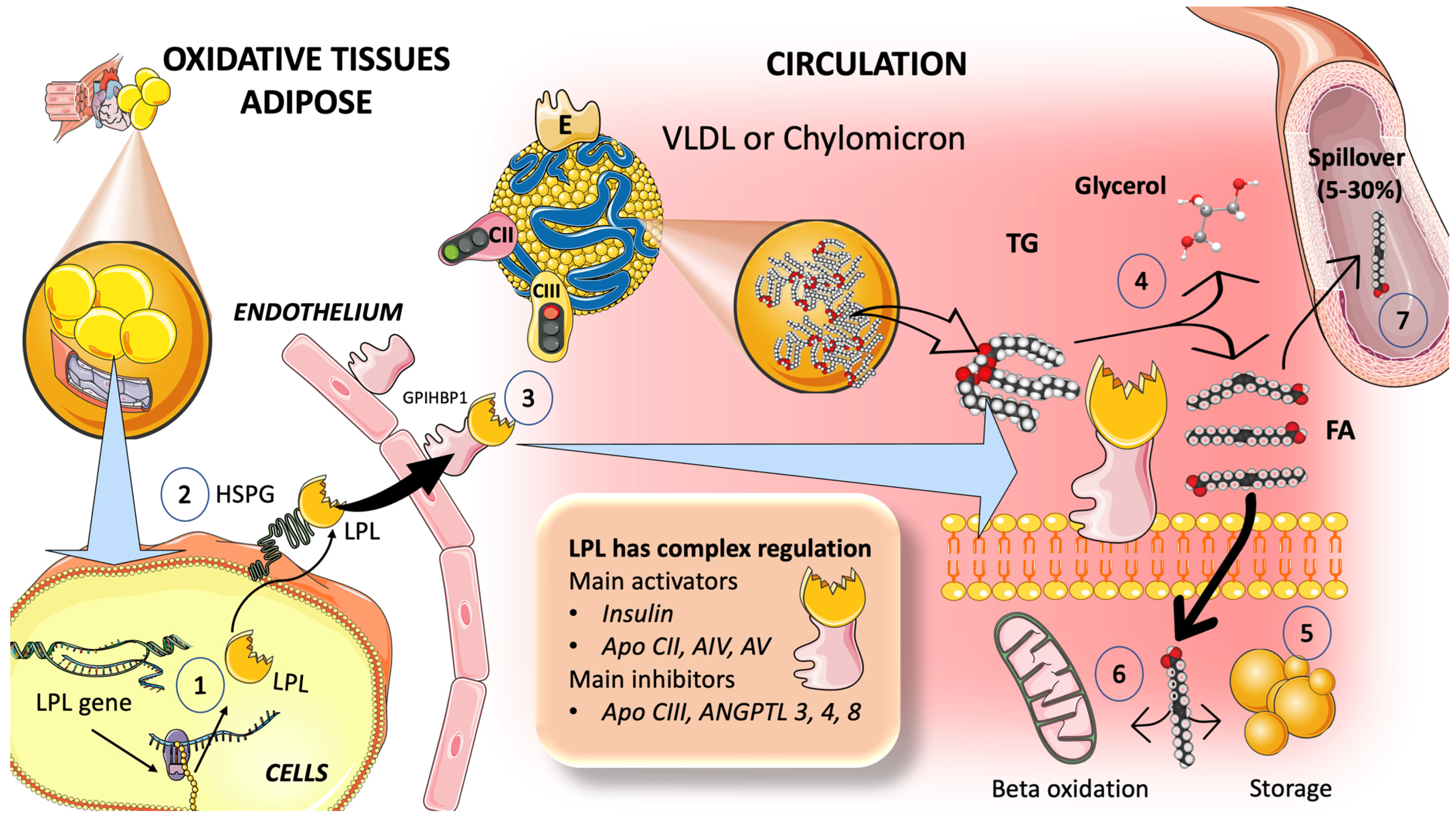
3.2. Catabolism of TRL
3.2.1. Multiple Modulators Work Together in the Intricate Regulation of LPL Synthesis and Activity
3.2.2. TRL and HDL Dynamic Intravascular Crosstalk
3.2.3. TRL Remnants Are at Least as Atherogenic as LDL
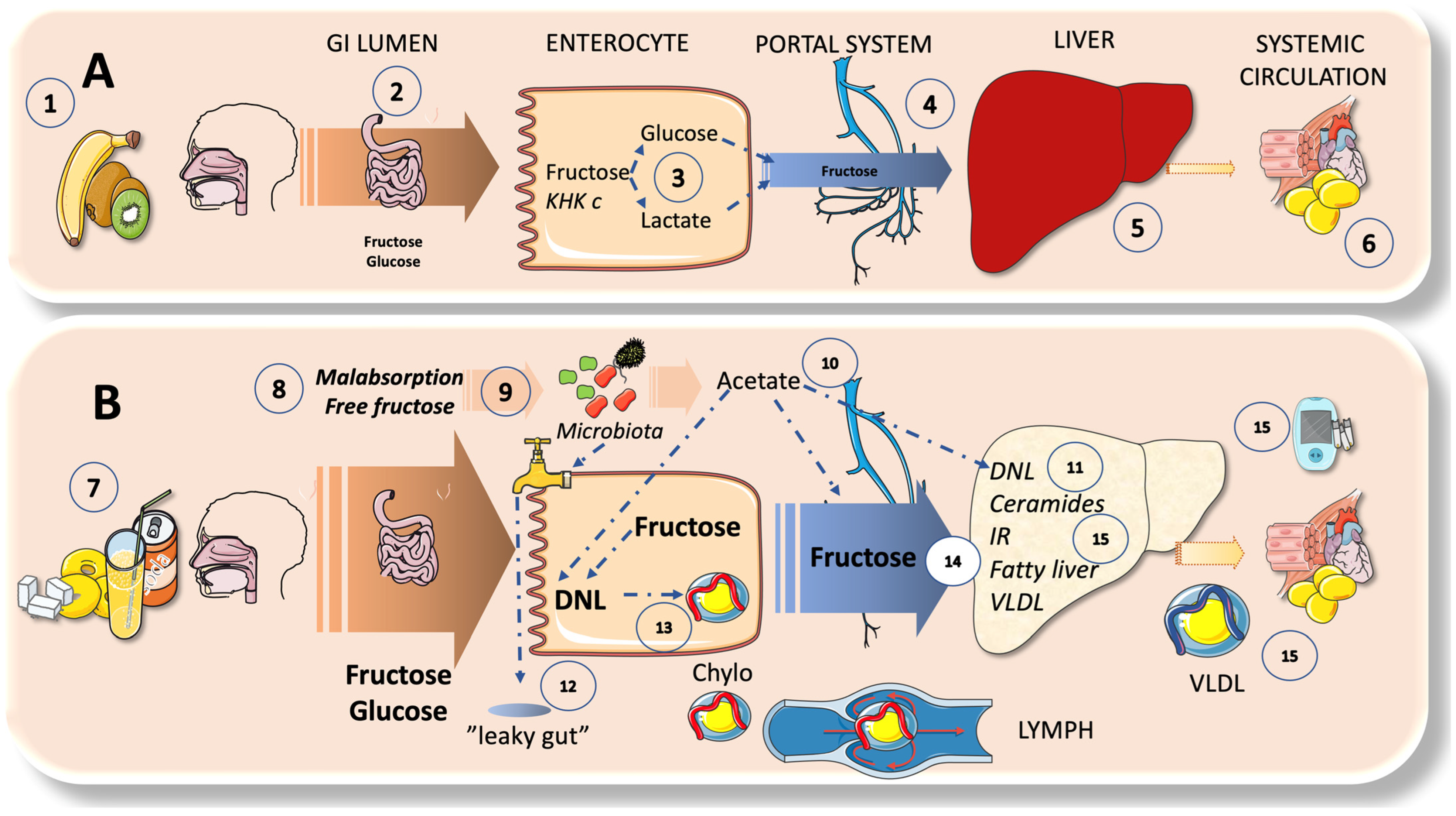
4. Sugar Metabolism: A Bird’s Eye View on the Axis of Enterocyte-Liver-Systemic Organs
4.1. Fructose Has a Very Low Serum Concentration; the Liver Acts as a Very Effective First-Pass Buffer
4.2. Enteral and Hepatic Fructose Metabolism
4.3. DNL Can Also Occur in the Intestines
5. Sugar and Increased TRL Production and the Key Role of the Liver
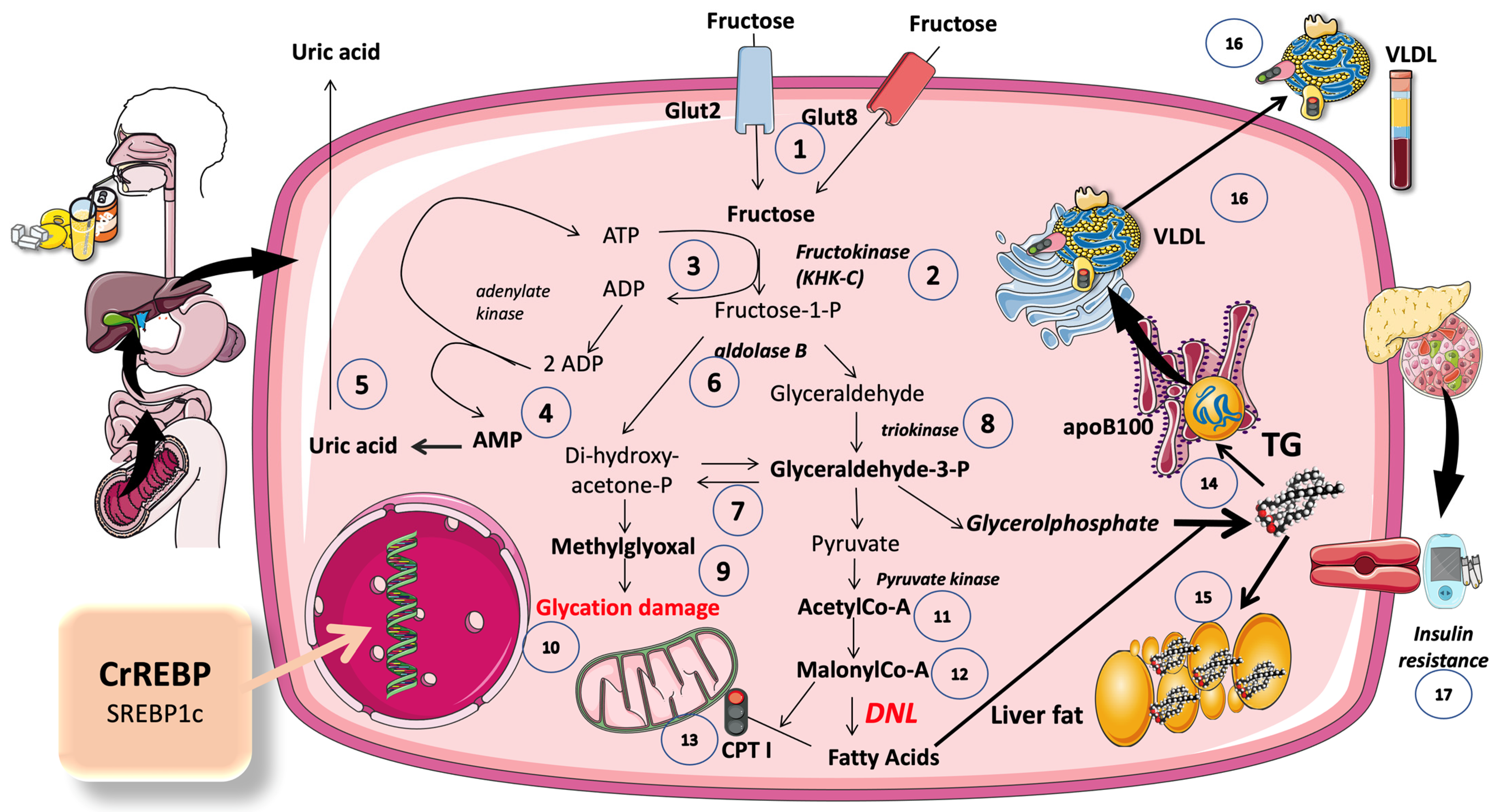
5.1. A Set of Three Unique Enzymes Are Used in the Non-Tightly Controlled, Primarily Hepatic, Metabolism of Fructose
5.2. An Ingenious Evolutionary Defense System Gone Awry
5.3. Excess Trioses Not Only Feed TG Production but also Produce Toxic Metabolites
5.4. DNL Is Greatly Enhanced When Glucose and Fructose Are Metabolized at the Same Time (Sugar or HFCS, Its Usual Source)
5.4.1. DNL Transforms Extra Dietary Carbohydrates (CHO) into Fatty Acids (FA)
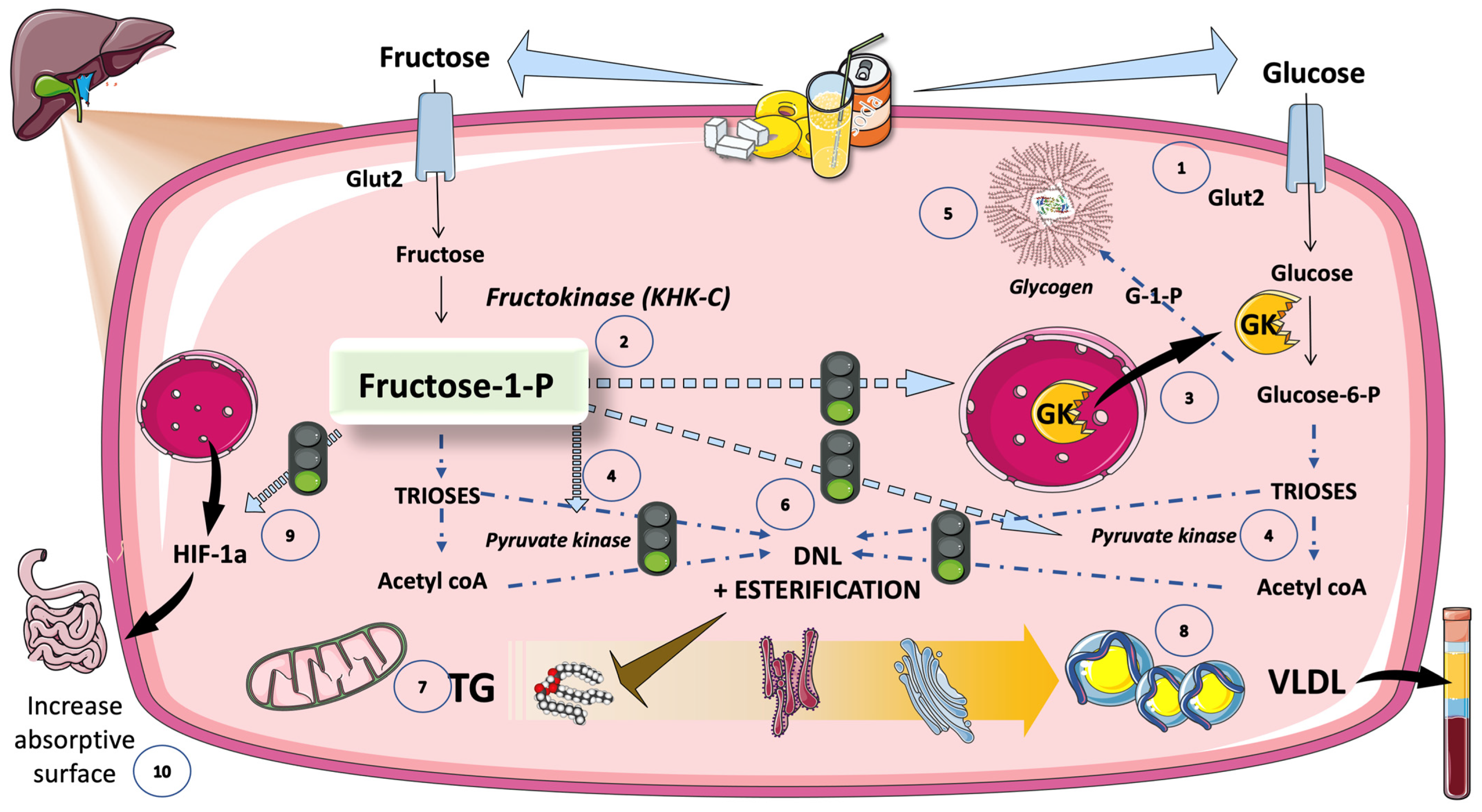
5.4.2. Inhibition of KHC-c Blocks Fat Synthesis
5.5. Cross-Talk between Fructose and Glucose Metabolism in the Liver: The Role of F-1-P as a Signaling Molecule of Abundance That Has Gone Awry in Current Dietary Habits
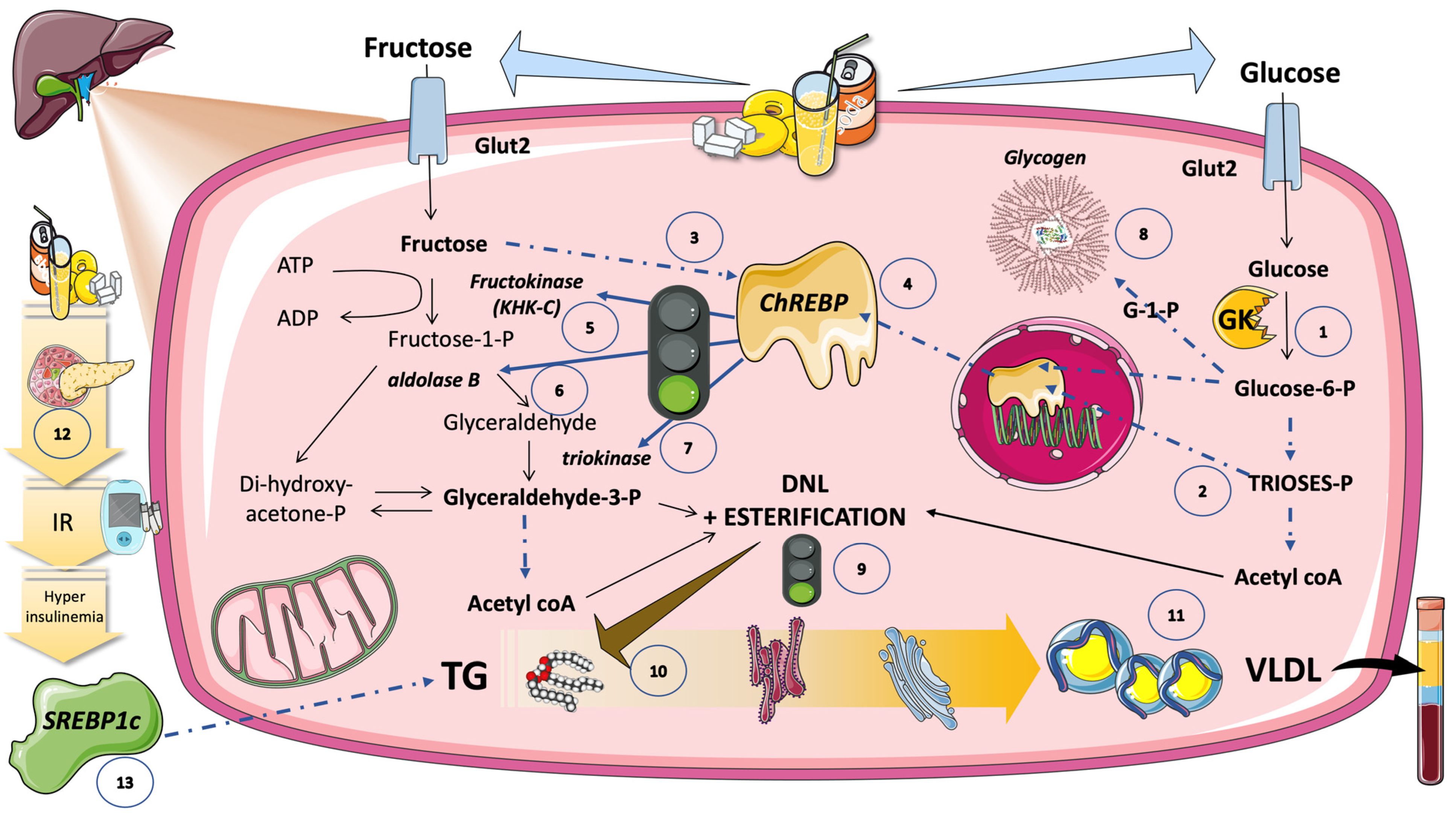
5.6. DNL Is Further Regulated by Transcription Factors Induced by Glucose and Fructose (Sugar or HFCS, the Usual Sources)
5.6.1. Hexose- and Triose-Phosphate Concentrations in the Hepatocyte Rise Because of Increasing Glycolytic Flow and Fructolytic Flow
5.6.2. ChREBP Does Not Work Alone
5.6.3. Carbons vs. Stimulus
6. Sugar and Increased Intestinal TRL Production: Fructose May Be Involved in Increased Production of Chylomicrons
New Insights into Human Chylomicron Metabolism Have Been Provided by Recent Chylomicron Purification Techniques
7. The Second Hit: Fructose and LPL, and the Role of the Main Inhibitors
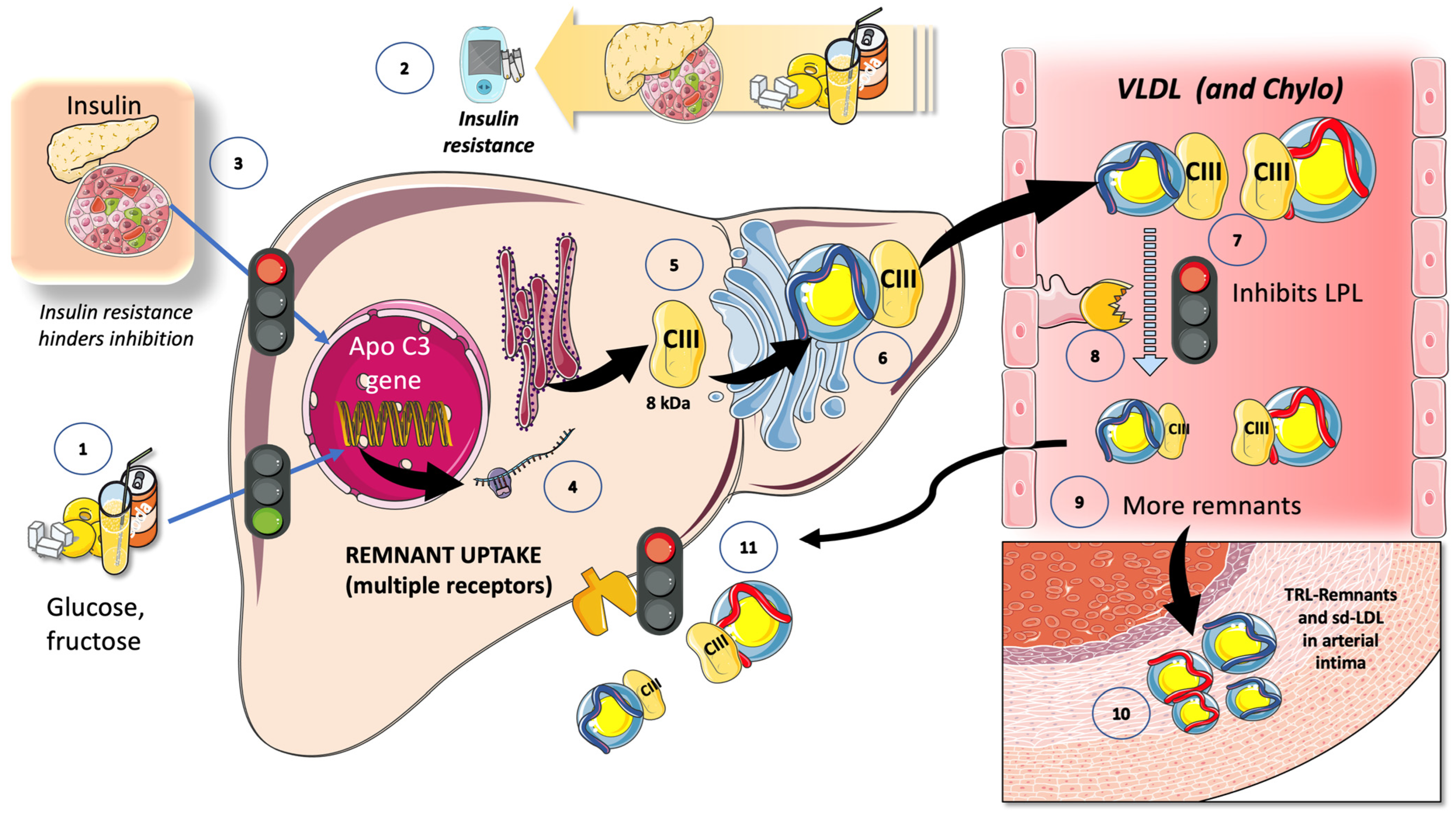
7.1. Fructose and apoCIII
7.2. Fructose and the Partition of TRL in the Fast-Fed Cycle
8. Fructose and TRL in the Postprandial Period
8.1. Atherogenesis as a Postprandial Phenomenon
8.2. Fructose Increases TRL Remnants: Their Role in “Residual Risk”
8.3. LPL Inhibitors Are the Targets of New Approaches to Residual Risk
9. Isocaloric Fructose Restriction Reverts Most of the Lipid Disturbances in Adolescents
10. High Sugar Diets Promote TRL Accumulation and Increase in Atherogenic Remnants by Interfering with Both Their Production and Catabolism: Recapitulation
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, G.A. How Bad Is Fructose? Am. J. Clin. Nutr. 2007, 86, 895–896. [Google Scholar] [CrossRef]
- Bray, G.A. Soft Drink Consumption and Obesity: It Is All about Fructose. Curr. Opin. Lipidol. 2010, 21, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A. Fructose: Pure, White, and Deadly? Fructose, by Any Other Name, Is a Health Hazard. J. Diabetes Sci. Technol. 2010, 4, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H. Fructose: Metabolic, Hedonic, and Societal Parallels with Ethanol. J. Am. Diet. Assoc. 2010, 110, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Isganaitis, E.; Lustig, R.H. Fast Food, Central Nervous System Insulin Resistance, and Obesity. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2451–2462. [Google Scholar] [CrossRef]
- Bremer, A.A.; Mietus-Snyder, M.; Lustig, R.H. Toward a Unifying Hypothesis of Metabolic Syndrome. Pediatrics 2012, 129, 557–570. [Google Scholar] [CrossRef]
- Bray, G.A.; Popkin, B.M. Dietary Sugar and Body Weight: Have We Reached a Crisis in the Epidemic of Obesity and Diabetes?: Health Be Damned! Pour on the Sugar. Diabetes Care 2014, 37, 950–956. [Google Scholar] [CrossRef]
- Bray, G.A.; Popkin, B.M. Calorie-Sweetened Beverages and Fructose: What Have We Learned 10 Years Later. Pediatr. Obes. 2013, 8, 242–248. [Google Scholar] [CrossRef]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of High-Fructose Corn Syrup in Beverages May Play a Role in the Epidemic of Obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [CrossRef]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The Role of Fructose in the Pathogenesis of NAFLD and the Metabolic Syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Bray, G.A. Fructose—How Worried Should We Be? MedGenMed 2008, 10, 157. [Google Scholar]
- Schwarz, J.M.; Acheson, K.J.; Tappy, L.; Piolino, V.; Muller, M.J.; Felber, J.P.; Jequier, E. Thermogenesis and Fructose Metabolism in Humans. Am. J. Physiol. Endocrinol. Metab. 1992, 262, E591–E598. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming Fructose-Sweetened, Not Glucose-Sweetened, Beverages Increases Visceral Adiposity and Lipids and Decreases Insulin Sensitivity in Overweight/Obese Humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Faeh, D.; Minehira, K.; Schwarz, J.M.; Periasami, R.; Seongsu, P.; Tappy, L. Effect of Fructose Overfeeding and Fish Oil Administration on Hepatic de Novo Lipogenesis and Insulin Sensitivity in Healthy Men. Diabetes 2005, 54, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Havel, P.J. Adverse Metabolic Effects of Dietary Fructose: Results from the Recent Epidemiological, Clinical, and Mechanistic Studies. Curr. Opin. Lipidol. 2013, 24, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.; Mulligan, K.; Schwarz, J.M. Dietary Carbohydrates and Fatty Liver Disease: De Novo Lipogenesis. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 277–282. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Griffen, S.C.; Bremer, A.A.; Vink, R.G.; Schaefer, E.J.; Nakajima, K.; Schwarz, J.M.; Beysen, C.; Berglund, L.; Keim, N.L.; et al. Metabolic Responses to Prolonged Consumption of Glucose- and Fructose-Sweetened Beverages Are Not Associated with Postprandial or 24-h Glucose and Insulin Excursions. Am. J. Clin. Nutr. 2011, 94, 112–119. [Google Scholar] [CrossRef]
- Lustig, R.H. Fructose: It’s “Alcohol without the Buzz”. Adv. Nutr. 2013, 4, 226–235. [Google Scholar] [CrossRef]
- Bains, Y.; Erkin-Cakmak, A.; Caccavello, R.; Mulligan, K.; Noworolski, S.; Schwarz, J.-M.; Lustig, R. Alejandro Gugliucci Isocaloric Fructose Restriction Improves Postprandial Chylomicron and VLDL Excursions in Adolescents With Obesity by Reducing Angiopoietin-Like Protein 3 and Apolipoprotein CIII. Circulation 2020, 142, A16511. [Google Scholar] [CrossRef]
- Mortera, R.R.; Bains, Y.; Gugliucci, A. Fructose at the Crossroads of the Metabolic Syndrome and Obesity Epidemics. Front. Biosci. Landmark 2019, 24, 186–211. [Google Scholar] [CrossRef]
- Lustig, R.H.; Mulligan, K.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Erkin-Cakmak, A.; Gugliucci, A.; Schwarz, J.M. Isocaloric Fructose Restriction and Metabolic Improvement in Children with Obesity and Metabolic Syndrome. Obesity 2016, 24, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Erkin-Cakmak, A.; Bains, Y.; Caccavello, R.; Noworolski, S.M.; Schwarz, J.M.; Mulligan, K.; Lustig, R.H.; Gugliucci, A. Isocaloric Fructose Restriction Reduces Serum D-Lactate Concentration in Children with Obesity and Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 3003–3011. [Google Scholar] [CrossRef] [PubMed]
- Siri-Tarino, P.W.; Krauss, R.M. Diet, Lipids, and Cardiovascular Disease. Curr. Opin. Lipidol. 2016, 27, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.A.; Price, C.A.; Graham, J.L.; Stanhope, K.L.; King, S.; Hung, Y.H.; Sethupathy, P.; Wong, S.; Hamilton, J.; Krauss, R.M.; et al. Fructose-Induced Hypertriglyceridemia in Rhesus Macaques Is Attenuated with Fish Oil or ApoC3 RNA Interference. J. Lipid Res. 2019, 60, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Astrup, A.; Magkos, F.; Bier, D.M.; Brenna, J.T.; de Oliveira Otto, M.C.; Hill, J.O.; King, J.C.; Mente, A.; Ordovas, J.M.; Volek, J.S.; et al. Saturated Fats and Health: A Reassessment and Proposal for Food-Based Recommendations: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Taskinen, M.R. Metabolism of Triglyceride-Rich Lipoproteins. Handb. Exp. Pharmacol. 2022, 270, 133–156. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Taskinen, M.R.; Björnson, E.; Packard, C.J. Metabolism of Triglyceride-Rich Lipoproteins in Health and Dyslipidaemia. Nat. Rev. Cardiol. 2022, 19, 577–592. [Google Scholar] [CrossRef]
- Régnier, M.; Carbinatti, T.; Parlati, L.; Benhamed, F.; Postic, C. The Role of ChREBP in Carbohydrate Sensing and NAFLD Development. Nat. Rev. Endocrinol. 2023, 19, 336–349. [Google Scholar] [CrossRef]
- Inci, M.K.; Park, S.H.; Helsley, R.N.; Attia, S.L.; Softic, S. Fructose Impairs Fat Oxidation: Implications for the Mechanism of Western Diet-Induced NAFLD. J. Nutr. Biochem. 2023, 114, 109224. [Google Scholar] [CrossRef]
- Olson, E.; Suh, J.H.; Schwarz, J.M.; Noworolski, S.M.; Jones, G.M.; Barber, J.R.; Erkin-Cakmak, A.; Mulligan, K.; Lustig, R.H.; Mietus-Snyder, M. Effects of Isocaloric Fructose Restriction on Ceramide Levels in Children with Obesity and Cardiometabolic Risk: Relation to Hepatic De Novo Lipogenesis and Insulin Sensitivity. Nutrients 2022, 14, 1432. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Noworolski, S.M.; Erkin-Cakmak, A.; Korn, N.J.; Wen, M.J.; Tai, V.W.; Jones, G.M.; Palii, S.P.; Velasco-Alin, M.; Pan, K.; et al. Effects of Dietary Fructose Restriction on Liver Fat, De Novo Lipogenesis, and Insulin Kinetics in Children With Obesity. Gastroenterology 2017, 153, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Sievenpiper, J.L. Fructose: Back to the Future? Am. J. Clin. Nutr. 2017, 106, 439–442. [Google Scholar] [CrossRef]
- Khan, T.A.; Sievenpiper, J.L. Controversies about Sugars: Results from Systematic Reviews and Meta-Analyses on Obesity, Cardiometabolic Disease and Diabetes. Eur. J. Nutr. 2016, 55, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Semnani-Azad, Z.; Khan, T.A.; Blanco Mejia, S.; De Souza, R.J.; Leiter, L.A.; Kendall, C.W.C.; Hanley, A.J.; Sievenpiper, J.L. Association of Major Food Sources of Fructose-Containing Sugars with Incident Metabolic Syndrome: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2020, 3, e209993. [Google Scholar] [CrossRef] [PubMed]
- Choo, V.L.; Viguiliouk, E.; Blanco Mejia, S.; Cozma, A.I.; Khan, T.A.; Ha, V.; Wolever, T.M.S.; Leiter, L.A.; Vuksan, V.; Kendall, C.W.C.; et al. Food Sources of Fructose-Containing Sugars and Glycaemic Control: Systematic Review and Meta-Analysis of Controlled Intervention Studies. BMJ 2018, 363, k4644. [Google Scholar] [CrossRef]
- Velicer, C.; St Helen, G.; Glantz, S.A. Tobacco Papers and Tobacco Industry Ties in Regulatory Toxicology and Pharmacology. J. Public Health Policy 2018, 39, 34–48. [Google Scholar] [CrossRef]
- Nguyen, K.H.; Glantz, S.A.; Palmer, C.N.; Schmidt, L.A. Tobacco Industry Involvement in Children’s Sugary Drinks Market. BMJ 2019, 364, l736. [Google Scholar] [CrossRef]
- Apollonio, D.E.; Glantz, S.A. Marketing with Tobacco Pack Onserts: A Qualitative Analysis of Tobacco Industry Documents. Tob. Control 2019, 28, 274–281. [Google Scholar] [CrossRef]
- Vreman, R.A.; Goodell, A.J.; Rodriguez, L.A.; Porco, T.C.; Lustig, R.H.; Kahn, J.G. Health and Economic Benefits of Reducing Sugar Intake in the USA, Including Effects via Non-Alcoholic Fatty Liver Disease: A Microsimulation Model. BMJ Open 2017, 7, e013543. [Google Scholar] [CrossRef]
- Hieronimus, B.; Griffen, S.C.; Keim, N.L.; Bremer, A.A.; Berglund, L.; Nakajima, K.; Havel, P.J.; Stanhope, K.L. Effects of Fructose or Glucose on Circulating ApoCIII and Triglyceride and Cholesterol Content of Lipoprotein Subfractions in Humans. J. Clin. Med. 2019, 8, 913. [Google Scholar] [CrossRef]
- Hieronimus, B.; Medici, V.; Bremer, A.A.; Lee, V.; Nunez, M.V.; Sigala, D.M.; Keim, N.L.; Havel, P.J.; Stanhope, K.L. Synergistic Effects of Fructose and Glucose on Lipoprotein Risk Factors for Cardiovascular Disease in Young Adults. Metabolism 2020, 112, 154356. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liang, Z.; Ma, J.; Hu, D.; Yao, F.; Qin, P. Total Sugar, Added Sugar, Fructose, and Sucrose Intake and All-Cause, Cardiovascular, and Cancer Mortality: A Systematic Review and Dose-Response Meta-Analysis of Prospective Cohort Studies. Nutrition 2023, 111, 112032. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Choi, H.K.; Lustig, R.H.; Hsu, C. yuan Sugar-Sweetened Beverages, Serum Uric Acid, and Blood Pressure in Adolescents. J. Pediatr. 2009, 154, 807–813. [Google Scholar] [CrossRef]
- Gugliucci, A.; Lustig, R.H.; Caccavello, R.; Erkin-Cakmak, A.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Mulligan, K.; Schwarz, J.M. Short-Term Isocaloric Fructose Restriction Lowers ApoC-III Levels and Yields Less Atherogenic Lipoprotein Profiles in Children with Obesity and Metabolic Syndrome. Atherosclerosis 2016, 253, 171–177. [Google Scholar] [CrossRef]
- Herman, M.A.; Birnbaum, M.J. Molecular Aspects of Fructose Metabolism and Metabolic Disease. Cell Metab. 2021, 33, 2329–2354. [Google Scholar] [CrossRef] [PubMed]
- Sindhunata, D.P.; Meijnikman, A.S.; Gerdes, V.E.A.; Nieuwdorp, M. Dietary Fructose as a Metabolic Risk Factor. Am. J. Physiol. Cell Physiol. 2022, 323, C847–C856. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Karin, M. “Sweet Death”: Fructose as a Metabolic Toxin That Targets the Gut-Liver Axis. Cell Metab. 2021, 33, 2316–2328. [Google Scholar] [CrossRef]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Bremer, A.A.; Berglund, L.; McGahan, J.P.; Havel, P.J.; et al. Consumption of Fructose-Sweetened Beverages for 10 Weeks Reduces Net Fat Oxidation and Energy Expenditure in Overweight/Obese Men and Women. Eur. J. Clin. Nutr. 2012, 66, 201–208. [Google Scholar] [CrossRef]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Bremer, A.A.; Berglund, L.; McGahan, J.P.; Keim, N.L.; et al. Consumption of Fructose- but Not Glucose-Sweetened Beverages for 10 Weeks Increases Circulating Concentrations of Uric Acid, Retinol Binding Protein-4, and Gamma-Glutamyl Transferase Activity in Overweight/Obese Humans. Nutr. Metab. 2012, 9, 68. [Google Scholar] [CrossRef]
- Dechristopher, L.R.; Tucker, K.L. Excess Free Fructose, Apple Juice, High Fructose Corn Syrup and Childhood Asthma Risk—The National Children’s Study. Nutr. J. 2020, 19, 60. [Google Scholar] [CrossRef]
- DeChristopher, L.R.; Uribarri, J.; Tucker, K.L. Intakes of Apple Juice, Fruit Drinks and Soda Are Associated with Prevalent Asthma in US Children Aged 2–9 Years. Public Health Nutr. 2016, 19, 123–130. [Google Scholar] [CrossRef]
- Gugliucci, A.; Bastos, D.H.M.; Schulze, J.; Souza, M.F.F. Caffeic and Chlorogenic Acids in Ilex Paraguariensis Extracts Are the Main Inhibitors of AGE Generation by Methylglyoxal in Model Proteins. Fitoterapia 2009, 80, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Bains, Y.; Gugliucci, A.; Caccavello, R. Advanced Glycation Endproducts Form during Ovalbumin Digestion in the Presence of Fructose: Inhibition by Chlorogenic Acid. Fitoterapia 2017, 120, 1–5. [Google Scholar] [CrossRef]
- Wierzbicki, A.S.; Clarke, R.E.; Viljoen, A.; Mikhailidis, D.P. Triglycerides: A Case for Treatment? Curr. Opin. Cardiol. 2012, 27, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Baratta, F.; Cocomello, N.; Coronati, M.; Ferro, D.; Pastori, D.; Angelico, F.; Ben, M. Del Cholesterol Remnants, Triglyceride-Rich Lipoproteins and Cardiovascular Risk. Int. J. Mol. Sci. 2023, 24, 4268. [Google Scholar] [CrossRef] [PubMed]
- Dallinga-Thie, G.M.; Kroon, J.; Borén, J.; Chapman, M.J. Triglyceride-Rich Lipoproteins and Remnants: Targets for Therapy? Curr. Cardiol. Rep. 2016, 18, 67. [Google Scholar] [CrossRef]
- Gugliucci, A. Triglyceride-Rich Lipoprotein Metabolism: Key Regulators of Their Flux. J. Clin. Med. 2023, 12, 4399. [Google Scholar] [CrossRef]
- Chait, A.; Ginsberg, H.N.; Vaisar, T.; Heinecke, J.W.; Goldberg, I.J.; Bornfeldt, K.E. Remnants of the Triglyceride-Rich Lipoproteins, Diabetes, and Cardiovascular Disease. Diabetes 2020, 69, 508–516. [Google Scholar] [CrossRef]
- Gugliucci, A. Beyond LDL: Understanding Triglyceride-Rich Lipoproteins to Tackle Residual Risk. J. Clin. Med. 2023, 12, 3991. [Google Scholar] [CrossRef]
- Borén, J.; Taskinen, M.R.; Olofsson, S.O.; Levin, M. Ectopic Lipid Storage and Insulin Resistance: A Harmful Relationship. J. Intern. Med. 2013, 274, 25–40. [Google Scholar] [CrossRef]
- Ginsberg, H.N.; Packard, C.J.; Chapman, M.J.; Borén, J.; Aguilar-Salinas, C.A.; Averna, M.; Ference, B.A.; Gaudet, D.; Hegele, R.A.; Kersten, S.; et al. Triglyceride-Rich Lipoproteins and Their Remnants: Metabolic Insights, Role in Atherosclerotic Cardiovascular Disease, and Emerging Therapeutic Strategies-a Consensus Statement from the European Atherosclerosis Society. Eur. Heart J. 2021, 42, 4791–4806. [Google Scholar] [CrossRef]
- Hsieh, J.; Hayashi, A.A.; Webb, J.; Adeli, K. Postprandial Dyslipidemia in Insulin Resistance: Mechanisms and Role of Intestinal Insulin Sensitivity. Atheroscler. Suppl. 2008, 9, 7–13. [Google Scholar] [CrossRef]
- Denechaud, P.D.; Dentin, R.; Girard, J.; Postic, C. Role of ChREBP in Hepatic Steatosis and Insulin Resistance. FEBS Lett. 2008, 582, 68–73. [Google Scholar] [CrossRef]
- Gugliucci, A. “Blinding” of AMP-Dependent Kinase by Methylglyoxal: A Mechanism That Allows Perpetuation of Hepatic Insulin Resistance? Med. Hypotheses 2009, 73, 921–924. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Björnson, E.; Matikainen, N.; Söderlund, S.; Rämö, J.; Ainola, M.M.; Hakkarainen, A.; Sihlbom, C.; Thorsell, A.; Andersson, L.; et al. Postprandial Metabolism of Apolipoproteins B48, B100, C-III, and E in Humans with APOC3 Loss-of-Function Mutations. JCI Insight 2022, 7, e160607. [Google Scholar] [CrossRef]
- Nakajima, K.; Nakano, T.; Tokita, Y.; Nagamine, T.; Inazu, A.; Kobayashi, J.; Mabuchi, H.; Stanhope, K.L.; Havel, P.J.; Okazaki, M.; et al. Postprandial Lipoprotein Metabolism: VLDL vs. Chylomicrons. Clin. Chim. Acta 2011, 412, 1306–1318. [Google Scholar] [CrossRef]
- Adiels, M.; Matikainen, N.; Westerbacka, J.; Söderlund, S.; Larsson, T.; Olofsson, S.O.; Borén, J.; Taskinen, M.R. Postprandial Accumulation of Chylomicrons and Chylomicron Remnants Is Determined by the Clearance Capacity. Atherosclerosis 2012, 222, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Rahmany, S.; Jialal, I. Biochemistry, Chylomicron. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Black, D.D. Development and Physiological Regulation of Intestinal Lipid Absorption. I. Development of Intestinal Lipid Absorption: Cellular Events in Chylomicron Assembly and Secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G519–G524. [Google Scholar] [CrossRef] [PubMed]
- Desmarchelier, C.; Borel, P.; Lairon, D.; Maraninchi, M.; Valéro, R. Effect of Nutrient and Micronutrient Intake on Chylomicron Production and Postprandial Lipemia. Nutrients 2019, 11, 1299. [Google Scholar] [CrossRef]
- Jones, G.M.; Caccavello, R.; Palii, S.P.; Pullinger, C.R.; Kane, J.P.; Mulligan, K.; Gugliucci, A.; Schwarz, J.M. Separation of Postprandial Lipoproteins: Improved Purification of Chylomicrons Using an ApoB100 Immunoaffinity Method. J. Lipid Res. 2020, 61, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Paola Gutiérrez Castro, K.; Patricia González, A.; Caccavello, R.; Garay-Sevilla, M.E.; Gugliucci, A. Lean Adolescents with Insulin Resistance Display Higher Angiopoietin like Protein 3, ApoC-III and Chylomicron Remnant Dyslipidemia. Clin. Chim. Acta 2022, 526, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Kim, K.; Choi, S.H. Lipoprotein Lipase: Is It a Magic Target for the Treatment of Hypertriglyceridemia. Endocrinol. Metab. 2022, 37, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Kristensen, K.K.; Ploug, M.; Winther, A.-M.L. The Importance of Lipoprotein Lipase Regulation in Atherosclerosis. Biomedicines 2021, 9, 782. [Google Scholar] [CrossRef]
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021, 32, 48–61. [Google Scholar] [CrossRef]
- He, P.P.; Jiang, T.; OuYang, X.P.; Liang, Y.Q.; Zou, J.Q.; Wang, Y.; Shen, Q.Q.; Liao, L.; Zheng, X.L. Lipoprotein Lipase: Biosynthesis, Regulatory Factors, and Its Role in Atherosclerosis and Other Diseases. Clin. Chim. Acta 2018, 480, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Chen, Y.Q.; Konrad, R.J. The Regulation of Triacylglycerol Metabolism and Lipoprotein Lipase Activity. Adv. Biol. 2022, 6, e2200093. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; Guo, D.M.; Li, S.Y.; Zeng, Y.X.; Liu, C.H.; Fu, R.; Huang, M.Q.; Xie, W. Triglyceride Metabolism and Angiopoietin-like Proteins in Lipoprotein Lipase Regulation. Clin. Chim. Acta 2020, 503, 19–34. [Google Scholar] [CrossRef]
- Abu-Farha, M.; Ghosh, A.; Al-Khairi, I.; Madiraju, S.R.M.; Abubaker, J.; Prentki, M. The Multi-Faces of Angptl8 in Health and Disease: Novel Functions beyond Lipoprotein Lipase Modulation. Prog. Lipid Res. 2020, 80, 101067. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Mortera, R.; Caccavello, R.; Garay-Sevilla, M.E.; Gugliucci, A. Higher ANGPTL3, ApoC-III, and ApoB48 Dyslipidemia, and Lower Lipoprotein Lipase Concentrations Are Associated with Dysfunctional Visceral Fat in Adolescents with Obesity. Clin. Chim. Acta 2020, 508, 61–68. [Google Scholar] [CrossRef]
- Rhainds, D.; Tardif, J.C. From HDL-Cholesterol to HDL-Function: Cholesterol Efflux Capacity Determinants. Curr. Opin. Lipidol. 2019, 30, 101–107. [Google Scholar] [CrossRef]
- Nicholls, S.J. CETP-Inhibition and HDL-Cholesterol: A Story of CV Risk or CV Benefit, or Both. Clin. Pharmacol. Ther. 2018, 104, 297–300. [Google Scholar] [CrossRef]
- Cho, K.H. The Current Status of Research on High-Density Lipoproteins (HDL): A Paradigm Shift from HDL Quantity to HDL Quality and HDL Functionality. Int. J. Mol. Sci. 2022, 23, 3967. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Fujita, M.; Ikewaki, K. HDL Functions—Current Status and Future Perspectives. Biomolecules 2023, 13, 105. [Google Scholar] [CrossRef] [PubMed]
- Feghaly, J.J.; Mooradian, A.D. The Rise and Fall “Ing” of the HDL Hypothesis. Drugs 2020, 80, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Riggs, K.A.; Rohatgi, A. HDL and Reverse Cholesterol Transport Biomarkers. Methodist Debakey Cardiovasc. J. 2019, 15, 39–46. [Google Scholar] [CrossRef]
- Krauss, R.M.; King, S.M. Remnant Lipoprotein Particles and Cardiovascular Disease Risk. Best Pract. Res. Clin. Endocrinol. Metab. 2022, 37, 101682. [Google Scholar] [CrossRef] [PubMed]
- Zilversmit, D.B. Atherogenic Nature of Triglycerides, Postprandial Lipidemia, and Triglyceride-Rich Remnant, Lipoproteins. Clin. Chem. 1995, 41, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Ishikawa, Y. Remnant Lipoproteins as Strong Key Particles to Atherogenesis. J. Atheroscler. Thromb. 2009, 16, 145–154. [Google Scholar] [CrossRef]
- Wang, K.; Wang, R.; Yang, J.; Liu, X.; Shen, H.; Sun, Y.; Zhou, Y.; Fang, Z.; Ge, H. Remnant Cholesterol and Atherosclerotic Cardiovascular Disease: Metabolism, Mechanism, Evidence, and Treatment. Front. Cardiovasc. Med. 2022, 9, 913869. [Google Scholar] [CrossRef]
- Tappy, L. Metabolism of Sugars: A Window to the Regulation of Glucose and Lipid Homeostasis by Splanchnic Organs. Clin. Nutr. 2021, 40, 1691–1698. [Google Scholar] [CrossRef]
- Jegatheesan, P.; Seyssel, K.; Stefanoni, N.; Rey, V.; Schneiter, P.; Giusti, V.; Lecoultre, V.; Tappy, L. Effects of Gastric Bypass Surgery on Postprandial Gut and Systemic Lipid Handling. Clin. Nutr. ESPEN 2020, 35, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Theytaz, F.; de Giorgi, S.; Hodson, L.; Stefanoni, N.; Rey, V.; Schneiter, P.; Giusti, V.; Tappy, L. Metabolic Fate of Fructose Ingested with and without Glucose in a Mixed Meal. Nutrients 2014, 6, 2632–2649. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Liu, T.; Guo, Q.; Wang, Z.; Chen, Y.; Dong, Y. Disruption of the Intestinal Mucosal Barrier Induced by High Fructose and Restraint Stress Is Regulated by the Intestinal Microbiota and Microbiota Metabolites. Microbiol. Spectr. 2023, 11, e0469822. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.-Y.; Qi, L.-W.; Chen, H.-F.; Gao, P.; Zhang, Q.; Leng, R.-X.; Fan, Y.-G.; Li, B.-Z.; Pan, H.-F.; Ye, D.-Q. The Interaction Between Dietary Fructose and Gut Microbiota in Hyperuricemia and Gout. Front. Nutr. 2022, 9, 890730. [Google Scholar] [CrossRef]
- Drożdż, K.; Nabrdalik, K.; Hajzler, W.; Kwiendacz, H.; Gumprecht, J.; Lip, G.Y.H. Metabolic-Associated Fatty Liver Disease (MAFLD), Diabetes, and Cardiovascular Disease: Associations with Fructose Metabolism and Gut Microbiota. Nutrients 2022, 14, 103. [Google Scholar] [CrossRef]
- Iizuka, K.; Takao, K.; Yabe, D. ChREBP-Mediated Regulation of Lipid Metabolism: Involvement of the Gut Microbiota, Liver, and Adipose Tissue. Front. Endocrinol. 2020, 11, 587189. [Google Scholar] [CrossRef]
- Kanbay, M.; Guler, B.; Ertuglu, L.A.; Dagel, T.; Afsar, B.; Incir, S.; Baygul, A.; Covic, A.; Andres-Hernando, A.; Sánchez-Lozada, L.G.; et al. The Speed of Ingestion of a Sugary Beverage Has an Effect on the Acute Metabolic Response to Fructose. Nutrients 2021, 13, 1916. [Google Scholar] [CrossRef]
- Johnson, R.J.; Wilson, W.L.; Bland, S.T.; Lanaspa, M.A. Fructose and Uric Acid as Drivers of a Hyperactive Foraging Response: A Clue to Behavioral Disorders Associated with Impulsivity or Mania? Evol. Hum. Behav. 2021, 42, 194–203. [Google Scholar] [CrossRef]
- Kanbay, M.; Altıntas, A.; Yavuz, F.; Copur, S.; Sanchez-Lozada, L.G.; Lanaspa, M.A.; Johnson, R.J. Responses to Hypoxia: How Fructose Metabolism and Hypoxia-Inducible Factor-1a Pathways Converge in Health and Disease. Curr. Nutr. Rep. 2023, 12, 181–190. [Google Scholar] [CrossRef]
- Rodriguez-Iturbe, B.; Johnson, R.J.; Lanaspa, M.A.; Nakagawa, T.; Garcia-Arroyo, F.E.; Sanchez-Lozada, L.G. Sirtuin Deficiency and the Adverse Effects of Fructose and Uric Acid Synthesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2022, 322, R347–R359. [Google Scholar] [CrossRef]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A. Fructose Surges Damage Hepatic Adenosyl-Monophosphate-Dependent Kinase and Lead to Increased Lipogenesis and Hepatic Insulin Resistance. Med. Hypotheses 2016, 93, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Mortera, R.; Luevano-Contreras, C.; Solorio-Meza, S.; Caccavello, R.; Bains, Y.; Garay-Sevilla, M.E.; Gugliucci, A. Higher D-Lactate Levels Are Associated with Higher Prevalence of Small Dense Low-Density Lipoprotein in Obese Adolescents. Clin. Chem. Lab. Med. 2018, 56, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A.; Caccavello, R. Optimized Sensitive and Inexpensive Method to Measure D-Lactate as a Surrogate Marker of Methylglyoxal Fluxes in Metabolically Relevant Contexts. Methods 2022, 203, 5–9. [Google Scholar] [CrossRef]
- Geidl-Flueck, B.; Gerber, P.A. Fructose Drives de Novo Lipogenesis Affecting Metabolic Health. J. Endocrinol. 2023, 257, e220270. [Google Scholar] [CrossRef] [PubMed]
- Steenson, S.; Shojaee-Moradie, F.; Whyte, M.B.; Jackson, K.G.; Lovegrove, J.A.; Fielding, B.A.; Margot Umpleby, A. The Effect of Fructose Feeding on Intestinal Triacylglycerol Production and De Novo Fatty Acid Synthesis in Humans. Nutrients 2020, 12, 1781. [Google Scholar] [CrossRef]
- Jung, S.; Bae, H.; Song, W.S.; Jang, C. Dietary Fructose and Fructose-Induced Pathologies. Annu. Rev. Nutr. 2022, 42, 45–66. [Google Scholar] [CrossRef]
- Steenson, S.; Umpleby, A.M.; Lovegrove, J.A.; Jackson, K.G.; Fielding, B.A. Role of the Enterocyte in Fructose-induced Hypertriglyceridaemia. Nutrients 2017, 9, 349. [Google Scholar] [CrossRef]
- Mamo, J.C.L.; Hirano, T.; James, L.; Szeto, L.; Steiner, G. Partial Characterization of the Fructose-Induced Defect in Very-Low-Density Lipoprotein Triglyceride Metabolism. Metabolism 1991, 40, 888–893. [Google Scholar] [CrossRef]
- Saito, H.; Saito, H.; Kato, M.; Yoshida, A.; Naito, M. The Ingestion of a Fructose-Containing Beverage Combined with Fat Cream Exacerbates Postprandial Lipidemia in Young Healthy Women. J. Atheroscler. Thromb. 2015, 22, 85–94. [Google Scholar] [CrossRef]
- DeChristopher, L.R.; Auerbach, B.J.; Tucker, K.L. High Fructose Corn Syrup, Excess-Free-Fructose, and Risk of Coronary Heart Disease among African Americans- the Jackson Heart Study. BMC Nutr. 2020, 6, 70. [Google Scholar] [CrossRef]
- Hieronimus, B.; Stanhope, K.L. Dietary Fructose and Dyslipidemia: New Mechanisms Involving Apolipoprotein CIII. Curr. Opin. Lipidol. 2020, 31, 20–26. [Google Scholar] [CrossRef]
- Price, C.A.; Argueta, D.A.; Medici, V.; Bremer, A.A.; Lee, V.; Nunez, M.V.; Chen, G.X.; Keim, N.L.; Havel, P.J.; Stanhope, K.L.; et al. Plasma Fatty Acid Ethanolamides Are Associated with Postprandial Triglycerides, ApoCIII, and ApoE in Humans Consuming a High-Fructose Corn Syrup-Sweetened Beverage. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E141–E149. [Google Scholar] [CrossRef]
- Brouwers, M.C.G.J. Fructose 1-Phosphate, an Evolutionary Signaling Molecule of Abundancy. Trends Endocrinol. Metab. 2022, 33, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Robichon, C.; Girard, J.; Postic, C. Can the Hyperactivity of Lipogenesis Cause Hepatic Steatosis? A Role for ChREBP. Medecine/Sciences 2008, 24, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Noworolski, S.M.; Wen, M.J.; Dyachenko, A.; Prior, J.L.; Weinberg, M.E.; Herraiz, L.A.; Tai, V.W.; Bergeron, N.; Bersot, T.P.; et al. Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat. J. Clin. Endocrinol. Metab. 2015, 100, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Prieto, P.; Postic, C. Carbohydrate Sensing Through the Transcription Factor ChREBP. Front. Genet. 2019, 10, 472. [Google Scholar] [CrossRef]
- Postic, C.; Dentin, R.; Denechaud, P.D.; Girard, J. ChREBP, a Transcriptional Regulator of Glucose and Lipid Metabolism. Annu. Rev. Nutr. 2007, 27, 179–192. [Google Scholar] [CrossRef]
- Carbinatti, T.; Régnier, M.; Parlati, L.; Benhamed, F.; Postic, C. New Insights into the Inter-Organ Crosstalk Mediated by ChREBP. Front. Endocrinol. 2023, 14, 1095440. [Google Scholar] [CrossRef]
- Iizuka, K.; Horikawa, Y. ChREBP. Nippon. Rinsho. Jpn. J. Clin. Med. 2006, 64 (Suppl. 9), 249–253. [Google Scholar]
- Iizuka, K. Recent Progress on Fructose Metabolism-Chrebp, Fructolysis, and Polyol Pathway. Nutrients 2023, 15, 1778. [Google Scholar] [CrossRef]
- Filhoulaud, G.; Guilmeau, S.; Dentin, R.; Girard, J.; Postic, C. Novel Insights into ChREBP Regulation and Function. Trends Endocrinol. Metab. 2013, 24, 257–268. [Google Scholar] [CrossRef]
- Iizuka, K. Recent Progress on the Role of ChREBP in Glucose and Lipid Metabolism. Endocr. J. 2013, 60, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.S.; Baumel-Alterzon, S.; Scott, D.K.; Herman, M.A. Adaptive and Maladaptive Roles for ChREBP in the Liver and Pancreatic Islets. J. Biol. Chem. 2021, 296, 100623. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Cha, J.Y. Recent Insights into the Role of ChREBP in Intestinal Fructose Absorption and Metabolism. BMB Rep. 2018, 51, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B. The Function of MondoA and ChREBP Nutrient-Sensing Factors in Metabolic Disease. Int. J. Mol. Sci. 2023, 24, 8811. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; So, J.S.; Park, J.G.; Lee, A.H. Transcriptional Control of Hepatic Lipid Metabolism by SREBP and ChREBP. Semin. Liver Dis. 2013, 33, 301–311. [Google Scholar] [CrossRef]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Bremer, A.A.; Berglund, L.; McGahan, J.P.; Keim, N.L.; et al. Circulating Concentrations of Monocyte Chemoattractant Protein-1, Plasminogen Activator Inhibitor-1, and Soluble Leukocyte Adhesion Molecule-1 in Overweight/Obese Men and Women Consuming Fructose- or Glucose-Sweetened Beverages for 10 Weeks. J. Clin. Endocrinol. Metab. 2011, 96, E2034–E2038. [Google Scholar] [CrossRef]
- Harder, N.H.O.; Hieronimus, B.; Stanhope, K.L.; Shibata, N.M.; Lee, V.; Nunez, M.V.; Keim, N.L.; Bremer, A.; Havel, P.J.; Heffern, M.C.; et al. Effects of Dietary Glucose and Fructose on Copper, Iron, and Zinc Metabolism Parameters in Humans. Nutrients 2020, 12, 2581. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Schutz, Y.; Piolino, V.; Schneider, H.; Felber, J.P.; Jequier, E. Thermogenesis in Obese Women: Effect of Fructose vs. Glucose Added to a Meal. Am. J. Physiol. Endocrinol. Metab. 1992, 262, E394–E401. [Google Scholar] [CrossRef]
- Neese, R.A.; Schwarz, J.M.; Faix, D.; Turner, S.; Letscher, A.; Vu, D.; Hellerstein, M.K. Gluconeogenesis and Intrahepatic Triose Phosphate Flux in Response to Fasting or Substrate Loads. Application of the Mass Isotopomer Distribution Analysis Technique with Testing of Assumptions and Potential Problems. J. Biol. Chem. 1995, 270, 14452–14463. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Béliard, S.; D’Erasmo, L.; Bruckert, E. Familial Chylomicronemia Syndrome (FCS): Recent Data on Diagnosis and Treatment. Curr. Atheroscler. Rep. 2020, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Packard, C.J.; Taskinen, M.-R. The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front. Endocrinol. 2020, 11, 474. [Google Scholar] [CrossRef] [PubMed]
- Tramontano, D.; Bini, S.; D’Erasmo, L.; Arca, M. Recent Apolipoprotein CIII Trials. Curr. Opin. Lipidol. 2022, 33, 309–318. [Google Scholar] [CrossRef]
- de la Parra Soto, L.G.; Gutiérrez-Uribe, J.A.; Sharma, A.; Ramírez-Jiménez, A.K. Is Apo-CIII the New Cardiovascular Target? An Analysis of Its Current Clinical and Dietetic Therapies. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 295–308. [Google Scholar] [CrossRef]
- Krauss, R.M. Small Dense Low-Density Lipoprotein Particles: Clinically Relevant? Curr. Opin. Lipidol. 2022, 33, 160–166. [Google Scholar] [CrossRef]
- Sylvers-Davie, K.L.; Davies, B.S.J. Regulation of Lipoprotein Metabolism by ANGPTL3, ANGPTL4, and ANGPTL8. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E493–E508. [Google Scholar] [CrossRef]
- Zhang, R. The ANGPTL3-4-8 Model, a Molecular Mechanism for Triglyceride Trafficking. Open Biol. 2016, 6, 150272. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, K. An Updated ANGPTL3-4-8 Model as a Mechanism of Triglyceride Partitioning between Fat and Oxidative Tissues. Prog. Lipid Res. 2022, 85, 101140. [Google Scholar] [CrossRef]
- Guo, C.; Wang, C.; Deng, X.; He, J.; Yang, L.; Yuan, G. ANGPTL8 in Metabolic Homeostasis: More Friend than Foe? Open Biol. 2021, 11, 210106. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, N.H. New Therapeutic Approaches to the Treatment of Dyslipidemia 1: ApoC-III and ANGPTL3. J. Lipid Atheroscler. 2023, 12, 23–36. [Google Scholar] [CrossRef]
- Zilversmit, D.B. Atherogenesis: A Postprandial Phenomenon. Circulation 1979, 60, 473–485. [Google Scholar] [CrossRef]
- Chong, M.F.F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the Acute Effect of Fructose on Postprandial Lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar] [CrossRef]
- DiDonna, N.M.; Chen, Y.Q.; Konrad, R.J. Angiopoietin-like Proteins and Postprandial Partitioning of Fatty Acids. Curr. Opin. Lipidol. 2022, 33, 39–46. [Google Scholar] [CrossRef]
- Vallejo-Vaz, A.J.; Corral, P.; Schreier, L.; Ray, K.K. Triglycerides and Residual Risk. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 95–103. [Google Scholar] [CrossRef]
- Reith, C.; Armitage, J. Management of Residual Risk after Statin Therapy. Atherosclerosis 2016, 245, 161–170. [Google Scholar] [CrossRef]
- Cho, K.I.; Yu, J.; Hayashi, T.; Han, S.H.; Koh, K.K. Strategies to Overcome Residual Risk during Statins Era. Circ. J. 2019, 83, 1973–1979. [Google Scholar] [CrossRef]
- Geladari, E.; Tsamadia, P.; Vallianou, N.G. ANGPTL3 Inhibitors: Their Role in Cardiovascular Disease through Regulation of Lipid Metabolism. Circ. J. 2019, 83, 267–273. [Google Scholar] [CrossRef]
- Ginsberg, H.N.; Goldberg, I.J. Broadening the Scope of Dyslipidemia Therapy by Targeting APOC3 (Apolipoprotein C3) and ANGPTL3 (Angiopoietin-Like Protein 3). Arterioscler. Thromb. Vasc. Biol. 2023, 43, 388–398. [Google Scholar] [CrossRef]
- Johnson, R.J.; Sautin, Y.Y.; Oliver, W.J.; Roncal, C.; Mu, W.; Gabriela Sanchez-Lozada, L.; Rodriguez-Iturbe, B.; Nakagawa, T.; Benner, S.A. Lessons from Comparative Physiology: Could Uric Acid Represent a Physiologic Alarm Signal Gone Awry in Western Society? J. Comp. Physiol. B 2009, 179, 67–76. [Google Scholar] [CrossRef]
- Kratzer, J.T.; Lanaspa, M.A.; Murphy, M.N.; Cicerchi, C.; Graves, C.L.; Tipton, P.A.; Ortlund, E.A.; Johnson, R.J.; Gaucher, E.A. Evolutionary History and Metabolic Insights of Ancient Mammalian Uricases. Proc. Natl. Acad. Sci. USA 2014, 111, 3763–3768. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gugliucci, A. Sugar and Dyslipidemia: A Double-Hit, Perfect Storm. J. Clin. Med. 2023, 12, 5660. https://doi.org/10.3390/jcm12175660
Gugliucci A. Sugar and Dyslipidemia: A Double-Hit, Perfect Storm. Journal of Clinical Medicine. 2023; 12(17):5660. https://doi.org/10.3390/jcm12175660
Chicago/Turabian StyleGugliucci, Alejandro. 2023. "Sugar and Dyslipidemia: A Double-Hit, Perfect Storm" Journal of Clinical Medicine 12, no. 17: 5660. https://doi.org/10.3390/jcm12175660
APA StyleGugliucci, A. (2023). Sugar and Dyslipidemia: A Double-Hit, Perfect Storm. Journal of Clinical Medicine, 12(17), 5660. https://doi.org/10.3390/jcm12175660